

Abstract
In a subset of inherited retinal degenerations (including cone, cone-rod, and macular dystrophies), cone photoreceptors are more severely affected than rods; ABCA4 mutations are the most common cause of this heterogeneous class of disorders. To identify retinal-disease-associated genes, we performed exome sequencing in 28 individuals with “cone-first” retinal disease and clinical features atypical for ABCA4 retinopathy. We then conducted a gene-based case-control association study with an internal exome data set as the control group. TTLL5, encoding a tubulin glutamylase, was highlighted as the most likely disease-associated gene; 2 of 28 affected subjects harbored presumed loss-of-function variants: c.[1586_1589delAGAG];[1586_1589delAGAG], p.[Glu529Valfs∗2];[Glu529Valfs∗2], and c.[401delT(;)3354G>A], p.[Leu134Argfs∗45(;)Trp1118∗]. We then inspected previously collected exome sequence data from individuals with related phenotypes and found two siblings with homozygous nonsense variant c.1627G>T (p.Glu543∗) in TTLL5. Subsequently, we tested a panel of 55 probands with retinal dystrophy for TTLL5 mutations; one proband had a homozygous missense change (c.1627G>A [p.Glu543Lys]). The retinal phenotype was highly similar in three of four families; the sibling pair had a more severe, early-onset disease. In human and murine retinae, TTLL5 localized to the centrioles at the base of the connecting cilium. TTLL5 has been previously reported to be essential for the correct function of sperm flagella in mice and play a role in polyglutamylation of primary cilia in vitro. Notably, genes involved in the polyglutamylation and deglutamylation of tubulin have been associated with photoreceptor degeneration in mice. The electrophysiological and fundus autofluorescence imaging presented here should facilitate the molecular diagnosis in further families.
Main Text
Retinal dystrophies are a clinically and genetically diverse group of inherited disorders that feature loss or dysfunction of photoreceptor cells as a primary or secondary event.1 Thorough structural and functional assessment of the retina can be performed with the use of optical coherence tomography,2 fundus autofluorescence imaging,3 and visual electrophysiology.4,5 The latter is critical to the accurate diagnosis of retinal dystrophies and can reveal the degree of associated cone and rod photoreceptor dysfunction. Disorders in which the cone photoreceptors are more severely affected than rods include cone and cone-rod dystrophies (central- and usually peripheral-cone involvement) and macular dystrophies (central-cone involvement). These disorders show clinical overlap, and central visual loss in the first decades of life is a common symptom. Genetic overlap is also observed; recessive mutations in ABCA4 (MIM 601691) are by far the most common cause of both cone-rod and macular dystrophy.6,7 ABCA4 retinopathy exhibits extensive clinical heterogeneity, but despite the range of phenotypes, the majority of affected individuals have suggestive features on fundus examination. These include yellow-white retinal flecks and/or sparing of retinal tissue around the optic disc (“peripapillary sparing”). It is easier to detect these abnormalities on fundus autofluorescence imaging, a noninvasive imaging modality that uses naturally occurring fluorescence from the retina to provide functional information about retinal cells.3
In order to gain insights into the molecular pathology of retinal dystrophies, we recruited 28 families from the inherited-retinal-disease clinics at Moorfields Eye Hospital in London (Table S1, available online). Inclusion criteria were (1) a retinal dystrophy phenotype with early cone photoreceptor involvement, (2) an unknown molecular diagnosis after previous genetic screening or no previous genetic testing, and (3) an absence of fundoscopic and fundus autofluorescence imaging suggestive of ABCA4-associated retinopathy. Data from a representative set of 22 of the 28 probands are presented in Figures 1A, 1B, and S1, which show a common phenotype regarding retinal topography on fundus autofluorescence imaging. The study was approved by the local ethics committee, and all investigations were conducted in accordance with the principles of the Declaration of Helsinki; informed consent was obtained from all study participants.
Figure 1.
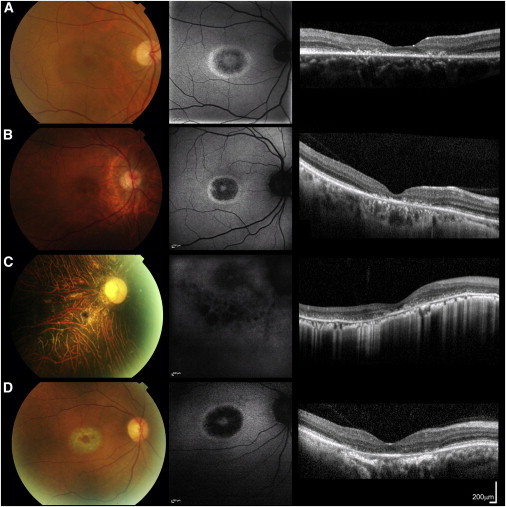
Color Fundus Photographs, Fundus Autofluorescence Images, and Foveal Optical Coherence Tomographs of the Right Eyes of Subjects CD1, CD2, CD3, and CD5
Images from subjects CD1 (aged 35 years; A), CD2 (aged 45 years; B), and CD5 (aged 53 years; D) are highly similar. Fundus autofluorescence imaging revealed a high-density concentric perifoveal ring surrounding irregular foveal autofluorescence in subjects CD1, CD2, and CD5; outside this ring, normal signal was observed (A, B, and D). In subject CD3 (aged 46 years; C), hypoautofluorescent patches were noted in the fovea and parafovea; this was combined with irregular autofluorescence outside the foveal region, suggesting more generalized retinal pigment epithelial dysfunction (C). Optical coherence tomography revealed abnormalities consistent with photoreceptor loss; they were either confined to the foveal region (subjects CD1, CD2, and CD5) or observed throughout the scan (subject CD3). Scale bars represent 200 μm.
DNA samples were collected and analyzed by high-throughput sequencing (exon capture by SureSelectXT Human All Exon V5, Agilent; sequencing by HiSeq2000, Illumina). To rank genes and prioritize follow-up, we then performed a gene-based case-control association study. This case-control approach compares the number of rare potentially deleterious alleles between case and control groups, hence making no specific assumption about the mode of inheritance (dominant or recessive); although more powerful models could be used if the inheritance model were known, the lack of information motivated this choice. The control samples (“UCL-exomes samples”) were collected by a research consortium based in the UK and, in particular, by University College London. This consortium was designed to share raw read-level data from multiple exome sequencing projects in order to facilitate case-control association studies.
Case-control comparison using calls generated from short-read high-throughput DNA sequencing is complicated by the nonnegligible frequency of variant-calling inaccuracies that result from limitations of existing technologies. This issue is compounded by the heterogeneity of sequence-capture kits (especially for the diverse UCL-exomes collection of control samples) and variant calling. As an example, such a technical issue arose for TTLL5 in the context of our comparison with the NHLBI Exome Sequencing Project Exome Variant Server (EVS, see below). To mitigate this problem, we used a multisample sequence-variant-calling strategy, including BAM file compression of redundant sequencing reads,8 for the 28 probands and 1,750 internal control samples on the basis of the Genome Analysis Toolkit guidelines (GATK version 2.7.4, Broad Institute).9 The variant-quality recalibration steps recommended by the GATK best practices were applied. Candidate variants were further filtered with ANNOVAR (OpenBioinformatics)10 on the basis of putative effect on protein and/or mRNA (presumed loss-of-function, nonsynonymous, and splice-altering changes were selected; Ensembl gene and transcript annotations were used).
Gene-based p values were computed with two strategies: a binomial test for excess of rare variants in the case group and the more general gene-based testing procedure Sequence Kernel Association Test (SKAT).11 In order to use the UCL-exomes control samples, (1) we inferred ancestry on the basis of the exome sequencing data, and using a principal-component analysis, we excluded samples that did not cluster with the bulk of the UCL-exomes samples, which are predominantly of European origin (Figure 2A; 5 out of 28 case samples were also removed); (2) we removed all samples with a history of retinal disease; and (3) when several samples were sequenced in a family, we kept a single sample per family to obtain unrelated control samples. After these exclusion steps, 1,465 control samples were left. For our binomial testing approach, it has been previously highlighted that association tests are biased when the same control cohort is also used for defining a minor-allele-frequency (MAF) threshold to flag candidate variants.12 To address this issue while still taking advantage of our technically and ethnically matched control samples, we divided the remaining 1,465 control samples into two subsets. The first subset included 25% of the samples (n = 366) and was used for defining a MAF threshold; a MAF < 0.3% (i.e., no more than two occurrences of the rare allele in 366 control samples) was utilized. The NHLBI Exome Sequencing Project EVS was also used for filtering rare candidate variants (with a frequency threshold of 0.1%). The second subset included the remaining 1,099 UCL-exomes control samples and was used directly for generating gene-based case-control binomial-test association statistics. This splitting of the control data set was not relevant for the SKAT gene-based testing.
Figure 2.
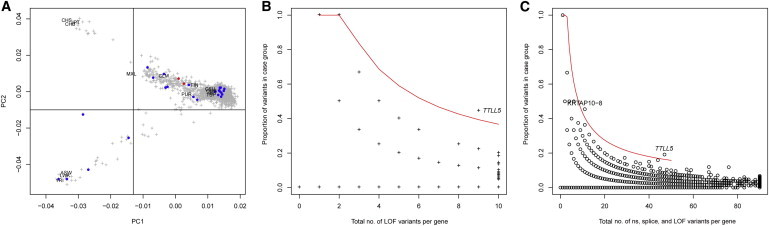
Case-Control Association Results
(A) Principal-component analysis (PCA) of an internal control cohort (“UCL-exomes samples”). PCA was estimated with 1,750 UCL-exomes samples combined with 1,092 samples from diverse ethnic backgrounds; data from the latter were generated as part of the 1000 Genomes Project. Samples selected for the case-control analysis are located in the top right corner of the plot (which includes the samples of European origin). Labels indicate the position of the 1000 Genomes subpopulations. Blue points indicate case samples, and red points indicate the two samples with presumed loss-of-function variants in TTLL5.
(B) Total number of presumed loss-of-function (LOF) alleles in case and control groups (x axis) and the proportion of these alleles in the 23 retinal dystrophy samples (y axis). The area above the red line corresponds to a gene-based p value threshold of p < 10−4.
(C) Same as (B) but for the total number of nonsynonymous (ns) variants (including presumed LOF variants) and splice-site variants (within 5 bp of a splice site). The red line corresponds to the p < 10−5 threshold.
The result of this genome-wide scan is shown in Figure 2B (for presumed loss-of-function variants) and Figure 2C (for nonsynonymous and splice-altering rare variants). Table 1 shows the list of autosomal genes ranked on the basis of the gene-based binomial p values that test for an excess of presumed loss-of-function candidate variants in case samples. Table S2 shows the larger set of nonsynonymous (including presumed loss-of-function) and splice-altering variants. The loss-of-function analysis flagged two hemizygous disease-causing variants in RPGR (MIM 300029), a gene previously associated with X-linked retinal dystrophy, and one homozygous presumed loss-of-function variant in another retinal-disease-related gene, CDH3 (MIM 114021; Table S1).
Table 1.
Top Five Most Significant Autosomal Genes: the Count of Presumed Loss-of-Function Rare Variants Was Compared between Probands with Retinal Dystrophy and Internal Control Samples
| Gene | Chr | Number of Presumed Loss-of-Function Variants in Probandsa(n = 23) | Number of Presumed Loss-of-Function Variants in UCL-Exomes Control Samplesb(n = 1,465) | SKAT p Value | Binomial p Value |
|---|---|---|---|---|---|
| TTLL5 | 14 | 4 | 5 | 8.51 × 10−4 | 2.05 × 10−5 |
| OR5AU1 | 14 | 2 | 0 | 0.0033 | 4.21 × 10−4 |
| CDH3 | 16 | 2 | 0 | 0.0031 | 4.21 × 10−4 |
| KRTAP3-3 | 17 | 2 | 0 | 0.0034 | 4.21 × 10−4 |
| FAM200B | 4 | 2 | 0 | 0.0033 | 4.21 × 10−4 |
Genes are ranked on the basis of the binomial p value test, which tests for equal proportion of presumed loss-of-function rare variants between case and control groups against the alternative of an excess of the same class of variants in the case group. To define “rare” variants, we utilized two cohorts: a subset of 25% of UCL-exomes control samples (366 unrelated samples, randomly sampled and not included directly in the case-control analysis; MAF < 0.3% was used) and the NHLBI Exome Sequencing Project EVS (MAF < 0.1% was used). The following abbreviation is used: Chr, chromosome.
The 28 probands had (1) retinal dystrophy with early cone photoreceptor involvement, (2) an unknown molecular diagnosis after previous genetic screening or no previous genetic testing, and (3) an absence of fundoscopic and fundus autofluorescence imaging suggestive of ABCA4 retinopathy. Five of these 28 subjects were excluded on the basis of ancestry (Figure 2A).
The 1,750 control samples were analyzed with the same sequence-variant-calling strategy as the 28 retinal dystrophy probands. After (1) inferring ancestry on the basis of the exome sequencing data and using a principal-component analysis to exclude samples that did not cluster with the bulk of the UCL-exomes samples (which are predominantly of European origin; Figure 2A), (2) removing all samples with a history of retinal disease, and (3) excluding related control samples, we were left with 1,465 unrelated control samples.
The most significant gene-based p value was obtained for TTLL5 (MIM 612268, RefSeq accession number NM_015072.4), a gene encoding tubulin tyrosine ligase-like family, member 5 (Tables 1 and S2). Two of 28 probands were found to harbor a pair of presumed loss-of-function variants in this gene. Subject CD1 (IV1, family gc18728 in Figure 3), a 38-year-old man born to consanguineous parents, had a homozygous 4 bp deletion (c.1586_1589delAGAG [p.Glu529Valfs∗2]). Furthermore, subject CD2 (IV:6, family gc19552 in Figure 3), a 45-year-old male with a very similar phenotype (Figures 1A and 1B; Table 2), had a 1 bp deletion and a nonsense mutation each in the heterozygous state (c.[401delT(;)3354G>A], p.[Leu134Argfs∗45(;)Trp1118∗]).
Figure 3.
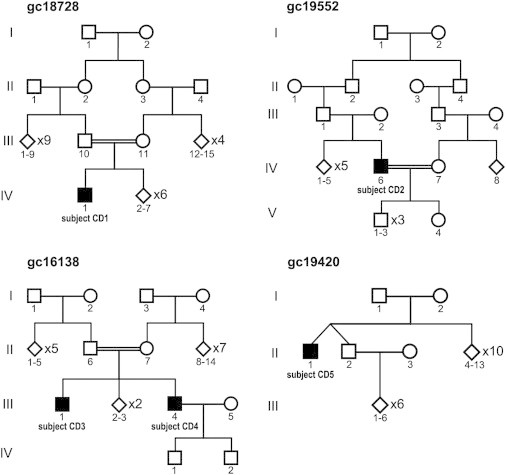
Pedigrees from Families Affected by TTLL5-Related Retinal Disease
The probands are subject CD1 (IV1, family gc1872; p.[Glu529Valfs∗2];[Glu529Valfs∗2]), subject CD2 (IV:6, family gc19552; p.[Leu134Argfs∗45(;)Trp1118∗]), subject CD3 (III:4, family gc16138; p.[Glu543∗];[Glu543∗]), and subject CD5 (II:1, family gc19420; p.[Glu543Lys];[Glu543Lys]). Interestingly, heterozygous variants were detected in subject CD2 despite his being born to consanguineous parents.
Table 2.
Clinical Characteristics and Molecular Pathology of Subjects with TTLL5-Associated Retinopathy
| Subject (Family ID) | Age at Presentation | Age at Examination |
LogMAR Visual Acuity |
Refraction |
Electrophysiology (Age when Tested) | TTLL5 Mutations and Predicted Protein Changes | Other Features | ||
|---|---|---|---|---|---|---|---|---|---|
| Right | Left | Right | Left | ||||||
| CD1 (gc18728) | 34 years | 38 years | 0.20 | 0.20 | −1.25 DS | −0.75/−075 × 180 | undetectable PERGs, normal DA ERGs, mildly subnormal LA ERGs (35 years) | c.[1586_1589delAGAG];[1586_1589delAGAG], p.[Glu529Valfs∗2];[Glu529Valfs∗2] | has two children |
| CD2 (gc19552) | 28 years | 45 years | 1.80 | 1.80 | emmetropia | emmetropia | undetectable PERGs, borderline DA ERGs, subnormal LA ERGs (39 years) | c.[401delT(;)3354G>A], p.[Leu134Argfs∗45(;)Trp1118∗] | has four children, one of whom is reported to have glaucoma |
| CD3 (gc16138) | <5 years | 46 years | 1.44 | 1.50 | −16.50/−3.50 × 20 | −16.50/−3.50 × 120 | undetectable PERGs, markedly subnormal DA ERGs, residual LA ERGs (39 years) | c.[1627G>T];[1627G>T], p.[Glu543∗];[Glu543∗] | bilateral mixed hearing loss and hearing aids since 42 years of age |
| CD4 (gc16138) | <5 years | 50 years | 2.00 | 1.50 | high myopia | high myopia | not tested | c.[1627G>T];[1627G>T], p.[Glu543∗];[Glu543∗] | right pseudophakia since 31 years of age |
| CD5 (gc19420) | 52 years | 53 years | 0.48 | 0.48 | +0.50/−3.00 × 85 | +1.00/−3.00 × 95 | undetectable PERGs, borderline DA ERGs, markedly subnormal LA ERGs (53 years) | c.[1627G>A];[1627G>A], p.[Glu543Lys];[Glu543Lys] | left amblyopia and convergent squint correction at 55 years of age |
Subjects CD1 and CD2 and the sibling pair of subjects CD3 and CD4 were born to consanguineous parents. All affected individuals presented with problems with central vision; subject CD1 also reported difficulty adapting from light to dark. Electroretinograms were performed according to the International Society for Clinical Electrophysiology of Vision minimum standards. The cDNA is numbered according to Ensembl transcript ENST00000298832 and RefSeq NM_015072.4. Abbreviations are as follows: DA, dark adapted; ERG, full-field electroretinogram; LA, light adapted; LogMAR, logarithm of the minimal angle of resolution; and PERG, pattern electroretinogram.
In addition to data from this cohort of 28 affected subjects with homogeneous clinical presentation, exome sequencing data from 63 molecularly unsolved families with retinal dystrophies were generated as part of an ongoing project at Moorfields Eye Hospital. The clinical diagnoses in these families were cone-rod dystrophy (n = 4), cone dystrophy (n = 3), macular dystrophy (n = 20), rod-cone dystrophy (n = 8 nonsyndromic and 11 syndromic), early-onset retinal dystrophy (n = 9), and Leber congenital amaurosis (n = 8). We reviewed the exome sequencing data from these families with the aim of identifying additional individuals with most likely disease-causing variants in TTLL5; a 44-year-old man (subject CD3; III:4, family gc16138 in Figure 3) with an early-onset cone-rod dystrophy phenotype (Figure 1C; Table 2) was found to harbor a homozygous nonsense variant (c.[1627G>T];[1627G>T], p.[Glu543∗];[Glu543∗]). Notably, he was born to consanguineous parents and has an older affected brother (subject CD4; III:1, family gc16138 in Figure 3) with the same genotype (Table S3). An unaffected sibling was heterozygous for the mutation.
Subsequently, 55 additional probands with “cone-first” retinal dystrophy were ascertained and tested for mutations in TTLL5 by Sanger sequencing of the coding region and intron-exon boundaries of the gene (primers and conditions are provided in Table S4). A 53-year-old man (subject CD5; II:1, family gc19420 in Figure 3) with an adult-onset cone dystrophy phenotype had a homozygous missense change (c.1627G>A [p.Glu543Lys]); this sequence alteration affects the same amino acid that is altered in the sibling pair of subjects CD3 and CD4.
Overall, four families affected by retinal dystrophy and most likely disease-causing variants in TTLL5 were identified. Two frameshift (p.Leu134Argfs∗45 and p.Glu529Valfs∗2), two nonsense (p.Glu543∗ and p.Trp1118∗), and one missense (p.Glu543Lys) change altering an amino acid conserved in all vertebrates (Figure S2) were found. In contrast, only five presumed loss-of-function variants were present in 1,465 unrelated UCL-exomes control samples (Table S5). In order to estimate the prevalence of disease caused by biallelic TTLL5 variants, we investigated the frequency of presumed loss-of-function alleles in a larger data set of 26,000 exomes assembled from a variety of complex-disease-sequencing consortia at the Broad Institute of Harvard and MIT in Boston. The Broad 26K exome data set includes the widely used NHLBI Exome Sequencing Project EVS but was reanalyzed with an optimized joint calling strategy similar to the one applied to UCL-exomes.9 Interestingly, two relatively common frameshift indels (up to 0.5% allele frequency) are listed in the NHLBI EVS. Excess of homozygous calls for these variants points to false-positive calls, and indeed, the optimized multisample calling approach excluded these calls as artifacts. Overall, the estimated frequency of presumed loss-of-function variants in the 26,000 exomes of the Broad 26K data set was 0.09% (Table S5), a number not statistically different (p > 0.05) from the frequency estimate in the smaller UCL-exomes control cohort (0.17%).
The clinical and electrophysiological phenotype in three of four families affected by TTLL5-related disease was almost identical: subjects CD1, CD2, and CD5 had central and peripheral cone dysfunction with preservation of rod photoreceptor function on electrophysiology (Figure 4; Table 2) and a similar appearance on fundus autofluorescence imaging (Figures 1A, 1B, and 1D). In contrast, the sibling pair of subjects CD3 and CD4 had a more severe phenotype with poor vision from the first years of life, severe generalized cone-system dysfunction, and additional significant involvement of rod photoreceptors (Figures 1C and 4; Table 2). This clinical heterogeneity cannot be easily explained by the TTLL5 genotype; notably, subject CD1 had presumed loss-of-function variants earlier in the protein than did subjects CD3 and CD4 (p.[Glu529Valfs∗2];[Glu529Valfs∗2]) and p.[Glu543∗];[Glu543∗], respectively).
Figure 4.
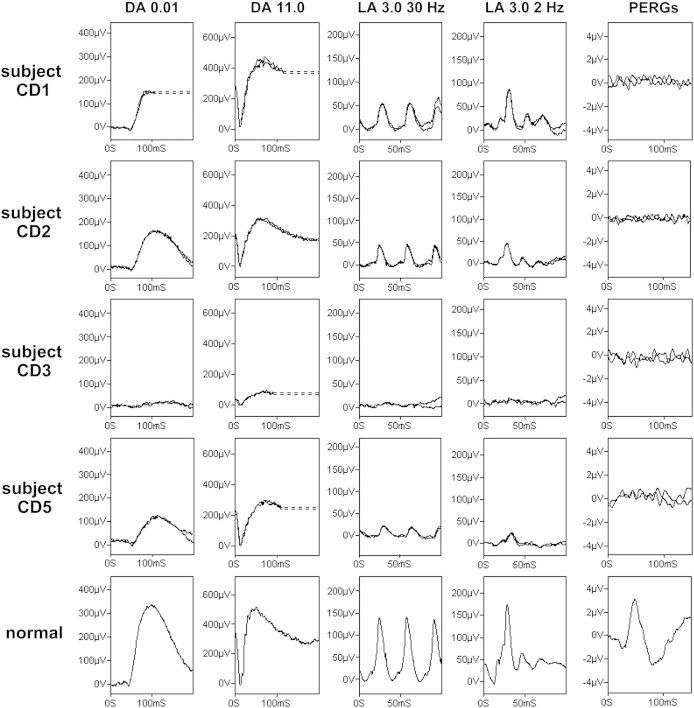
Electroretinography in TTLL5-Associated Retinal Disease
Full-field electroretinograms (ERGs) and pattern ERGs (PERGs) from subjects CD1 (aged 35 years; row 1), CD2 (aged 39 years; row 2), CD3 (aged 39 years; row 3), and CD5 (aged 53 years; row 4). Representative normal traces are shown for comparison (row 5). Dark-adapted (DA) responses are shown for flash strengths of 0.01 cd.s/m2 (DA 0.01) and 11.0 cd.s/m2 (DA 11.0). Light-adapted (LA) ERGs are shown for flash strength 3.0 cd.s/m2 (LA 3.0 30 Hz and LA 3.0 2 Hz). The pattern ERGs assessed macular function. Broken lines replace blink artifacts that occurred after ERGs had attained maximum amplitudes. All responses show a high degree of interocular symmetry and are for one eye only. See the main text and Table 2 for further explanation.
TTLL5 is a 32-exon gene with high expression in heart and skeletal muscle and lower expression in many other tissues, including the eye (Unigene) and brain.13,14 It encodes a 1,281 amino acid protein that is localized to the cytoplasm and nucleus.13 This protein is the largest of 13 members of the tubulin tyrosine ligase-like (TTLL) superfamily and contains the highly homologous core tubulin tyrosine ligase domain in its N terminus. In addition, TTLL5 has a C-terminal coactivator-interaction domain and three C-terminal receptor-interaction domains.15,16 Multiple activities have been implicated for TTLL5. First, it is thought to play an important role in the polyglutamylation of primary cilia.17,18 Polyglutamylation is a posttranslation modification associated with sequential attachment of glutamic acids (up to 20 units) to an internal glutamate residue of the target protein.19,20 The main target of polyglutamylation is thought to be the glutamate-rich C terminus of tubulins (building blocks of microtubules),21 and TTLL5 is thought to be a key initiator of polyglutamylation in α-tubulin.17 Second, TTLL5 has been found to be essential for the correct function of sperm flagella.16 Mutant mice that retain the TTLL domain but lack the C-terminal extension that is thought to be responsible for a variety of transcriptional cofactor activities (including glucocortocoid-mediated gene induction)13,22 have been previously generated.16 These mice (Stamptm/tm), despite having either no TTLL5 or markedly reduced levels of a prematurely terminated protein (roughly half TTLL5 will be missing), only demonstrate a sex-dependent effect on fertility. Female mice are normal, whereas male mice are infertile and have defective sperm structure and motility.16 Third and finally, a recent study has shown that TTLL5 has no unique function for ciliary stability or beating in brain ependymal cilia.14
The tubulin tyrosine ligase domain in human TTLL5 is predicted to be between amino acids 62 and 407 (UniProt). It has been shown in other TTLLs that added sequences of 100–150 amino acids on either side of the core tubulin tyrosine ligase domain are required for full polyglutamylation activity.23 Thus, it can be speculated that four (p.Leu134Argfs∗45, p.Glu529Valfs∗2, p.Glu543∗, and p.Glu543Lys) of the five most likely disease-causing changes identified here might result in reduced levels of polyglutamylation. Subjects CD1 (p.[Glu529Valfs∗2];[Glu529Valfs∗2]) and CD3 and CD4 (p.[Glu543∗];[Glu543∗]) would be expected to have a molecular defect similar to that of the Stamptm/tm mice. It is therefore of interest that subject CD3 has two unaffected children (Figure 3; paternity has not been confirmed). Analysis of the sperm from affected individuals might provide further insights.
It is not clear how defects in TTLL5 can cause central- and peripheral-cone dysfunction. It has been previously reported that defects in fleer, a regulator of tubulin glutamylation and glycylation of cilia microtubules, result in photoreceptor outer-segment defects in zebrafish.24,25 Furthermore, mice lacking one of the enzymes that catalyze deglutamylation of α-tubulin (Agtpbp1pcd mutant mice), an essential subunit of cilia microtubules, have been shown to have retinal degeneration.26,27 Interestingly, around 25% of previously reported retinal-dystrophy-related genes are associated with the structure or function of the photoreceptor connecting cilium, a specialized nonmotile primary sensory cilium that represents the light-sensitive outer segments.1,28 Additionally, although polyglutamylation was initially considered a tubulin-specific modification, it is now well recognized as a much more widespread posttranslational modification.17 Further experiments using proteomic approaches might demonstrate new substrates for polyglutamylation in the retina.
To study the localization of TTLL5 in the retina, we stained a donor human retina from a 56-year-old healthy individual (from the Department of Ophthalmology, University of Mainz [Germany]) and cryofixed BI6 mouse eye sections with TTLL5 antibodies as previously described.29 TTLL5 was detected in rod and cone photoreceptors of mouse and human retinae; in the human retina, TTLL5 staining was more prominent in cones (Figure 5). Furthermore, TTLL5 localized to the base of the connecting cilium between the basal body (mother centriole) and the adjacent daughter centriole of the cilium. There, TTLL5 might be responsible for the tubulin polyglutamination in the microtubule triplets of the centrioles, increasing the centriole stability, as previously reported.30 Notably, as in other primary cilia, the periciliary region of the photoreceptor cilium harbors the molecular modules for the regulation of delivery into the ciliary compartment, namely the connecting cilium (transition zone) and the photosensitive outer segment.31,32 It is worthy of speculation that polyglutamylation of tubulin molecules destined for the cilium might occur in this strategic site at the base of the cilium. In any case, the microtubules of the photoreceptor cilia apparatus are stabilized against mechanical forces. Given that cones are characterized by open membrane disks lacking the complete sheath of the plasma membrane present in rods, the absence of TTLL5, which should result in the destabilization of the microtubule cytoskeleton of photoreceptor cells, might affect the maintenance of cones more than rods. In the periciliary compartment, the products of other ciliopathy genes can be found.33–37
Figure 5.
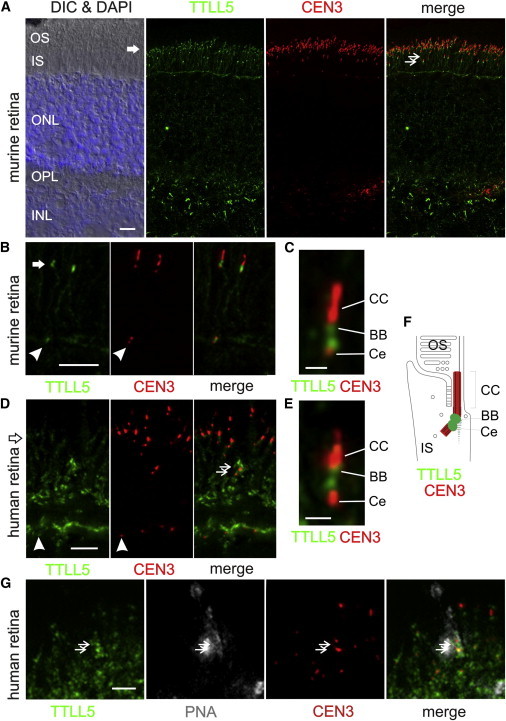
TTLL5 Localization to the Ciliary Base of Photoreceptor Cells
(A) Mouse retina cryosections were stained for TTLL5 (green; Abnova) and counterstained for the ciliary marker centrin-3 (CEN3, red) and DAPI (blue) for nuclear stain of the outer nuclear layer (ONL) and inner nuclear layer (INL). The merged image reveals substantial colocalization of TTLL5 and CEN3 in the ciliary region of photoreceptor cells (arrow). Double arrows indicate cone photoreceptor cells located in the lower portion of the photoreceptor layer. Other abbreviations are as follows: DIC, differential interference contrast microscopy; OS, outer segment; IS, inner segment; OPL, outer plexiform layer; and IPL, inner plexiform layer.
(B and C) Higher magnification of double labeling of TTLL5 (green) and CEN3 (red) in the photoreceptor layer of the mouse retina demonstrated TTLL5 localization in the periciliary region at the proximal poles of the adjacent daughter centriole (Ce) and the basal body (BB, mother centriole) of the connecting cilium (CC) of rod photoreceptor cells.
(D) Human retina cryosections through the photoreceptor layer were stained for TTLL5 (green) and counterstained for CEN3 (red). Double arrows indicate cone photoreceptor cells located in the lower portion of the photoreceptor layer.
(E) Higher magnification of double labeling of TTLL5 (green) and CEN3 (red) in the ciliary region of human photoreceptor cells revealed a nearly identical staining pattern.
(F) Schematic illustration of the localization of CEN3 (red) and TTLL5 (green) in the ciliary compartment of mouse and human photoreceptor cells. In addition to localizing to ciliary centrioles, TTLL5 was found at the centrioles of centrosomes in other retinal cell types (arrowheads).
(G) Cryosections through the basal portion of human photoreceptor layer were triple labeled for TTLL5 (green), CEN3 (red), and fluorescein-tagged peanut agglutinin (PNA, Sigma Aldrich; magenta), a molecular marker for the specialized extracellular sheath of cone photoreceptor cells. TTLL5 labeling was concentrated in cones (double arrow). The coefficients for double staining of PNA and TTLL5 were calculated by application of the ImageJ plugin JACoP; Pearson’s coefficient was r = 0.862, and Manders’s coefficients were M1 = 0.825 and M2 = 0.717. These indicate colocalization of both signals.
Scale bars represent 10 μm (A), 5 μm (B and D), 1 μm (C and E), and 2.5 μm (G).
We have shown that TTLL5, encoding a member of the TTLL superfamily, is associated with human disease. Other genes encoding members of the TTLL family have been shown to cause a variety of disorders in animal models; these include primary ciliary dyskinesia in Ttll1 mutant mice38 and defective olfactory cilia structures in ttll6 mutant zebrafish.24,25 The human phenotype observed in the present study would be consistent with some degree of functional redundancy among some of these glutamylating enzymes in humans.
To date, mutations in over 200 genes have been shown to cause retinal degeneration (Retinal Information Network, see Web Resources). The identification of genes associated with these disorders is a major challenge, particularly because they are likely to be less prevalent and less obvious candidates than those already known. We have performed exome sequencing in 28 individuals with a similar disease phenotype and subsequently used a case-control approach to identify mutations in TTLL5 as a cause of recessive retinal dystrophy. This powerful approach facilitates the identification of disease-causing alleles among the background of nonpathogenic genomic variation and sequencing errors. Overall, three families affected by presumed loss-of-function variants and one proband with a homozygous missense change were identified. The electrophysiological and fundus autofluorescence imaging reported in the present series should hopefully facilitate the identification of further families.
Acknowledgments
We acknowledge the cooperation and help provided by the family members in this study. We are grateful to colleagues who referred affected individuals to us at Moorfields Eye Hospital and to those who contributed to the assembly of the cone-rod and macular dystrophy panel, particularly Kaoru Fujinami, Alice Davidson, Alan Bird, Michel Michaelides, Genevieve Wright, Sophie Devery, Ravinder Chana, Beverley Scott, and Naushin Waseem. We thank our colleagues from the cornea bank of the Department of Ophthalmology at Johannes Gutenberg University Mainz for providing the human donor eye and our colleagues at University College London for kindly contributing to the UCL-exomes control panel. We acknowledge the following sources of funding: Macular Society UK (www.maculardisease.org), the UK National Institute for Health Research (Biomedical Research Centre, Moorfields Eye Hospital, and Institute of Ophthalmology), RP Fighting Blindness, Fight for Sight (including the Mercer Fund), the US Foundation Fighting Blindness, the UK Medical Research Council, the Wellcome Trust, the German Ministry of Education and Research (“HOPE2”), FAUN-Stiftung, and the European Commission Seventh Framework Program (EC FP7/2009/241955, SYSCILIA). D.G.M. and M.L. were funded by the NIH National Institute of General Medical Sciences under award 1R01GM104371.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase1/
ClustalW2, http://www.ebi.ac.uk/Tools/msa/clustalw2/
Ensembl Genome Browser, http://www.ensembl.org/
Genome Analysis Toolkit (GATK), http://www.broadinstitute.org/gatk/
HGVS Nomenclature for the description of sequence variations, http://www.hgvs.org/mutnomen/
Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/
ImageJ, http://rsbweb.nih.gov/ij/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
R statistical software, http://www.r-project.org/
Retinal Information Network (RetNet), http://www.sph.uth.tmc.edu/retnet/
SAMtools, http://samtools.sourceforge.net/
Unigene, http://www.ncbi.nlm.nih.gov/UniGene/
UniProt, http://www.uniprot.org/
References
- 1.Wright A.F., Chakarova C.F., Abd El-Aziz M.M., Bhattacharya S.S. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010;11:273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- 2.Drexler W., Fujimoto J.G. State-of-the-art retinal optical coherence tomography. Prog. Retin. Eye Res. 2008;27:45–88. doi: 10.1016/j.preteyeres.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Schmitz-Valckenberg S., Holz F.G., Bird A.C., Spaide R.F. Fundus autofluorescence imaging: review and perspectives. Retina. 2008;28:385–409. doi: 10.1097/IAE.0b013e318164a907. [DOI] [PubMed] [Google Scholar]
- 4.Marmor M.F., Fulton A.B., Holder G.E., Miyake Y., Brigell M., Bach M., International Society for Clinical Electrophysiology of Vision ISCEV Standard for full-field clinical electroretinography (2008 update) Doc. Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 5.Bach M., Brigell M.G., Hawlina M., Holder G.E., Johnson M.A., McCulloch D.L., Meigen T., Viswanathan S. ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc. Ophthalmol. 2013;126:1–7. doi: 10.1007/s10633-012-9353-y. [DOI] [PubMed] [Google Scholar]
- 6.Sheffield V.C., Stone E.M. Genomics and the eye. N. Engl. J. Med. 2011;364:1932–1942. doi: 10.1056/NEJMra1012354. [DOI] [PubMed] [Google Scholar]
- 7.Maugeri A., Klevering B.J., Rohrschneider K., Blankenagel A., Brunner H.G., Deutman A.F., Hoyng C.B., Cremers F.P. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 2000;67:960–966. doi: 10.1086/303079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu M.C., Lee S., Cai T., Li Y., Boehnke M., Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pearson R.D. Bias due to selection of rare variants using frequency in controls. Nat. Genet. 2011;43:392–393. doi: 10.1038/ng.816. author reply 394–395. [DOI] [PubMed] [Google Scholar]
- 13.He Y., Simons S.S., Jr. STAMP, a novel predicted factor assisting TIF2 actions in glucocorticoid receptor-mediated induction and repression. Mol. Cell. Biol. 2007;27:1467–1485. doi: 10.1128/MCB.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosch Grau M., Gonzalez Curto G., Rocha C., Magiera M.M., Marques Sousa P., Giordano T., Spassky N., Janke C. Tubulin glycylases and glutamylases have distinct functions in stabilization and motility of ependymal cilia. J. Cell Biol. 2013;202:441–451. doi: 10.1083/jcb.201305041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janke C., Rogowski K., Wloga D., Regnard C., Kajava A.V., Strub J.M., Temurak N., van Dijk J., Boucher D., van Dorsselaer A. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science. 2005;308:1758–1762. doi: 10.1126/science.1113010. [DOI] [PubMed] [Google Scholar]
- 16.Lee G.S., He Y., Dougherty E.J., Jimenez-Movilla M., Avella M., Grullon S., Sharlin D.S., Guo C., Blackford J.A., Jr., Awasthi S. Disruption of Ttll5/stamp gene (tubulin tyrosine ligase-like protein 5/SRC-1 and TIF2-associated modulatory protein gene) in male mice causes sperm malformation and infertility. J. Biol. Chem. 2013;288:15167–15180. doi: 10.1074/jbc.M113.453936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Dijk J., Miro J., Strub J.M., Lacroix B., van Dorsselaer A., Edde B., Janke C. Polyglutamylation is a post-translational modification with a broad range of substrates. J. Biol. Chem. 2008;283:3915–3922. doi: 10.1074/jbc.M705813200. [DOI] [PubMed] [Google Scholar]
- 18.Backer C.B., Gutzman J.H., Pearson C.G., Cheeseman I.M. CSAP localizes to polyglutamylated microtubules and promotes proper cilia function and zebrafish development. Mol. Biol. Cell. 2012;23:2122–2130. doi: 10.1091/mbc.E11-11-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eddé B., Rossier J., Le Caer J.P., Desbruyères E., Gros F., Denoulet P. Posttranslational glutamylation of alpha-tubulin. Science. 1990;247:83–85. doi: 10.1126/science.1967194. [DOI] [PubMed] [Google Scholar]
- 20.Janke C., Rogowski K., van Dijk J. Polyglutamylation: a fine-regulator of protein function? ‘Protein Modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:636–641. doi: 10.1038/embor.2008.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janke C., Bulinski J.C. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2011;12:773–786. doi: 10.1038/nrm3227. [DOI] [PubMed] [Google Scholar]
- 22.Awasthi S., Simons S.S., Jr. Separate regions of glucocorticoid receptor, coactivator TIF2, and comodulator STAMP modify different parameters of glucocorticoid-mediated gene induction. Mol. Cell. Endocrinol. 2012;355:121–134. doi: 10.1016/j.mce.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Dijk J., Rogowski K., Miro J., Lacroix B., Eddé B., Janke C. A targeted multienzyme mechanism for selective microtubule polyglutamylation. Mol. Cell. 2007;26:437–448. doi: 10.1016/j.molcel.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 24.Pathak N., Austin C.A., Drummond I.A. Tubulin tyrosine ligase-like genes ttll3 and ttll6 maintain zebrafish cilia structure and motility. J. Biol. Chem. 2011;286:11685–11695. doi: 10.1074/jbc.M110.209817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pathak N., Obara T., Mangos S., Liu Y., Drummond I.A. The zebrafish fleer gene encodes an essential regulator of cilia tubulin polyglutamylation. Mol. Biol. Cell. 2007;18:4353–4364. doi: 10.1091/mbc.E07-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rogowski K., van Dijk J., Magiera M.M., Bosc C., Deloulme J.C., Bosson A., Peris L., Gold N.D., Lacroix B., Bosch Grau M. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell. 2010;143:564–578. doi: 10.1016/j.cell.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 27.Marchena M., Lara J., Aijón J., Germain F., de la Villa P., Velasco A. The retina of the PCD/PCD mouse as a model of photoreceptor degeneration. A structural and functional study. Exp. Eye Res. 2011;93:607–617. doi: 10.1016/j.exer.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 28.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 29.Overlack N., Kilic D., Bauss K., Märker T., Kremer H., van Wijk E., Wolfrum U. Direct interaction of the Usher syndrome 1G protein SANS and myomegalin in the retina. Biochim. Biophys. Acta. 2011;1813:1883–1892. doi: 10.1016/j.bbamcr.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 30.Bobinnec Y., Khodjakov A., Mir L.M., Rieder C.L., Eddé B., Bornens M. Centriole disassembly in vivo and its effect on centrosome structure and function in vertebrate cells. J. Cell Biol. 1998;143:1575–1589. doi: 10.1083/jcb.143.6.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roepman R., Wolfrum U. Protein networks and complexes in photoreceptor cilia. Subcell. Biochem. 2007;43:209–235. doi: 10.1007/978-1-4020-5943-8_10. [DOI] [PubMed] [Google Scholar]
- 32.Nachury M.V., Seeley E.S., Jin H. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu. Rev. Cell Dev. Biol. 2010;26:59–87. doi: 10.1146/annurev.cellbio.042308.113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Estrada-Cuzcano A., Roepman R., Cremers F.P., den Hollander A.I., Mans D.A. Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum. Mol. Genet. 2012;21(R1):R111–R124. doi: 10.1093/hmg/dds298. [DOI] [PubMed] [Google Scholar]
- 34.Evans R.J., Schwarz N., Nagel-Wolfrum K., Wolfrum U., Hardcastle A.J., Cheetham M.E. The retinitis pigmentosa protein RP2 links pericentriolar vesicle transport between the Golgi and the primary cilium. Hum. Mol. Genet. 2010;19:1358–1367. doi: 10.1093/hmg/ddq012. [DOI] [PubMed] [Google Scholar]
- 35.Chakarova C.F., Khanna H., Shah A.Z., Patil S.B., Sedmak T., Murga-Zamalloa C.A., Papaioannou M.G., Nagel-Wolfrum K., Lopez I., Munro P. TOPORS, implicated in retinal degeneration, is a cilia-centrosomal protein. Hum. Mol. Genet. 2011;20:975–987. doi: 10.1093/hmg/ddq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zach F., Grassmann F., Langmann T., Sorusch N., Wolfrum U., Stöhr H. The retinitis pigmentosa 28 protein FAM161A is a novel ciliary protein involved in intermolecular protein interaction and microtubule association. Hum. Mol. Genet. 2012;21:4573–4586. doi: 10.1093/hmg/dds268. [DOI] [PubMed] [Google Scholar]
- 37.Maerker T., van Wijk E., Overlack N., Kersten F.F., McGee J., Goldmann T., Sehn E., Roepman R., Walsh E.J., Kremer H., Wolfrum U. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum. Mol. Genet. 2008;17:71–86. doi: 10.1093/hmg/ddm285. [DOI] [PubMed] [Google Scholar]
- 38.Ikegami K., Sato S., Nakamura K., Ostrowski L.E., Setou M. Tubulin polyglutamylation is essential for airway ciliary function through the regulation of beating asymmetry. Proc. Natl. Acad. Sci. USA. 2010;107:10490–10495. doi: 10.1073/pnas.1002128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.