

Abstract
Currently there is great interest in detecting associations between complex traits and rare variants. In this report, we describe Variant Association Tools (VAT) and the VAT pipeline, which implements best practices for rare-variant association studies. Highlights of VAT include variant-site and call-level quality control (QC), summary statistics, phenotype- and genotype-based sample selection, variant annotation, selection of variants for association analysis, and a collection of rare-variant association methods for analyzing qualitative and quantitative traits. The association testing framework for VAT is regression based, which readily allows for flexible construction of association models with multiple covariates and weighting themes based on allele frequencies or predicted functionality. Additionally, pathway analyses, conditional analyses, and analyses of gene-gene and gene-environment interactions can be performed. VAT is capable of rapidly scanning through data by using multi-process computation, adaptive permutation, and simultaneously conducting association analysis via multiple methods. Results are available in text or graphic file formats and additionally can be output to relational databases for further annotation and filtering. An interface to R language also facilitates user implementation of novel association methods. The VAT's data QC and association-analysis pipeline can be applied to sequence, imputed, and genotyping array, e.g., “exome chip,” data, providing a reliable and reproducible computational environment in which to analyze small- to large-scale studies with data from the latest genotyping and sequencing technologies. Application of the VAT pipeline is demonstrated through analysis of data from the 1000 Genomes project.
Main Text
Despite substantial research efforts to identify associations between genetic variations and complex disease, the scope of association studies was previously limited to testing the common disease, common variant hypothesis. Although association analysis of common variants has been highly successful, most of the identified complex-trait associations explain only a small fraction of total heritability. A number of population-based sequencing studies demonstrate the involvement of rare variants in the genetic etiology of complex traits.1–4 To date, there has been great interest in further elucidating the role of rare variants in complex-trait etiology by performing association analysis with data from whole-genome sequencing, exome sequencing, and exome genotyping arrays to test the common disease, rare variant hypothesis. For rare-variant association studies, exome sequencing is currently used more frequently than whole-genome sequencing because it is substantially less expensive, and it allows integration of genomic regions of potentially high functional importance. However, exome sequencing of thousands of samples can still be prohibitively expensive. In order to address this problem, researchers have developed exome genotype arrays (“exome chips”) as an affordable alternative.5,6 Additionally, genome-wide complex-trait association studies that genotyped arrays consisting mainly of common variants are imputing rare variants from resources such as the 1000 Genomes project in order to test for rare-variant associations.
Currently, several large-scale exome sequencing studies, including Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium sequencing project,7 the National Heart, Lung and Blood Institute (NHLBI) Exome Sequencing Project (ESP),8 and the T2D sequencing project,9 are ongoing. These collaborative studies have generated exome data on millions of variant sites for thousands of individuals. Upstream pipelines for sequence alignment and variant calling are well established as a result of years of whole-genome sequencing efforts.10,11 However, downstream association analysis of whole-genome and exome sequence data poses new computational and statistical challenges. There is a necessity for well-designed computational analysis tools that can facilitate quality control (QC) and association analysis of sequence data.
This report describes Variant Association Tools (VAT), a software pipeline for QC and association analysis of sequence, imputed, and genotype array (e.g., “exome chip” array) data. VAT provides a simple and efficient way to handle large data sets consisting of genome, exome, and region-specific variants. It is optimized with a high-performance data-management infrastructure that is scalable for analyzing thousands of samples. VAT facilitates the creation of versatile and efficient association-analysis pipelines for QC, selection and filtering of variant sites, calculation of genotype and sample summary statistics, annotation, and association analysis under a flexible association-testing framework, and it provides a unified interface to most commonly used rare-variant association methods.12–21 Here we describe major features of VAT along with our best-practices data QC and association-analysis pipeline by using sequence data from the 1000 Genomes project (version 3.0, modified April 30th, 2012). The data, in variant-call format (VCF), consist of whole-genome sequence data for 1,092 subjects from 14 populations (Table S1 in the Supplemental Data available with this article online). To demonstrate the VAT pipeline, we focused on single-nucleotide variant (SNV) sites obtained from the exome-capture arrays. All samples are used for initial evaluation of sequence-data properties and QC, and European and Asian samples are used for population-specific analyses.
VAT has an integrated data-management system, powered by variant tools,22 that stores variants, samples, and annotation information in relational databases. With a flexible configuration system, projects are built by extraction of selected information from input files generated by various variant-calling and/or annotation pipelines; file formats include VCF, CASAVA, Complete Genomics, and for genotyping arrays, the PLINK file format.23 It is possible to merge and manage multiple partially overlapping data from different sources, even those using different genomic builds. In addition, a number of project-management operations, including splitting large projects into smaller subsets (e.g., by selection of samples or genomic regions), annotating, and tracking subsets of variants and samples, are provided. The system allows for efficient selection and filtering on variant sites, genotype calls, variant annotations, and sample phenotypes on the basis of user-specified QC criteria.
The VAT QC and association-analysis pipeline is illustrated in Figure 1, and an outline of best practices is provided in Figure 2. Variant-, genotype-, and sample-level QC can flag or remove variant sites, calls, or samples according to various QC metrics that are provided with the sequence data; such metrics include allelic balance, base quality, depth of coverage, and soft QC filters from machine learning algorithms.10 In an analysis of 15,206 genes represented in the 1000 Genomes data, 365,042 single-nucleotide variants (SNVs) and 17,226 indels on exome capture targets passed variant-level quality filters. Additionally, 7,012 SNVs were flagged for having a nonreference allele with frequency of > 50%, and 46,923 SNVs were flagged for having an ancestral allele which is not the reference allele. VAT can also efficiently generate a number of summary statistics that can be used for QC. For example, transition-transversion ratios (Ti/Tv) can be calculated and assessed by empirical rules, e.g., ∼2 for whole-genome variants, ∼2.7 for novel exome variants, and ∼3.4 for known exome variants. Although most sequence data have read-depth information for genotype calls that are typically used for QC, the 1000 Genomes VCF files do not provide this annotation. Of the variants included in the VCF file, 0.34% of the genotype calls are imputed, and 1,793 (0.5%) of the SNV sites have at least one imputed genotype call. As part of QC, imputed genotypes and those variant sites missing more than 10% of their variant calls were removed. The Ti/Tv ratio is 2.72 for novel and 3.44 for known exome variant sites before QC and is 2.76 and 3.50 after QC, where novel variants are those submitted to dbSNP only by 1000 Genomes. By comparing the Ti/Tv ratios before and after QC procedures, researchers can establish protocols to properly clean variant sites. If duplicate samples are available, researchers can calculate the genotype concordance rate before and after applying different quality filters to determine the procedure that maximizes the concordance. However, caution should be used because stringent thresholds that maximize Ti/Tv ratios and concordance between duplicate pairs can remove many true-positive variant sites and/or genotype calls, and this can adversely impact the power of VAT to detect associations. Other useful summary statistics that VAT provides for QC include missing genotype call rates, homozygous and heterozygous genotype counts, allele or genotype frequencies, synonymous/nonsynonymous ratios (S/NS), total and average depth of coverage, and statistics on genotype properties such as minimum, maximum, and mean genotype-quality scores.
Figure 1.
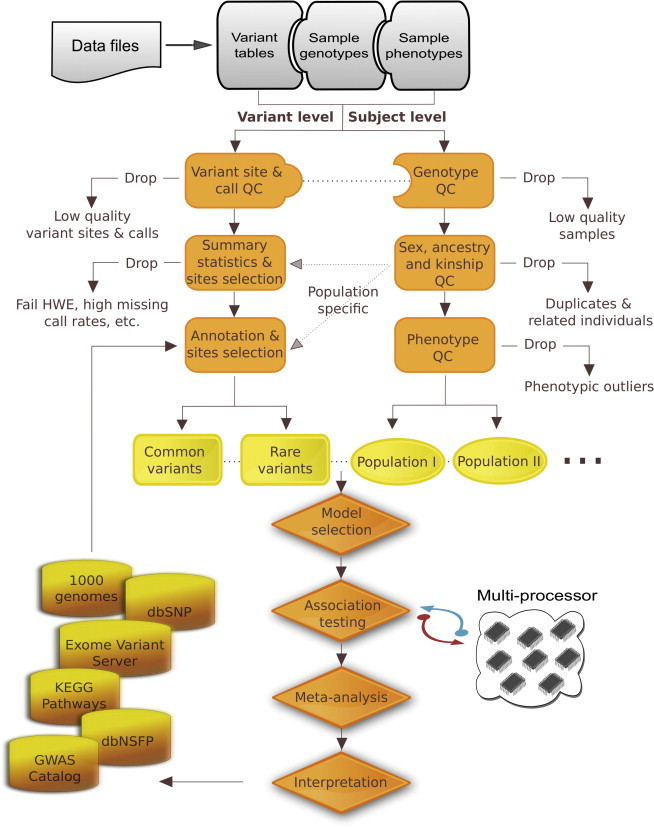
Variant Association Tools Pipeline for Quality Control and Association Analysis.
Figure 2.
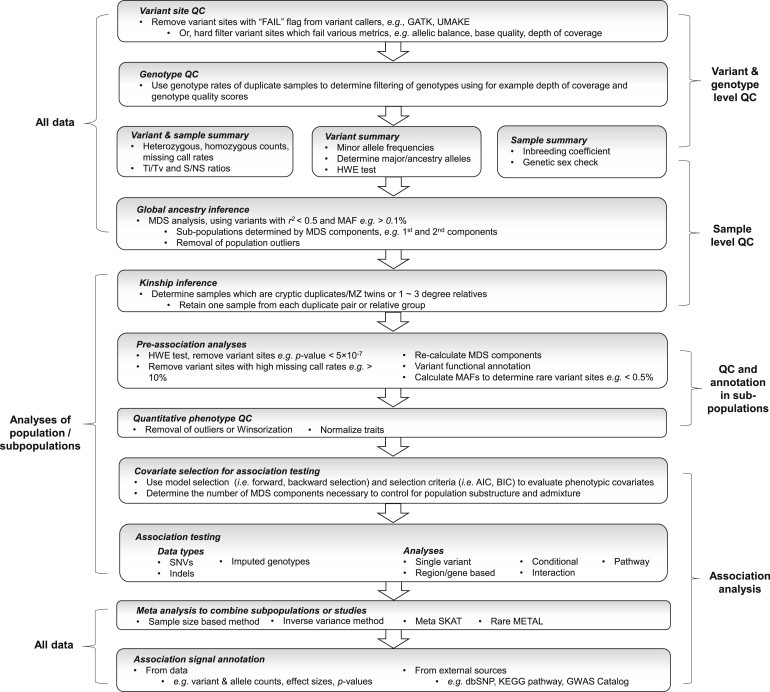
Best Practices for Implementing the Variant Association Tools Pipeline.
Calculations of QC summary statistics can be flexible and creative, allowing for construction of customized queries for specific needs. Summary statistics can be evaluated at variant or sample levels, so that it is possible to condition on other variant, genotype, or sample attributes. For example, for the 1000 Genomes data, missing genotype call rates were calculated per variant site (mean 0.55%, SD 2.7%, max 9.1%, min 0.09%, and median 0.18%) and per sample (mean 0.34%, stdev 0.41%, max 3.35%, min 0.09%, and median 0.21%). Ti/Tv and S/NS ratios can be obtained for all variant sites (Ti/Tv 3.22, S/NS 0.73) or for variants belonging to a specific individual or individuals (Ti/Tv: mean 3.31, SD 0.045, max 3.45, min 3.16, and median 3.31; S/NS mean 1.29, SD 0.015, max 1.35, min 1.24, and median 1.29). Researchers can use information on sample-level missing data and Ti/Tv ratios to determine whether the variant calls of particular samples are of low quality. With the use of more than a dozen built-in annotation databases for exome and whole-genome variants, additional statistics can be generated to aid in a better understanding of the data or can be used for QC procedures.
The VAT association-analysis pipeline provides population-ancestry analysis and detection of related individuals for QC purposes. Instead of relying on self-reported ancestry, analyses should assess genetic ancestry from available genotype data. VAT incorporates multidimensional scaling (MDS) to perform population-structure inference, and it allows for redesignation of ancestry or removal of individuals from analysis. We performed global ancestry and kinship inference for individuals of European (N = 379) and Asian (N = 286) ancestry by using SNVs that had a minor-allele frequency (MAF) of >5% and that were not in intermarker linkage disequilibrium (LD) (r2 < 0.5). An MAF cut-off of 5% was selected because of the small sample size; for larger samples, a lower MAF cut-off, e.g., 0.1%, should be used. Figure 3 displays results from the MDS analysis; the first and second components are plotted. It can be observed that the European and the Asian populations are clearly separated (Figure 3A). When the MDS components for only Europeans are plotted, it is observed that the Finnish (FIN) cluster is separated from the other European groups, and there is the greatest overlap between the Utah residents with Northern and Western European ancestry (CEU) and the British (GBR) (Figure 3B). It is also observed for Asians that the two Chinese populations (CHB and CHS) cluster separately from the Japanese (JPT) (Figure 3C). These observations are expected on the basis of the population histories. However, for both Europeans and Asians we observe subclusters within each population (Figures 3B and 3C). We suspected that such a pattern might have been due to batch effects because sequencing of 1000 Genomes data was performed on three platforms (Illumina, ABI Solid, and LS454). Therefore, we performed MDS analysis with this information and found that, indeed, the subclusters could be attributed to the use of different sequencing platforms (data not shown).
Figure 3.
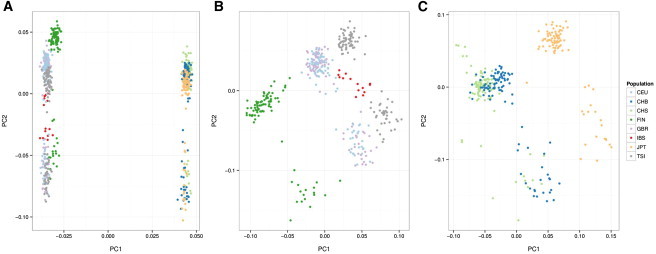
Global Ancestry Inference
Ancestry inference was performed using MDS analysis with graphic presentation of projection of samples to the first two MDS components. Color codes represent the region of collection or self-reported ancestry. MDS analysis was performed for all European and Asian samples (A); European samples (B); and Asian samples (C).
Within each population group, one can perform kinship analysis to identify and remove cryptically related and duplicate samples. VAT incorporates a robust pair-wise relationship inference algorithm to estimate kinship coefficients and output pairs of samples that are duplicate or MZ twins or first-degree, second-degree, or third-degree relatives.24 In practice, only one sample from each duplicate pair or relative group should be retained in association analysis unless empirical p values for association tests are obtained or mixed models are used. Subjects to be retained in the analysis should be determined by availability of phenotype data and quality of sequence data. Kinship inference indicates that, of the 379 DNA samples from Europeans, there were four cryptically related individuals, whereas of the 286 individuals from Asian populations, one individual is a child of a trio and eight individuals are cryptically related. It should be noted that performing kinship inference requires the use of caution because subpopulations can cause individuals to appear to be closely related, e.g., third-degree relatives who in reality are either more distantly related or unrelated. We therefore performed kinship inference separately not only for Asians and Europeans but also for each subpopulation listed in Table S1 and also for each sequencing platform (data not shown). The 375 unrelated European and 277 unrelated Asian exomes were used for further analysis.
Researchers can determine whether the reported sex is consistent with genetic data by examining the heterozygosity of markers on the X chromosome and presence of Y chromosomal variants. Inconsistencies are often due to sample swaps, but they can also occur for unreported cases of Turner or Klinefelter’s syndrome. In the situation where it is believed that inconsistencies are due to sample swaps, the samples in question should be removed because other phenotypic data might also be inconsistent. In the 1000 Genomes data, no inconsistencies between reported and genomic sex were detected.
It is advisable to remove those sites that deviate from Hardy-Weinberg equilibrium (HWE) because the deviation can be caused by genotyping error.25 VAT provides an efficient exact test of HWE.26 For case-control data, only controls should be tested for deviations from HWE because for cases, sites associated with disease status can deviate from HWE. Additionally, because population substructure can cause deviations from HWE, if more than one population is analyzed, each population should be tested separately. For the 1000 Genomes data, 345 SNV sites for Europeans and 340 SNV sites for Asians deviated from HWE (p value < 5 × 10−7) and were removed from further analysis.27
The remaining 170,245 SNV sites (106,920 European and 96,593 Asian SNV sites) were annotated with the built-in ANNOVAR pipeline. A number of summary statistics, including variant counts categorized by functional type and MAF, were computed for the European and Asian exome data. For European and Asian samples combined, Figure 4A displays the number and proportion of synonymous, missense, and nonsense variant sites, and Figure 4B displays the number and proportion of variant sites by MAF for synonymous, missense, and nonsense variants. It can be observed that the greatest number of variant sites are missense and that the least number are nonsense (Figures 4A and 4B). For synonymous, missense, and nonsense variants, most variant sites have frequencies of <1%, and the majority are singletons. The greatest proportion of singletons can be found in nonsense variants (67.5%), followed by missense (52.6%) and synonymous (42.2%) variants (Figure 4B). Figure 4C displays the average number of variant sites per individual for Europeans, Asians, and both. Asians have on average 18,583 variant sites per individual, which is greater than the 17,883 variant sites for Europeans (two-sample t test, p value = 1.06 × 10−67). The majority of per-individual variant sites are synonymous (Asian [56.1%], European [56.1%]), followed by missense (Asian [43.6%], European [43.6%]) and then nonsense (Asian [0.3%], European [0.3%]). Figure 4D shows the average number of singletons per individual. Although there are more singletons for Asians (N = 163 ± 33) than Europeans (N = 112 ± 35), their proportions by functional type are approximately the same for the two populations. Additionally, it is possible to investigate at the variant level which sites are unique to a population or shared between populations. Table 1 summarizes the shared and unique exonic variant sites between Europeans and Asians. For variant sites unique to the two populations, the proportion of singleton sites is greater in Asians than in Europeans for all functional types (p values < 2 × 10−16). Proportions of shared variants are significantly different between functional types (p values < 2 × 10−16). Of the 33,268 exonic SNVs shared by the two populations, 33.6% of the variant sites show a significant difference in MAF (p < 1.5 × 10−6, which is a p value of 0.05, for which a Bonferroni correction is performed for testing ∼33,000 variant sites). The results are obtained with a Fisher’s exact test provided in VAT, which can also be used to evaluate simple hypotheses such as those regarding the difference in MAF between groups of samples and heterozygote excess and deficiency.
Figure 4.
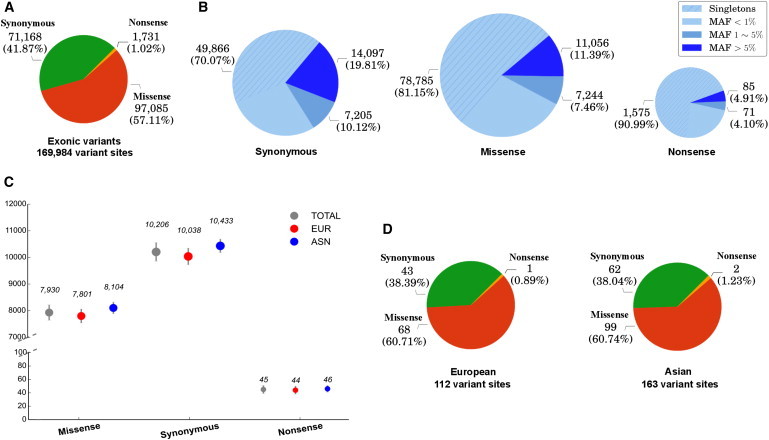
Variant- and Sample-Level Summary Statistics for European and Asian Samples by Variant Functional Type: Missense, Synonymous, and Nonsense
Variant-level statistics are displayed as the number and percentage of variant sites by functional type (A) and the frequency of variant sites by functional type (B) for Europeans and Asians combined. Sample-level statistics are displayed as average number of variants per individual by functional type for Europeans, Asians, and the two populations combined (C) and the average number of singletons per individual by functional type for European and Asians separately (D). (C) Error bars represent standard error of the mean.
Table 1.
Shared and Population-Specific Variant Sites for Europeans and Asians
|
European Specific |
Asian Specific |
Europeans and Asians Shared | Proportion of Shared Variants | |||
|---|---|---|---|---|---|---|
| Singletons | Non-singletons | Singletons | Non-singletons | |||
| All exonic variants | 33,725 | 39,927 | 38,688 | 24,637 | 33,268 | 0.19 |
| Synonymous | 11,704 | 17,255 | 13,882 | 10,380 | 17,947 | 0.25 |
| Missense | 21,416 | 22,311 | 24,203 | 14,034 | 15,121 | 0.15 |
| Nonsense | 546 | 302 | 540 | 200 | 143 | 0.08 |
VAT can also perform QC of the phenotypic data. Individuals to be included or excluded from analysis can be selected on the basis of information from multiple phenotypes, e.g., for the study of hypertension, controls with evaluated systolic and/or diastolic blood pressure can be excluded. If covariates are to be included in the analysis, missing covariate values can be replaced with the mean covariate value so that individuals with missing values do not need to be removed from the analysis. Quantitative phenotypes should be examined for outliers. If outliers are present, Winsorization should be applied unless the outliers are deemed unreliable, in which case they should be removed. For association analysis of quantitative traits, many tests require trait values to be normally distributed. Therefore, VAT can perform square root, logarithmic, and quantile normal (also called rank-based inverse normal) transformations. VAT can generate graphic summaries of phenotypic data to allow investigation of the normality of quantitative traits, outliers, and variability between samples, e.g., phenotype distributions in different study groups. In Figure 5A the left panel displays the simulated body mass index (BMI) phenotypes for Europeans; we generated phenotype data separately for Europeans and Asians by first simulating height and weight values under normal distribution and then calculating BMI with the equation BMI = Weight/Height2. The center panel displays the BMI phenotype data after log10 transformation, and the right panel displays the results after quantile normalization.
Figure 5.
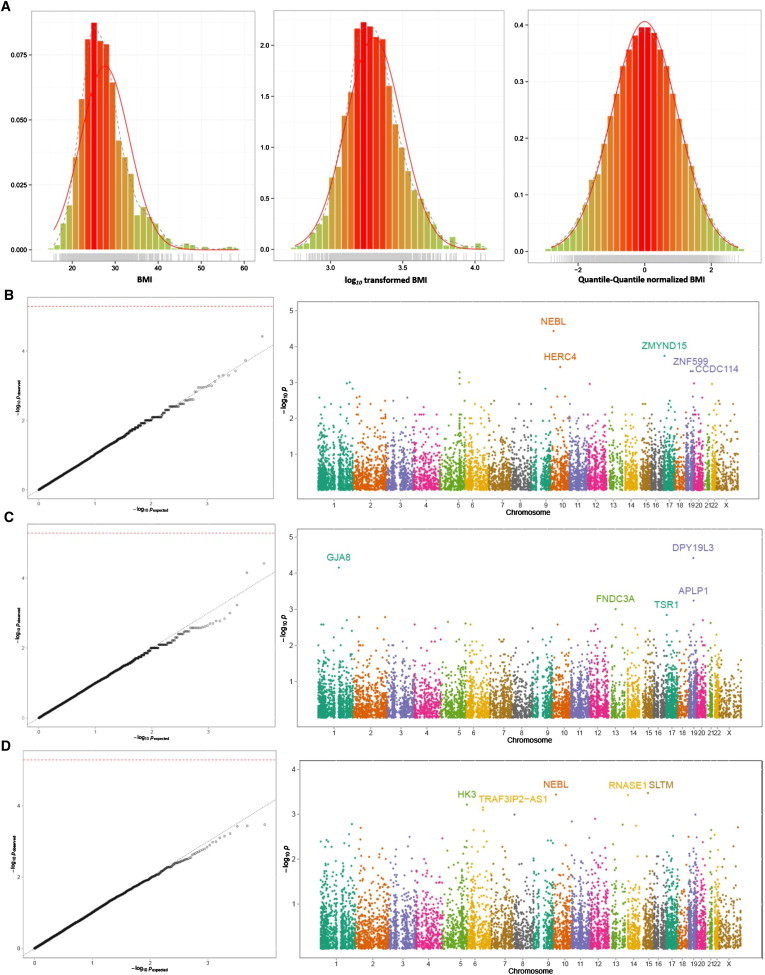
Distribution of BMI and Rare-Variant Association Analysis via the CMC Method
(A) The simulated BMI values for Europeans (left), BMI values after log10 transformation (center), and quantile normal transformation (right). Analysis of whole-exome association was performed for the quantile-normal-transformed BMI phenotype via the CMC method. For panels (B) to (D), results are represented by p values at the −log10 scale and displayed in quantile-quantile (QQ) (left) and Manhattan (right) plots. Analysis of Europeans (B) and Asians (C) and meta-analysis of European and Asian results (D) was performed via the sample-size-based method.
Association testing between individual variants and complex traits is standard practice in genome-wide association studies of common variants, and VAT can perform single-variant association analysis. However, it is well established that single-variant tests are underpowered to detect rare-variant associations. Instead, rare-variant association analysis aggregates variants within a region, which is usually a gene, to perform association tests. A number of aggregation approaches have been developed to exploit statistical information from genetic regions where multiple rare variants are harbored.12–21 Such analysis typically focuses on variants that are most likely to be functional, e.g., such variants include missense, nonsense, and splicing sites. Additionally, researchers should apply an MAF threshold to determine which variants to include in aggregated analysis. Aggregation of noncausal, higher-frequency variants can greatly increase type II error. Also, it is often of interest to detect association signals solely from rare variants rather than from a mixture of rare and common variants. The definition of “rare” is arbitrary, although an MAF of <0.5% or <1% is commonly used. These cut-offs are typically applied to rare-variant association methods by use of a fixed MAF threshold. However, for association methods that (1) compute optimized statistics over subsets of variants,15,18 (2) weight variants by frequency, where higher-frequency variants are down-weighted,13 or (3) are robust to noncausal variants,28 a higher MAF cut-off, e.g., 5%, can be used if desired.
During rare-variant association tests there must be at least two variants present within the tested region. However, it is often desirable to use more stringent criteria, e.g., a minimum of three variants, and/or to require that regions have a minimum cumulative MAF, e.g., 0.5%. Using these criteria ensures that regions where there is insufficient power to detect an association are not tested and thus reduces the number of tests that need to be performed. Usually, for exome analysis a family-wise type I error of 0.05 implies a significance level of 2.5 × 10−6 per test, after Bonferroni correction, for an analysis of 20,000 genes. The per-test significance level can be less stringent if fewer tests are performed.
It has been demonstrated that the relative power of rare-variant association tests depends greatly on the allelic architecture. Of the many rare-variant association tests that have been developed, no one method is uniformly the most powerful, and methods tend to be more powerful if their assumptions closely match those of the underlying genetic etiology.29–31 Implementation of some rare-variant methods are available as R packages,14,16 standalone programs,32 or commercial software, e.g., Golden Helix, but these implementations focus on a small collection of methods and lack a complete analysis pipeline or can only handle a very limited number of samples, both of which limit their usage. Additionally, as a result of the large size of association data consisting of thousands of samples and the fact that many methods depend on permutation to estimate valid p values, scalability and efficiency are crucial to high-quality computational tools for rare-variant association analysis. VAT offers a comprehensive collection of rare-variant analysis methods, including combined multivariate and collapsing (CMC),12 weighted-sum statistic (WSS),13 kernel-based adaptive cluster (KBAC),14 variable threshold (VT),15 RareCover,18 gene- or region-based analysis of variants of intermediate and low frequency (GRANVIL),19 burden of rare variants (BRV),21 adaptive sum test (AST),20 C-alpha,17 replication-based test (RBT),33 sequence kernel association test (SKAT),16 and estimated regression coefficients (EREC).32 In addition to implementing published association methods, we have made a number of optimizations and extensions to improve the power and computational efficiency of existing methods. For example, to reduce analysis time, we developed a “p value aware” VT procedure, which calculates analytical p values pi for each MAF that is evaluated. The minimum of pi, pm = min({pi}) gives an estimate of the smallest possible p value that can be obtained. If pm is larger than a given threshold, e.g., pm > 0.05, a permutation-based p value will not be obtained because the test is nonsignificant. For the AST method, instead of fitting the computationally intensive multivariate logistic regression, VAT uses a Fisher’s exact test on each variant site to determine potentially protective variants. More details on implementation and modifications of rare-variant association methods can be found in the VAT online documentation. Caution is advised during the application of different association tests because a multiple-testing penalty reduces rather than increases power. Although a Bonferroni correction could be applied, it is overly conservative because the results from rare association tests can be correlated. To correct for performing multiple rare-variant association tests, VAT provides a resampling-based p value adjustment for this particular multiple-testing problem (see Appendix A).
It has been demonstrated that the type I error for rare-variant association methods can be increased if there are differential missing rates between case and control genotype data.21 This same increase in type I error can occur for quantitative traits if there is more missing data for those individuals with either high or low quantitative-trait values. To control type I error, VAT replaces missing genotypes by a dosage that is based on the observed allele frequencies for that variant site.
All rare-variant association methods, with the exception of C-alpha, are incorporated in a regression framework, allowing covariates to be included in the analysis so that confounding is controlled for and increased type I and II errors are avoided. Covariates that might be potential confounders include age, sex, and population ancestry information provided as MDS components. Researchers can apply model selection procedures to determine which covariates should be included in the association analysis model, e.g., researchers can apply step-wise model selection algorithms by using the Akaike information criterion (AIC) or Bayes information criterion (BIC).34 Additionally, the genomic control λ can be used for evaluation of the number of MDS components to be included in association analysis.35,36
For analysis of different populations, e.g., Asians and Europeans, it is not appropriate to analyze all samples together even if MDS components are used to control for population substructure. Instead, each population should be analyzed separately, and the results should be combined via meta-analysis. Popular meta-analysis methods include the sample-size-based method or the inverse variance method,37 Meta-SKAT,38 and Rare METAL.39
In the analysis of 1000 Genomes data, association tests were performed for the quantile-normalized BMI values for European and Asian samples separately. It should be noted that the BMI phenotype data were generated under the null hypothesis without associations to specific genes. We controlled for population substructure by including two MDS components, estimated separately in Europeans and Asians, in the linear regression model. Sex was also included as a covariate in the analysis. For the simulated BMI phenotype, association analyses were performed with the CMC (Figure 5), BRV (Figure S1), VT (Figure S2), WSS (Figure S3), and SKAT (Figure S4) methods. Analyses were performed separately for Europeans and Asians and then combined via meta-analysis. All p values were assessed empirically with the exception of SKAT, where p values were obtained analytically because of the computational burden of this method. Only missense and nonsense variants with a MAF of <1% were analyzed, with the exception of those variants analyzed with the VT methods, where a cut-off of MAF < 5% was used. Only those genes with more than three variant sites (8,933 genes for Europeans, 8,495 genes for Asians) were analyzed. Although no significant associations with the simulated phenotype were found, as expected in light of the fact that the phenotype data were generated under the null hypothesis, the results can be used for comparing rare-variant association methods. Table 2 displays the comparison of the five association methods by the top association signals from each method in European data. Of the 28 gene associations listed, none is detected by all of the five methods. More than half (N = 16) of the genes are only detected by one method. Detection consistency is higher among fixed-threshold burden tests (CMC, BRV, and WSS) than in the variable threshold test (VT) and is substantially lower between burden tests and variance component test (SKAT). Analysis of the Asian data produced similar results (data not shown). In addition to displaying association results as p values, VAT association analysis provides test-region-specific statistics such as variants and allele counts, cumulative MAF, number of samples analyzed per region, missing-data rates, association test statistics, effect size values (β values), and standard errors. VAT performs meta-analysis across different studies or ethnic groups by combining p values via the sample-size based and inverse-variance methods37 (Figure 5D; Figures S1C, S2C, and S3C), as well as the Meta-SKAT method38 (Figure S4C). Results of both meta-analysis and study-specific analysis are stored and managed within the project database system, making it easy to access specific association results, e.g., results with p values below certain thresholds, and also to readily select and compare results between association studies. Annotation of association signals is provided through external databases: A catalog of published genome-wide association studies40 is used for annotating association signals that were previously detected in genome-wide association studies; Catalogue of somatic mutations in cancer (COSMIC)41 is used for annotating genes that were reported in cancer studies to have somatic mutations; and the Kyoto encyclopedia of genes and genomes (KEGG)42 database is used for determining whether detected association signals are involved in molecular interaction or reaction network. Users can also upload other external databases of interest in order to provide additional annotations.
Table 2.
Detection of Association Signals with Five Rare-Variant-Association Methods for European Data
| Association Methods | CMC | BRV | VT | WSS | SKAT |
|---|---|---|---|---|---|
| Association Detection Consistency | |||||
| CMC | 100% | 80% | 30% | 50% | 30% |
| BRV | 80% | 100% | 30% | 60% | 20% |
| VT | 30% | 30% | 100% | 40% | 10% |
| WSS | 50% | 60% | 40% | 100% | 0% |
| SKAT | 30% | 20% | 10% | 0% | 100% |
| Top Ten Gene Associations Detected by Each Methoda | |||||
| STON1-GTF2A1L | 1.13 × 10−4 | 1.05 × 10−4 | 7.14 × 10−4 | 9.20 × 10−5 | - |
| ST8SIA5 | 7.14 × 10−4 | 2.26 × 10−4 | 4.76 × 10−4 | 2.11 × 10−4 | - |
| ZFP30 | 3.08 × 10−4 | 3.00 × 10−4 | 3.75 × 10−4 | - | 2.27 × 10−3 |
| RASIP1 | 4.55 × 10−4 | 4.55 × 10−4 | - | - | 1.84 × 10−3 |
| NEBL | 4.20 × 10−5 | 3.80 × 10−5 | - | 1.11 × 10−4 | - |
| RBFA | 6.25 × 10−4 | 3.04 × 10−4 | - | 8.33 × 10−4 | - |
| MTF1 | 3.75 × 10−4 | 6.67 × 10−4 | - | 5.00 × 10−4 | - |
| SLC5A1 | 6.67 × 10−4 | 4.76 × 10−4 | - | - | - |
| COL19A1 | - | - | 6.60 × 10−5 | 4.55 × 10−4 | - |
| ALDH4A1 | - | 5.00 × 10−4 | - | 2.69 × 10−4 | - |
| NXNL1 | 1.11 × 10−3 | - | - | - | 8.66 × 10−4 |
| BTBD3 | - | - | 1.65 × 10−4 | 8.33 × 10−4 | - |
| MICALL2 | - | - | - | - | 1.16 × 10−3 |
| SDCCAG8 | - | - | - | - | 1.87 × 10−3 |
| MGST2 | - | - | - | - | 1.84 × 10−3 |
| SLC25A18 | - | 1.00 × 10−3 | - | - | - |
| DNAH10 | 1.00 × 10−3 | - | - | - | - |
| NLRP7 | - | - | - | - | 2.45 × 10−3 |
| FAM167A | - | - | 8.33 × 10−4 | - | - |
| TMCC2 | - | - | - | 5.00 × 10−4 | - |
| ZNF263 | - | - | 8.63 × 10−5 | - | - |
| PCDHA13 | - | - | 7.14 × 10−4 | - | - |
| NAT10 | - | - | 2.20 × 10−5 | - | - |
| PSRC1 | - | - | - | - | 1.60 × 10−3 |
| AARS | - | - | - | - | 4.27 × 10−4 |
| C8orf12 | - | - | - | 6.67 × 10−4 | - |
| MTRR | - | - | - | - | 2.23 × 10−3 |
| HCN3 | - | - | 3.75 × 10−4 | - | - |
Total = 28 genes.
Because every association method has its strengths and weaknesses, it is appealing to design a mechanism that takes advantage of different association strategies and can incorporate as much information as possible. Motivated by the fact that most burden tests for rare-variant association analysis differ only in genotype coding and weighting, we developed the VAT stacking algorithm as a unified association analysis framework. VAT stacking is a regression-based algorithm that applies likelihood-ratio statistical inference to test for associations between (1) qualitative and/or quantitative traits and (2) genotypic numeric coding themes, which can incorporate aggregation and weighting based on MAF or functional annotation as well as genotype-specific weights. Commonly used external weights are based on protein-function prediction tools such as PolyPhen2,43 SIFT,44 GREP,45 and CADD,46 which are available from built-in annotation databases. The VAT stacking algorithm is outlined in Appendix A. Association testing via VAT stacking with 1000 Genomes data was performed under the same settings as those previously described, and results are shown for analyses incorporating KBAC weights and KBAC weights stacked on the VT algorithm (Figure S5).
Researchers can use VAT to perform pathway analysis by collapsing selected variants from multiple genes within a pathway into one unit or by collapsing variants within each gene into separate groups and performing multivariate analysis. Annotations for pathway analysis from several databases, including KEGG, are provided.42 One can also use the regression-based framework to test for the presence of gene-gene or gene-environment interactions. For interaction analysis, one must specify the null hypothesis in order to test for interactions or to jointly test for both main effects and interactions. To determine whether an association signal is being driven solely by a gene of interest or is due to linkage disequilibrium between variants within or outside the gene, one can perform conditional association analysis with respect to other genes or individual variants.
Association analysis of indels can also be performed with VAT. Indels are first annotated, and potentially functional indels are then analyzed via rare-variant association methods through aggregation of either indels or both indel and SNVs within a region.
When analyzing whole-genome sequence data, in addition to analyzing coding regions, one can also analyze variants within predicted functional regions, e.g., enhancer regions, transcription-factor sites, or DNase-I-hypersensitivity sites, by using rare-variant association methods. Annotations for indels and noncoding variants are available from the built-in ANNOVAR47 annotation pipeline, which uses RefSeq48 and ENCODE data.
Imputation is commonly performed with genome sequencing data, e.g., 1000 Genomes. After imputation of rare variants with readily available software such as MaCH,49 Beagle,50 or IMPUTE2,51 VAT can also be used for analysis of imputed variants. A variety of information measures with values lying in the range 0–1 can help to determine which imputed variant sites are of good quality.52 Take, for example, the R2 generated by MaCH. Generally, only those variant sites with a high R2 value, e.g., ≥0.8, are analyzed so that the variant calls are of high quality. R2 can be relaxed so that a greater number of variants within a region are included in the analysis, although caution should be used because poorly imputed SNVs can increase both type I and II errors. For imputed data, a dosage that takes into account the probabilities of each of the possible three genotypes is analyzed.53 Imputed variants can be analyzed individually, but it is also possible to analyze these dosages by using aggregate association methods such as BRV, WSS, and VT. Additionally, a variety of weighting schemes in VAT stacking can be applied during analysis of imputed variants.
Implementation of association methods in VAT is highly optimized for computational efficiency and scalability. Researchers can apply parallel computation to analyze exomes or genomes by using either one or many different rare-variant association methods. Flexible permutation routines are available for efficient evaluation of empirical p values. It is possible to permute either the phenotype or genotype predictors for conditional association analysis. Additionally, two techniques can boost the efficiency of the permutation routine: (1) “adaptive” permutation, in which the use of fewer permutations allows researchers to obtain p value estimates for nonsignificant results and (2) “timeout” permutation, which ceases when the specified time limit per test expires; intermediate p values are reported with a flag so that analysis can be resumed after the entire association scan is complete. These permutation techniques are particularly useful in situations where access to high-performance computing resources is limited. With these optimizations in action, applying the CMC method to perform association analysis for 15,206 genes of 1,092 individuals takes around half an hour to provide empirical p values via permutation on a computer with AMD Opteron 6220 (16 threads at 3.0GHz) CPU and WD Black (7200 RPM) hard drive.
The VAT association pipeline can be further customized through the implementation of user-provided association methods written in the R language. Researchers can incorporate novel association methods in the VAT pipeline and thus take advantage of various features such as the ability to handle various input formats, e.g., VCF files, annotation of variant sites, parallel processing, timeout permutation, etc. Using the R interface in VAT, researchers can quickly convert new association methods into a computational tool that is readily applicable to real-world data.
In summary, VAT is a user-friendly, all-in-one software pipeline package for rare-variant association analysis. The data-management system makes it possible for researchers to constantly update projects as new information is generated or imported, preventing generation of numerous intermediate text files during analysis. With a collection of generic and versatile commands that select, update, and execute various queries on variants, genotypes, and samples, VAT allows researchers to personalize their analysis without having to write numerous scripts. Most importantly, in addition to being a powerful tool for data QC and exploration, the association testing framework in VAT is the most comprehensive, flexible, and extensible suite to date. The VAT pipeline provides a standard protocol for association analysis of sequence or genotyping array data via an elegant computational interface and data management that aids in reproducibility. The VAT package, documentation, tutorial, and data resources are publicly available online (see Web Resources).
Acknowledgments
We would like to thank Mengyuan Kan, Kristine Bucasas, Biao Li, and Regie Santos-Cortez for their useful suggestions. This work was supported by National Institutes of Health grants HL102926 (to S.M.L), MD005964 (to S.M.L), HG006493 (to S.M.L) and 1R01HG005859 (to B.P). Additional support for this work was provided by the Lyda Hill Foundation (to B.P.).
Appendix A
The VAT Stacking Algorithm
We adapted CMC, WSS, GRANVIL, BRV, VT, KBAC, and RBT methods to a generalized regression model , where XL represents sample genotype information and Z represents covariates. Let XL be the generic coding for one sample across a genetic region L and Gi be the genotype value of locus i (Gi ∈ {0,1,2} or Gi ∈ {0,1} under a dominant or recessive mode of inheritance, respectively). Coding for CMC and BRV are straightforward. The original form of the VT statistic is implemented as a least-squares estimate maximized over all minor-allele frequencies , which with equivalent VAT stacking notation, is used for computation of and maximization of the test statistic over all possible MAFs. One can adapt weighting for WSS, KBAC, and RBT to analyze either case-control data or quantitative traits. Weighting for the WSS involves calculation of variant-site-specific weights on the basis of allele frequencies, and individual genotype scores can be represented as the weighted sum of multilocus genotype values, , where the weight is based on the entire sample or a subset of the sample, e.g., on controls. Similarly, the RBT weights can be directly applied to the equation where .33 For the KBAC test, the genotype can be coded as , where the probability is defined by a hypergeometric kernel . Additionally, variant functional information from the VAT annotation pipeline is incorporated in VAT stacking as additional possible weights on the generic genotype matrix, yielding to replace the original genotype coding.
VAT stacking uses a resampling-based method for p value adjustment when multiple tests are applied to the same genetic region. For every tth permutation under the VAT stacking framework, the score statistic from each association test involved is compared; the statistic that implies the strongest evidence of association is kept [ for a one-sided test and for a two-sided test]. From the original data set, zm is also obtained. The adjusted p value is the number of permutations when is greater than zm divided by the total number of permutations. The additional computational burden involved in obtaining p values adjusted for multiple testing is negligible because their calculations are performed in parallel with estimation of p values for each association test.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
A catalog of published genome-wide association studies, http://www.genome.gov/gwastudies/index.cfm?pmid=20190752
Catalogue of somatic mutations in cancer (COSMIC), http://cancer.sanger.ac.uk/cancergenome/projects/cosmic
CASAVA, http://support.illumina.com/sequencing/sequencing_software/casava/documentation.ilmn
Complete Genomics, http://www.completegenomics.com/customer-support/documentation
Exome Chip Design, http://genome.sph.umich.edu/wiki/Exome_Chip_Design
Golden Helix, http://www.goldenhelix.com
Kyoto encyclopedia of genes and genomes (KEGG), http://www.genome.jp/kegg
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Variant Association Tools (VAT), http://varianttools.sourceforge.net/VAT
References
- 1.Cohen J.C., Kiss R.S., Pertsemlidis A., Marcel Y.L., McPherson R., Hobbs H.H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 2.Chang H., Jackson D.G., Kayne P.S., Ross-Macdonald P.B., Ryseck R.-P., Siemers N.O. Exome sequencing reveals comprehensive genomic alterations across eight cancer cell lines. PLoS ONE. 2011;6:e21097. doi: 10.1371/journal.pone.0021097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ji W., Foo J.N., O’Roak B.J., Zhao H., Larson M.G., Simon D.B., Newton-Cheh C., State M.W., Levy D., Lifton R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johansen C.T., Wang J., Lanktree M.B., Cao H., McIntyre A.D., Ban M.R., Martins R.A., Kennedy B.A., Hassell R.G., Visser M.E. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat. Genet. 2010;42:684–687. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huyghe J.R., Jackson A.U., Fogarty M.P., Buchkovich M.L., Stančáková A., Stringham H.M., Sim X., Yang L., Fuchsberger C., Cederberg H. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat. Genet. 2013;45:197–201. doi: 10.1038/ng.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peloso G.M., Auer P.L., Bis J.C., Voorman A., Morrison A.C., Stitziel N.O., Brody J.A., Khetarpal S.A., Crosby J.R., Fornage M., NHLBI GO Exome Sequencing Project Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am. J. Hum. Genet. 2014;94:223–232. doi: 10.1016/j.ajhg.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Psaty B.M., O’Donnell C.J., Gudnason V., Lunetta K.L., Folsom A.R., Rotter J.I., Uitterlinden A.G., Harris T.B., Witteman J.C.M., Boerwinkle E., CHARGE Consortium Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agarwala V., Flannick J., Sunyaev S., Altshuler D., GoT2D Consortium Evaluating empirical bounds on complex disease genetic architecture. Nat. Genet. 2013;45:1418–1427. doi: 10.1038/ng.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Challis D., Yu J., Evani U.S., Jackson A.R., Paithankar S., Coarfa C., Milosavljevic A., Gibbs R.A., Yu F. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012;13:8. doi: 10.1186/1471-2105-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li B., Leal S.M. Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. Am. J. Hum. Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madsen B.E., Browning S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D.J., Leal S.M. A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet. 2010;6:e1001156. doi: 10.1371/journal.pgen.1001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price A.L., Kryukov G.V., de Bakker P.I.W., Purcell S.M., Staples J., Wei L.-J., Sunyaev S.R. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu M.C., Lee S., Cai T., Li Y., Boehnke M., Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neale B.M., Rivas M.A., Voight B.F., Altshuler D., Devlin B., Orho-Melander M., Kathiresan S., Purcell S.M., Roeder K., Daly M.J. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhatia G., Bansal V., Harismendy O., Schork N.J., Topol E.J., Frazer K., Bafna V. A covering method for detecting genetic associations between rare variants and common phenotypes. PLoS Comput. Biol. 2010;6:e1000954. doi: 10.1371/journal.pcbi.1000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris A.P., Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet. Epidemiol. 2010;34:188–193. doi: 10.1002/gepi.20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han F., Pan W. A data-adaptive sum test for disease association with multiple common or rare variants. Hum. Hered. 2010;70:42–54. doi: 10.1159/000288704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auer P.L., Wang G., Leal S.M. Testing for rare variant associations in the presence of missing data. Genet. Epidemiol. 2013;37:529–538. doi: 10.1002/gepi.21736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.San Lucas F.A., Wang G., Scheet P., Peng B. Integrated annotation and analysis of genetic variants from next-generation sequencing studies with variant tools. Bioinformatics. 2012;28:421–422. doi: 10.1093/bioinformatics/btr667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leal S.M. Detection of genotyping errors and pseudo-SNPs via deviations from Hardy-Weinberg equilibrium. Genet. Epidemiol. 2005;29:204–214. doi: 10.1002/gepi.20086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wigginton J.E., Cutler D.J., Abecasis G.R. A note on exact tests of Hardy-Weinberg equilibrium. Am. J. Hum. Genet. 2005;76:887–893. doi: 10.1086/429864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., Ouwehand W.H., Samani N.J., Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung Y.H., Wang G., Leal S.M., Wang S. A fast and noise-resilient approach to detect rare-variant associations with deep sequencing data for complex disorders. Genet. Epidemiol. 2012;36:675–685. doi: 10.1002/gepi.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basu S., Pan W. Comparison of statistical tests for disease association with rare variants. Genet. Epidemiol. 2011;35:606–619. doi: 10.1002/gepi.20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ladouceur M., Dastani Z., Aulchenko Y.S., Greenwood C.M.T., Richards J.B. The empirical power of rare variant association methods: results from sanger sequencing in 1,998 individuals. PLoS Genet. 2012;8:e1002496. doi: 10.1371/journal.pgen.1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ladouceur M., Zheng H.-F., Greenwood C.M.T., Richards J.B. Empirical power of very rare variants for common traits and disease: results from sanger sequencing 1998 individuals. Eur. J. Hum. Genet. 2013;21:1027–1030. doi: 10.1038/ejhg.2012.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin D.-Y., Tang Z.-Z. A general framework for detecting disease associations with rare variants in sequencing studies. Am. J. Hum. Genet. 2011;89:354–367. doi: 10.1016/j.ajhg.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ionita-Laza I., Buxbaum J.D., Laird N.M., Lange C. A new testing strategy to identify rare variants with either risk or protective effect on disease. PLoS Genet. 2011;7:e1001289. doi: 10.1371/journal.pgen.1001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kutner M.H. McGraw-Hill Irwin; Boston: 2005. Applied linear statistical models. [Google Scholar]
- 35.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 36.Devlin B., Roeder K., Bacanu S.-A. Unbiased methods for population-based association studies. Genet. Epidemiol. 2001;21:273–284. doi: 10.1002/gepi.1034. [DOI] [PubMed] [Google Scholar]
- 37.Willer C.J., Li Y., Abecasis G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee S., Teslovich T.M., Boehnke M., Lin X. General framework for meta-analysis of rare variants in sequencing association studies. Am. J. Hum. Genet. 2013;93:42–53. doi: 10.1016/j.ajhg.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu D.J., Peloso G.M., Zhan X., Holmen O.L., Zawistowski M., Feng S., Nikpay M., Auer P.L., Goel A., Zhang H. Meta-analysis of gene-level tests for rare variant association. Nat. Genet. 2014;46:200–204. doi: 10.1038/ng.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forbes S.A., Bhamra G., Bamford S., Dawson E., Kok C., Clements J., Menzies A., Teague J.W., Futreal P.A., Stratton M.R. The Catalogue of Somatic Mutations in Cancer (COSMIC) Curr. Protoc. Hum. Genet. 2008;Chapter 10:11. doi: 10.1002/0471142905.hg1011s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 45.Cooper G.M., Goode D.L., Ng S.B., Sidow A., Bamshad M.J., Shendure J., Nickerson D.A. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat. Methods. 2010;7:250–251. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pruitt K.D., Tatusova T., Klimke W., Maglott D.R. NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res. 2009;37(Database issue):D32–D36. doi: 10.1093/nar/gkn721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Browning B.L., Browning S.R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Howie B.N., Donnelly P., Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 53.Li Y., Willer C., Sanna S., Abecasis G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.