

Abstract
BMS-791325 is an allosteric inhibitor that binds to thumb site 1 of the hepatitis C virus (HCV) NS5B RNA-dependent RNA polymerase. BMS-791325 inhibits recombinant NS5B proteins from HCV genotypes 1, 3, 4, and 5 at 50% inhibitory concentrations (IC50) below 28 nM. In cell culture, BMS-791325 inhibited replication of HCV subgenomic replicons representing genotypes 1a and 1b at 50% effective concentrations (EC50s) of 3 nM and 6 nM, respectively, with similar (3 to 18 nM) values for genotypes 3a, 4a, and 5a. Potency against genotype 6a showed more variability (9 to 125 nM), and activity was weaker against genotype 2 (EC50, 87 to 925 nM). Specificity was demonstrated by the absence of activity (EC50s of >4 μM) against a panel of mammalian viruses, and cytotoxic concentrations (50%) were >3,000-fold above the HCV EC50. Resistance substitutions selected by BMS-791325 in genotype 1 replicons mostly mapped to a single site, NS5B amino acid 495 (P495A/S/L/T). Additive or synergistic activity was observed in combination studies using BMS-791325 with alfa interferon plus ribavirin, inhibitors of NS3 protease or NS5A, and other classes of NS5B inhibitor (palm site 2-binding or nucleoside analogs). Plasma and liver exposures in vivo in several animal species indicated that BMS-791325 has a hepatotropic disposition (liver-to-plasma ratios ranging from 1.6- to 60-fold across species). Twenty-four hours postdose, liver exposures across all species tested were ≥10-fold above the inhibitor EC50s observed with HCV genotype 1 replicons. These findings support the evaluation of BMS-791325 in combination regimens for the treatment of HCV. Phase 3 studies are ongoing.
INTRODUCTION
Chronic infection with hepatitis C virus (HCV) is estimated to affect 130 to 170 million people worldwide, and its long-term sequelae represent a major and increasing public health concern (1). The virus—a member of the Hepacivirus genus of the Flaviviridae—has six major genotypes and is highly mutable (2). Acute HCV infection is often asymptomatic, and approximately 80% of acute infections progress to chronic disease. Of these, some 10 to 20% will ultimately develop cirrhosis and/or complications of chronic liver disease within 20 to 30 years, and 1 to 5% will develop hepatocellular carcinoma (3–7). The World Health Organization estimates that, globally, 350,000 people die of HCV-related complications each year (8).
Chronic HCV infection is curable in many patients, but the existing standard of care has a number of deficiencies. Treatment is largely—though no longer exclusively—based on parenteral administration of pegylated alfa interferon (IFN-α) together with the oral broad-spectrum antiviral ribavirin (RBV), for a period of up to 48 weeks depending on HCV genotype (9, 10). However, IFN-α/RBV efficacy is not equal across HCV genotypes. Genotype (GT) 1, which predominates in Europe, Japan, and the United States, is particularly difficult to treat, and rates of sustained virologic response (SVR) posttreatment seldom exceed 50% for GT 1 with IFN-α/RBV alone (9, 10). Two direct-acting antiviral (DAA) inhibitors of HCV NS3 protease, boceprevir and telaprevir, were approved in 2011 for use in combination with IFN-α/RBV for GT 1 treatment. The use of either of these inhibitors with IFN-α/RBV elevates SVR rates to approximately 70% in treatment-naive patients (11, 12). Two further DAA inhibitors were approved by the U.S. Food and Drug Administration in late 2013. Sofosbuvir, a nucleoside analog inhibitor of the HCV NS5B polymerase (discussed below), is indicated for use with IFN-α/RBV for treatment of GT 1 and GT 4 and with RBV alone for GT 2 and GT 3. Simeprevir, another NS3 protease inhibitor, is indicated for use with IFN-α/RBV for GT 1 treatment. Both these newer agents in combination with IFN-α/RBV have further improved GT 1 treatment responses, with treatment-naive SVR rates of approximately 80% for simeprevir (13, 14) and approximately 90% for sofosbuvir (15). However, both IFN-α and RBV have severe and treatment-limiting side effects that result in discontinuations and contraindications for a significant minority of patients (9, 10, 16, 17), and this adverse-event burden may be further elevated by the addition of a DAA—for example, telaprevir and boceprevir are associated with a number of toxicities such as rash, pruritus (telaprevir), and the exacerbation of treatment-related anemia (12). Thus, although the combination of IFN-α/RBV with a DAA represents an improvement in patient therapy for the more refractory HCV genotypes, there is still an unmet medical need for new agents and treatment regimens with improved efficacy and tolerability profiles.
The HCV life cycle presents a number of opportunities for chemotherapeutic intervention. Three HCV proteins have been the main focus for development of direct-acting small-molecule HCV antivirals: the NS3 protease, the multifunctional NS5A protein, and the NS5B RNA-dependent RNA polymerase. In addition to the four currently approved for treatment, a number of other DAAs are in clinical development, and their approval is anticipated to establish a new standard of care based on all-oral regimens—including ribavirin-free oral regimens—that offer shorter treatment durations with significantly improved convenience, tolerability, and efficacy across HCV genotypes.
The HCV NS5B polymerase is a particularly attractive target for intervention. As the catalytic core of the replicase complex, NS5B polymerase is critical for viral proliferation (18–20). It has the right-handed finger-palm-thumb domain structure typical of polymerases (21) with four individually targetable binding sites for allosteric small-molecule inhibitors: two sites in the thumb region (thumb sites 1 and 2) that are separate and two in the palm (palm sites 1 and 2) that partially overlap (22, 23). In addition, competitive inhibition by nucleoside analogs is also possible (24). One NS5B inhibitor—the nucleoside analog sofosbuvir (Sovaldi; GS-7977) (25)—is already approved for the treatment of HCV GTs 1 to 4 and a second, dasabuvir (ABT-333; a palm site 1 inhibitor) (26), is currently in phase 3 development. A third inhibitor, deleobuvir (BI 207127; thumb site 1) (27), was recently withdrawn from development. Each of these NS5B inhibitors has been combined with DAAs targeting other HCV proteins and/or combined with RBV to deliver high rates of SVR, a milestone closely associated with cure.
BMS-791325 (Fig. 1) is currently in phase 3 clinical development. It binds to thumb site 1 of the NS5B polymerase and is one of the most potent NS5B inhibitors in clinical development. As with other thumb site 1 inhibitors, binding of BMS-791325 forces the polymerase to adopt a catalytically inactive “open” conformation by displacing the Δ1 loop, part of the fingers domain, from a lipophilic pocket in the thumb (28, 29). We report here the preclinical characterization of BMS-791325 that supported its selection for clinical development, including its selectivity for HCV NS5B polymerase, its in vitro resistance profile, and its antiviral activity, both alone and in combination with other HCV antivirals.
FIG 1.
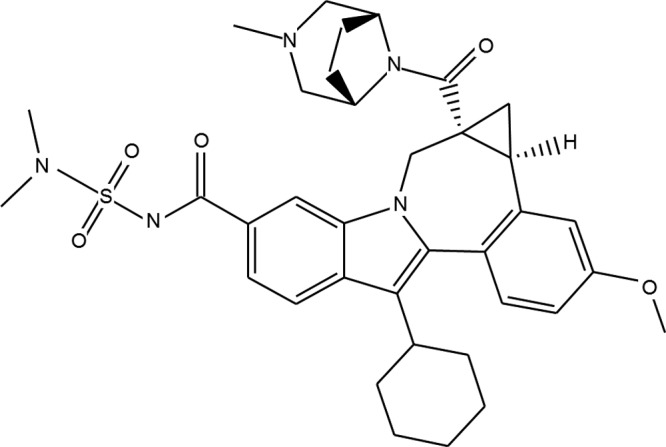
Structure of BMS-791325.
MATERIALS AND METHODS
Cell lines, viruses, and HCV inhibitors.
Huh-7 cells were obtained from Ralf Bartenschlager of the University of Heidelberg, Germany. MT-2 cells were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program. Vero, HeLa, MDBK, MRC5, and HEK293 cells were obtained from the American Type Culture Collection (ATCC). Huh-7 and MRC5 cells were propagated in Dulbecco's modified Eagle's medium (DMEM) containing 2 mM l-glutamine, 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Vero and MDBK cells were propagated in minimum essential medium (MEM), and MT-2 cells were propagated in RPMI 1640, supplemented as described above. Bovine viral diarrhea virus (BVDV) and GT 1a and 1b HCV replicon cell lines have been described previously (30, 31) and were propagated in DMEM containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, with or without 0.3 to 0.5 mg/ml Geneticin (G418). To construct a subgenomic 2a replicon clone, recombinant PCR was used to place the NS3 to 3′ untranslated region (UTR) sequences of the JFH-1 2a infectious clone (32) into the GT 1b replicon backbone described above. Human influenza virus (A/WSN/33), human rhinovirus 2, human coronavirus, poliovirus, and coxsackie virus A21 were obtained from the ATCC.
BMS-791325, daclatasvir (DCV; an investigational NS5A replication complex inhibitor) (33), and asunaprevir (ASV; an investigational NS3 protease inhibitor) (34) were synthesized by Bristol-Myers Squibb, as were HCV reference inhibitors HCV-796, a palm site 2 nonnucleoside inhibitor of NS5B (35), and NM-283, a prodrug of the anti-NS5B ribonucleoside analog 2′-C-methylcytidine (36). Recombinant alfa-2a interferon (Intron A) was obtained from Myoderm Medical Supply (Norristown, PA, USA).
Construction of hybrid HCV replicons.
Recombinant HCV hybrid replicon clones were constructed, which replaced Con1 or H77c NS5B with clinical isolate sequences from HCV GTs 1 to 6. A GT 1b shuttle replicon was prepared by introducing unique SpeI and SnaBI restriction sites using the QuikChange mutagenesis protocol (Stratagene, La Jolla, CA, USA). The SpeI site was located at the end of NS5A, approximately 20 nucleotides upstream of the start of NS5B, resulting in an alanine-to-threonine alteration, which has no effect on replicon replication efficiency or compound susceptibility. The SnaBI site was created 6 bp downstream of the NS5B stop codon. For GT 1a, a 1a shuttle replicon with unique restriction sites SpeI and ClaI was generated.
Patient sera were obtained from Cliniqa Corporation (Fallbrook, CA, USA) for GTs 1a and 1b and from Boca Biolistics (Coconut Creek, FL, USA) for GTs 2 to 5. GT 6 patient sera were obtained from SeraCare Life Sciences (Milford, MA, USA) or provided by Huy Trinh. Viral RNA was isolated using a QIAamp MinElute virus vacuum kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer's instructions. First-strand cDNA synthesis was performed using random primers and the Superscript III reverse transcriptase (RT) kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. NS5B regions were amplified using degenerate primers designed by examination of published HCV NS5B sequences. Patient PCR products were sequenced and used to replace the NS5B gene of the GT 1a shuttle replicon for GT 3a and the GT 1b shuttle replicon for GTs 2b to 6 using standard cloning techniques. In addition, for HCV GTs 2b, 4a, and 5a, patient-derived NS5B sequences known to generate viable chimeric replicons (37) were synthesized by DNA 2.0 and cloned into the GT 1b shuttle replicon.
To generate stable cell lines, replicon clones were linearized with restriction enzymes and transcribed in vitro using the Promega T7 RiboMax RNA production kit (Madison, WI, USA) according to the manufacturer's directions. Transcribed RNA (5 μg) was electroporated into 5 × 106 Huh-7 cells, and after 24 h, selective medium containing 0.25 mg/ml G418 was added. Medium was changed every 3 to 5 days. After approximately 3 weeks of selection, individual colonies were expanded for further analysis.
Cell culture assays.
To evaluate antiviral activity, HCV replicon cells were incubated in 96-well plates in the presence of compound for 3 days. For replicons containing a luciferase reporter gene, Renilla luciferase activity was then assayed using a Renilla luciferase assay system or a Dual-Glo luciferase assay system (Promega Corporation, Madison, WI, USA), according to the manufacturer's directions. Plates were read on a TopCount NXT microplate scintillation and luminescence counter (Packard Instrument Company, Meriden, CT, USA). The antiviral activity of test compounds, expressed as the 50% effective concentration (EC50), was determined as described previously (38). To study the effect of human serum (HS) on compound efficacy, the standard 10% fetal bovine serum in cell culture experiments was supplemented with 40% human serum.
To evaluate selectivity and cytotoxicity, BVDV assays were performed as described previously (31). Susceptibility of HIV, herpes simplex virus (HSV), influenza virus, and canine parainfluenza virus (CPIV) was determined by incubation with serial dilutions of compound. For recombinant HIVs expressing Renilla luciferase, antiviral activity was evaluated by measuring the production of luciferase in infected cells 5 days postinfection. For the CPIV and influenza virus assays, viral neuraminidase activity was used as a measure of viral production. Susceptibility of HSVs to compounds was determined using a multicycle HSV growth assay. For human coronavirus, poliovirus, coxsackie virus, and rhinovirus, MRC5 cells were infected at a multiplicity of infection of 0.1, incubated for 5 days with or without dilutions of compound, and then treated with alamarBlue to quantitate protection and cytotoxicity. To evaluate cytotoxicity for all other cell types, cells were incubated in the presence of serially diluted compounds for 3 to 4 days and cell viability was quantitated using either an XTT [2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide] assay for MT-2 cells or an alamarBlue assay for Huh-7, MDBK, and Vero cells. All 50% cytotoxic concentration (CC50) values were calculated using the median effect equation.
Transient replication assays.
Replicon clones were linearized with ScaI and transcribed in vitro using the Ambion T7 MegaScript kit (Ambion, Austin, TX, USA) according to the manufacturer's directions. Transcribed RNA (3 to 5 μg) was transfected into cured Huh-7 cells (∼2 × 106 cells in 60-mm dishes) with DMRIE-C reagent (Invitrogen) according to the manufacturer's protocol. After 4 to 6 h, transfected cells were transferred to 96-well assay plates (104 cells/well) and incubated in the presence of inhibitors for 72 h. Renilla luciferase assays were performed as described above.
Resistance selection and analysis.
For selection of BMS-791325-resistant replicons, HCV GT 1a and 1b replicon cells were passaged in medium containing 0.5 mg/ml G418 and BMS-791325 at a concentration of 5 to 20 EC50s (up to 0.2 μM for GT 1b and 0.1 μM for GT 1a). Cells were selected with dimethyl sulfoxide (DMSO) in parallel as a control. Cells were split or fed with fresh medium containing DMSO or BMS-791325 twice weekly to maintain a subconfluent monolayer. After approximately 5 weeks, selected cells were expanded for resistance testing and reverse transcription-PCR (RT-PCR) analysis. RNA was isolated from populations of resistant cells using TRIzol (Invitrogen Corp., Carlsbad, CA, USA) in accordance with the manufacturer's directions. First-strand cDNA synthesis was performed on 1 to 3 μg of total RNA using Superscript III reverse transcriptase (Invitrogen) primed with random hexamers. PCR was performed on the cDNA using pairs of primers flanking the NS5B gene. PCR products were sequenced, and mutations were identified relative to DMSO-treated control populations. For clonal analysis, PCR products were purified and cloned using TOPO PCR cloning methods (Invitrogen).
To generate drug-resistant replicons for analysis, point mutations were introduced with the QuikChange II XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions and confirmed by sequencing.
Cell-based inhibitor combination assays.
For combination studies, inhibitors were each tested at 11 concentrations. The compounds were tested as monotherapies and in combinations at various concentration ratios. A defined set of drug concentrations and ratios was achieved in a final concentration of 0.5% DMSO. The final drug concentration range for each compound as monotherapy was as follows: 1.7 × 10−5 to 1 μM for BMS-791325, 1.7 × 10−5 to 1 nM for DCV, 4 × 10−6 to 0.25 μM for ASV, 4.2 × 10−5 to 2.5 μM for HCV-796, 0.017 to 1,000 U/ml for IFN-α, 8 × 10−3 to 500 μM for ribavirin, and 8 × 10−4 to 50 μM for BMS-790453. For individual drug studies (with the exception of ribavirin), the EC50 was selected as the midpoint for the concentration range tested. When the compound was used in double combinations at a ratio of 2.5 relative to the second inhibitor, the concentration range of the compound was increased by 2.5-fold. Cells were exposed to compounds for 3 days, and the amount of HCV inhibition was then determined using the Dual-Glo luciferase assay system as described above. The potential cytotoxicities of these combined agents were also analyzed in parallel by alamarBlue staining. The degree of antagonism, additivity, or synergy was determined from combination dose-response curves, which were fitted to assess the antiviral effects of the drug treatment combinations. The concentration ratios were analyzed using the method of Chou (39). All estimates were computed using SAS Proc NLIN biostatistical software and a four-parameter logistic. Combination indices (50%, 75%, and 90% effective) were tested for departure from additivity using isobologram methods. Asymptotic confidence intervals were also calculated for each of the combination indices. These intervals are used to test for departure from additivity by comparing the bounds to 1: a lower bound of the interval greater than 1 indicates antagonism, an upper bound of less than 1 indicates synergism, and a value of 1 contained in the interval indicates additivity. Mixed results, where synergy is established for some combination indices and/or inhibitor ratios and additivity is established for others, are reported as “synergy/additivity.”
HCV NS5B polymerase cloning, expression, and purification.
The cDNAs encoding HCV NS5B proteins of GTs 1a (H77c), 1b (Con1), 2a (JFH-1), 2a (HC-J6), 2b (database consensus), 3a (database consensus), 4a (database consensus and two patient sequences), 5a (database consensus), and 6a (database consensus) were cloned into the pET21a expression vector (database consensus sequences are provided in the supplemental material). Each untagged protein was expressed with an 18-amino-acid C-terminal truncation that enhances polymerase solubility. The Escherichia coli competent cell line BL21(DE3) was used for expression of the protein. Proteins were purified using heparin-Sepharose and poly(U)-Sepharose chromatography as described elsewhere (40).
HCV NS5B polymerase assays.
RNA synthesis was measured by detecting the incorporation of radiolabeled nucleotides. Regardless of the assay format used to measure enzyme activity, inhibition by BMS-791325 was detected as a decrease in the incorporation of radiolabeled nucleotides compared with an untreated control. One format of the polymerase reaction mixture contained pGpG primer (8.6 μM), homopolymeric C template (0.35 nM), NS5B enzyme (2.8 nM), and 1 to 5 μM GTP (depending on the genotype of the enzyme) plus [33P]GTP substrate (1 μCi, 3,000 Ci/mmol; Amersham, GE Healthcare, Piscataway, NJ). After a ≥1-h preincubation of NS5B polymerase, template, and compound (serially diluted 1:3 in DMSO and transferred to 96-well assay plates [Corning] for a final DMSO concentration of 2%), RNA synthesis was initiated by the addition of primer and GTP. Reaction mixtures (total volume of 0.06 ml) were incubated at 30°C for 15 to 50 min, depending on the genotype of the NS5B polymerase tested. The newly synthesized RNA product was precipitated with 10% trichloroacetic acid (TCA), harvested onto 96-well GF/B filter plates (PerkinElmer, Shelton, CT, USA), and quantified on a Packard TopCount NXT gamma counter. Another format of the polymerase reaction has been described previously in detail (40). Briefly, the reaction mixture contained biotinylated oligo(dT)12 primer precaptured on streptavidin-coupled polystyrene imaging beads (GE Healthcare) by incubating primer and beads in assay buffer (20 mM HEPES, pH 7.5, 2.5 mM KCl, 2.5 mM MgCl2, 1 mM dithiothreitol [DTT], 1.6 U RNase inhibitor [Promega], 0.1 mg/ml bovine serum albumin [BSA] [Promega], 2% glycerol) at room temperature for 1 h. Unbound primer was removed with the supernatant after centrifugation. The primer-bound beads were suspended in 20 mM HEPES buffer, pH 7.5, and used in the assay at final concentrations of 15 nM primer and 0.33 mg/ml beads. Compounds were serially diluted 1:3 in DMSO and transferred to 384-well assay plates (Corning) to give a final DMSO concentration of 2%. The order of addition was 0.01 ml water, 0.01 ml enzyme (7 nM) diluted in assay buffer, 0.01 ml template (0.2 nM), and [3H]UTP (0.3 mCi, 0.29 mM); concentrations are final. After a preincubation period of 24 h, the polymerase reaction was initiated by the addition of primer-bound beads. Reactions were allowed to proceed overnight at 30°C and terminated by the addition of 0.01 ml EDTA (50 mM). After ≥15 min, plates were read on a LEADseeker multimodality imaging system (Amersham).
The 50% inhibition value (IC50) was calculated using a four-parameter logistic equation:
where A and B denote minimal and maximal % inhibition, respectively; C is the IC50; D is the Hill slope; x represents compound concentration; and Y is the signal observed.
Polymerase selectivity assays.
The selectivity of BMS-791325 was evaluated with a panel of enzymes unrelated to HCV NS5B, including bovine polymerase α, human polymerase β and γ, BVDV polymerase, Klenow polymerase, HCV protease, HIV reverse transcriptase, and HIV integrase. Compounds were also tested for nonspecific DNA and RNA binding.
Bovine polymerase α (Chimerx, Milwaukee, WI, USA), human polymerase β (Chimerx), Klenow polymerase (New England BioLabs, Ipswich, MA, USA), and HIV-1 reverse transcriptase (Amersham Biosciences) were assayed by scintillation counting of 33P-labeled, TCA-precipitated primer extension products essentially as described by the manufacturers.
Human polymerase γ was obtained from W. C. Copeland at the National Institute of Environmental Health Sciences (Research Triangle Park, NC, USA) and assayed essentially as described previously (41). Due to enzyme instability, no preincubation of enzyme and inhibitor was performed. The assay measures the incorporation of radiolabeled [3H]TTP. The reaction mixture contains 0.25 μg poly(A) template, biotinylated dT primer bound to streptavidin scintillation proximity assay (SPA) beads, [3H]dTTP, and 10 ng human polymerase γ in a reaction volume of 35 μl.
BVDV polymerase was isolated and assayed as described for HCV NS5B, except that the assay concentration of unlabeled GTP was increased to a final concentration of 20 μM.
HIV integrase was assayed by strand transfer activity of purified integrase enzyme as described previously (42).
DNA binding was tested in a competition assay using the fluorescent ligand Picogreen (Molecular Probes, Invitrogen). Compounds titrated in DMSO were incubated in 50 mM Tris buffer (pH 7.4) in the presence and absence of 10 μg/ml DNA (1-kb ladder from Invitrogen) for 30 min at room temperature. The final concentration of DMSO was 2%. Picogreen commercial stock was diluted 1 to 200, and 50 μl was added to each well. Plates were read with a Gemini fluorescence plate reader (485-nm excitation and 530-nm emission).
RNA binding was tested according to the protocol described for a 96-well format in a Ribogreen kit (Molecular Probes, Invitrogen).
In vivo exposure studies.
Plasma and liver tissue exposure to BMS-791325 was assessed in rats, dogs, and monkeys. Male Sprague-Dawley rats (280 to 330 g; n = 18; Hilltop Lab Animals, Inc., Scottdale, PA, USA) were administered BMS-791325 (10 mg/kg of body weight) in polyethylene glycol 400 (PEG-400) by oral gavage, and blood and liver samples were obtained at 10 min, 30 min, and 1, 2, 4, 6, 8, 24, and 48 h postdose (two rats per time point). Male beagle dogs (≈9 to 12 kg; n = 6; Marshall Farms USA Inc., North Rose, NY, USA) were orally dosed with BMS-791325 (3 mg/kg) in a mixture of PEG-400 (95%, vol/vol), povidone K-30 (2%), d-α-tocopherol polyethylene glycol succinate (TPGS; 2%), and Tween 80 (1%), and blood and liver samples were obtained at 1, 4, 6, 24, 48, and 72 h postdose (one dog per time point). Male cynomolgus monkeys (2.5 to 4 kg; n = 4; Charles River Biomedical Research Foundation, Houston, TX, USA) were orally administered BMS-791325 (3 mg/kg) in a mixture of sodium phosphate buffer (0.1 M; 93%, wt/wt), hydroxypropylcellulose-SL (5%), and TPGS (2%), and blood and tissue samples were taken at 1, 4, 8, and 24 h postdose (one monkey per time point). Vehicles were optimized for each species based on both specific dosing volume and tolerability requirements and optimization of formulation properties as the compound proceeded through pharmacokinetic testing. All animal procedures were performed under protocols approved by the Institutional Animal Care and Use Committee of the test facility. Plasma samples were stored at −20°C until analyzed. Liver samples (approximately 2 to 3 g per animal) were rinsed, blotted dry, weighed, and stored frozen. BMS-791325 was assayed in plasma and tissue using a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) methodology using standard curves fitted by weighted (1/concentration2) linear regression over the range of 2 to 10,000 nM. Liquid chromatography-tandem mass spectrometry conditions were as follows. We used a Cohesive Aria high-pressure liquid chromatography (HPLC) system consisting of 8 Shimadzu LC-10ADvp pumps with 2 SCL-10Avp system controllers (Columbia, MD) and a dual-arm CTC Analytics HTS PAL autosampler (Switzerland) equipped with a cooling stack that maintained samples at 10°C during analysis. The online extraction column used was a Cyclone-P mixed polymer column (0.5 mm by 50 mm, 60 μM particles; Cohesive Technologies, Franklin, MA) at room temperature. The analytical column used was a Supelguard Ascentis C18 column (3.0 mm by 20 mm, 5 μM particles; Supelco, Bellefonte, PA) at room temperature. The mobile phase for the online extraction consisted of 10 mM ammonium acetate in water (A) and 40/40/20 acetonitrile/isopropanol/acetone (B) and was delivered as a gradient at a flow rate of 2.0 ml/min to the turbulent-flow chromatography (TFC) extraction column. The mobile phases for the analytical column consisted of 0.1% formic acid in water (A) and 0.1% formic acid acetonitrile (B) and were delivered as a gradient at a flow rate of 1.5 ml/min. The TFC column was interfaced with an API4000 LC-MS/MS system (AB Sciex Instruments, Toronto, Canada) equipped with an Ionspray ionization interface operating in the positive ionization mode. The source temperature was 600°C. Detection of the analyte was achieved through selected reaction monitoring. Standards were analyzed in duplicate. Quality control (QC) samples in a blank biological matrix at concentrations within the standard curve were analyzed as replicates within each analytical set. The predicted concentrations of more than 83.7% of the QCs from various different matrices were within 20% of nominal values, indicating acceptable assay performance. Standard pharmacokinetic parameters (24-h concentration [C24] and area under the concentration-time curve at 24 h [AUC24] or extrapolated to infinity [AUCinf]) were derived from concentration-time data using noncompartmental methods in KINETICA version 4.2 (InnaPhase Corporation, Philadelphia, PA, USA).
RESULTS
BMS-791325 antiviral activity, cytotoxicity, and specificity for HCV NS5B.
The in vitro potency, specificity, and therapeutic index of BMS-791325 were evaluated in a series of experiments testing antiviral activity against purified enzymes, replicons, and a panel of RNA and DNA viruses in cell culture. In addition, a number of cell lines derived from various tissue origins were used to assess its cytotoxicity.
BMS-791325 demonstrated potent and selective inhibition of HCV GT 1a and 1b replicons, with EC50s of 3 nM and 6 nM, respectively (Table 1). It did not significantly affect the replication of a panel of other RNA and DNA viruses, including the closely related BVDV, with EC50s of >4 μM, whereas control inhibitors for each virus gave the expected EC50s (Table 1). BMS-791325 cytotoxicity (CC50) was ≥14 μM in a panel of cell lines representing liver, T lymphocytes, lung, and kidney following incubation for 3 to 4 days (Table 2). Its antiviral activity against HCV (GT 1b; EC50, 6 nM) in Huh-7 replicon cells was more than 3,000-fold below cytotoxic concentrations.
TABLE 1.
Antiviral activity and selectivity of BMS-791325 in cell culture assays
| Replicon/virusa | Cell line | EC50 (μM) | Selectivity indexb |
|---|---|---|---|
| HCV replicon 1a (H77c) | Huh-7 | 0.003 ± 0.001c | |
| HCV replicon 1b (Con1) | Huh-7 | 0.006 ± 0.002c | |
| BVDV replicon | Huh-7 | >14 | >2,333 |
| BVDV | MDBK | 11 | 1,833 |
| HIV | MT-2 | >14 | >2,333 |
| HSV-1 and -2 | Vero | >4 | >667 |
| Influenza virus | MDBK | >29 | >4,833 |
| CPIV | Vero | >32 | >5,333 |
| Human rhinovirus | MRC5 | >16 | >2,667 |
| Coxsackie virus | MRC5 | >16 | >2,667 |
| Poliovirus | MRC5 | >16 | >2,667 |
| Human coronavirus | MRC5 | >16 | >2,667 |
Positive controls run in the experiments: BVDV (EC50 of UPS-1453, 0.6 μM), HIV (EC50 of BMS-035, 1.5 nM), HSV-1 and -2 (EC50s of acyclovir, 0.08 and 0.31 μM, respectively), influenza virus (EC50 of BMS-659, 9 μM), CPIV (EC50 of BMS-011, 0.1 μM), human rhinovirus (EC50 of rupintrivir, 9 nM), coxsackie virus (EC50 of rupintrivir, 135 nM), and poliovirus (EC50 of rupintrivir, 82 nM). Compounds BMS-035, BMS-659, and BMS-011 are Bristol-Myers Squibb proprietary compounds that inhibit HIV, influenza virus, and CPIV, respectively.
EC50 for virus or replicon/EC50 for HCV replicon 1b.
Means ± standard deviations from two or more independent experiments.
TABLE 2.
Cytotoxicity of BMS-791325 in cell culture assays
| Cell line | Tissue | CC50 (μM)a |
|---|---|---|
| Huh-7 | Liver | 20, 26 |
| Vero | Kidney | 29, 32 |
| MDBK | Kidney | 30, 43 |
| MRC5 | Lung fibroblasts | 16, 18 |
| MT-2 | T lymphocytes | 14, 14 |
Results from two independent experiments.
The effect of human serum binding on antiviral activity was evaluated. Results showed a modest 4.4-fold decrease in susceptibility in GT 1b replicons supplemented with 40% human serum during the assay period relative to unsupplemented controls.
BMS-791325 inhibitory activity was also highly selective for the HCV NS5B polymerase when tested against a panel of unrelated microbial and mammalian enzymes (Table 3). The IC50 against isolated HCV NS5B (GT 1b) was 3 nM, more than 8,000-fold lower than that for any other polymerase tested or for HIV-1 integrase. In contrast, control inhibitors used for each assay inhibited the enzymes with the expected IC50s (Table 3). In addition, BMS-791325 did not display any nonspecific DNA or RNA binding (IC50, >20 μM) in competition assays against sensitive nucleic acid-binding probes (Table 3).
TABLE 3.
In vitro enzyme inhibition and nucleic acid binding by BMS-791325
| Enzyme or assay typea | BMS-791325 IC50 (μM)b |
|---|---|
| HCV NS5B 1b (Con1) | 0.003 |
| Bovine polymerase α | >25 |
| Human polymerase β | >25 |
| Human polymerase γ | >25 |
| BVDV polymerase | >25 |
| HIV reverse transcriptase | >25 |
| Klenow polymerase | >25 |
| HIV integrase | >40 |
| DNA binding | >25 |
| RNA binding | >20 |
Positive controls were run in each experiment: mammalian DNA polymerases (IC50 of ddTTP, 5 to 20 mM), BVDV polymerase (IC50 of BMS-514, 6.3 ± 2.6 mM), HIV RT (IC50 of Sustiva, 0.017 ± 0.017 mM), Klenow polymerase and nucleoside triphosphate binding (IC50 of actinomycin D, 1.9 ± 0.4 mM), and HIV integrase (IC50 of BMS-035, 0.004 ± 0.001 mM). Compounds BMS-514 and BMS-035 are Bristol-Myers Squibb proprietary compounds that inhibit BVDV polymerase and HIV integrase, respectively.
From two or more independent experiments.
Genotype-specific activity of BMS-791325.
The sequence divergence among the major HCV genotypes is significant (>30%) (2) and may result in substantial intergenotype variability in the activity of an inhibitor. Similarly, sequence diversity and the adaptive potential of the virus under in vivo selective pressures may result in intragenotype variability between clinical isolates. Since replicon culture systems are not available for all six of the major HCV genotypes, the cross-genotype activity of BMS-791325 was investigated by evaluating antiviral activity against hybrid replicons bearing clinical NS5B sequences and enzymatic inhibition of purified NS5Bs from GTs 1 to 6.
The results of the two sets of experiments show similar profiles of genotypic coverage by BMS-791325 (Table 4). For clinical NS5B sequences in hybrid replicons, EC50s ranged from 1.6 to 5.3 nM for GT 1a and from 3.5 to 9.5 nM for GT 1b, similar to values obtained from the H77c and Con1 lab strains (3.2 nM and 6.0 nM, respectively). The GT 1a patient-derived NS5Bs were 97 to 98% identical to H77c NS5B at the amino acid level, whereas the GT 1b NS5Bs were 96 to 97% identical to Con1 NS5B. This level of conservation is similar to that observed among other HCV GT 1a and 1b NS5B database sequences. The H77c and Con1 NS5Bs are only 88% identical, yet BMS-791325 shows similar potencies on GT 1a and 1b, suggesting that BMS-791325 should exhibit good activity against HCV GT 1-infected patients. BMS-791325 also displayed potency similar to that for GT 1 against hybrid replicons containing clinical NS5B sequences from GTs 3a, 4a, and 5a (EC50s, 0.8 to 18.0 nM). GT 2 replicons, however, were approximately 1 to 2 orders of magnitude less susceptible to BMS-791325 than were the other genotypes evaluated. BMS-791325 exhibited high potency against GT 6a hybrid replicons generated from two patients (pt-752 and pt-tt003), with EC50s (8.6 nM and 9.7 nM) similar to those observed for GT 1. However, a GT 6a replicon generated from a third patient (pt-hn001) was approximately 9-fold less susceptible to BMS-791325.
TABLE 4.
BMS-791325 activity against major HCV genotypes in patient-derived chimeric replicons and in in vitro NS5B polymerase assays
| NS5B genotype | Isolate | Chimeric replicon EC50 (nM)a,b | Enzyme IC50 (nM)a |
|---|---|---|---|
| 1a | H77c | 3.2 ± 2.4 | 3.3 ± 1.1 |
| pt-5-22 | 1.6 ± 0.3 | NDh | |
| pt-6-14 | 5.3 ± 2.3 | ND | |
| pt-8-3 | 4.3 ± 1.5 | ND | |
| pt-2-1 | 4.5 ± 2.1 | ND | |
| 1b | Con1 | 6 ± 1.5 | 4.2 ± 2.1 |
| pt-7-4 | 3.5 ± 0.7 | ND | |
| pt-10-5 | 9.5 ± 0.7 | ND | |
| 2a | JFH-1 | 87 ± 2.8 | 165 ± 133 |
| J6 | 498 ± 229 | 228 ± 72 | |
| 2b | Patient consensusc | ND | 164 ± 39 |
| pt-H6 | 480 ± 327 | ND | |
| pt-308 | >1,000 | ND | |
| 3a | Patient consensusd | ND | 1.8 ± 0.1 |
| pt-002 | 4.7 ± 1.5 | ND | |
| pt-007 | 3.5 ± 2.1 | ND | |
| pt-341 | 9.5 ± 3.5 | ND | |
| pt-342 | 7.5 ± 0.7 | ND | |
| 4a | Database consensuse | ND | 19.9 ± 9.6 |
| pt-H2 | 6 ± 2.9 | ND | |
| pt-A | 3 ± 0.6 | 7.6 ± 1.9 | |
| pt-B | 18 ± 2.5 | 27.1 ± 13.1 | |
| 5a | Database consensusf | ND | 4.8 ± 2.4 |
| pt-h3 | 4.3 ± 0.6 | ND | |
| pt-010 | 0.8 ± 0.3 | ND | |
| pt-011 | 1.2 ± 0.7 | ND | |
| pt-019 | 2.8 ± 1.7 | ND | |
| 6a | Patient consensusg | ND | 61.6 ± 30.3 |
| pt-752 | 8.6 ± 2.5 | ND | |
| pt-tt003 | 9.7 ± 3.5 | ND | |
| pt-hn001 | 79.5 ± 24.5 | ND |
Mean (± standard deviation) from three or more independent experiments.
Antiviral activity against HCV GTs 1, 3, 4, 5, and 6 was 250- to 25,000-fold below cytotoxic concentrations, while activity against GT 2 was <20- to 230-fold below cytotoxic concentrations.
Consensus from chimp infected with patient serum HC-J8 (97.4% identical to European HCV database GT 2b consensus).
Consensus from chimp infected with patient serum S52 (97.8% identical to European HCV database GT 3a consensus).
Synthesis based on previous GenBank consensus from 11 sequences (99.8% identical to European HCV database GT 4a consensus).
Synthesis based on previous GenBank consensus from 5 sequences (98.3% identical to European HCV database GT 5a consensus).
Consensus from patient serum (97.2% identical to the current European HCV database GT 6a consensus).
ND, not determined.
Consistent with the replicon results, in vitro polymerase assays gave BMS-791325 IC50s that ranged from 1.8 nM to 27.1 nM for GT 1, 3, 4, and 5 NS5B and were approximately 60 nM for GT 6 and 164 to 228 nM for GT 2a and GT 2b. The good concordance observed between BMS-791325 potency in the in vitro polymerase and cell-based replicon assays confirms that inhibition of viral RNA replication is indeed due to inhibition of NS5B polymerase activity. Results from both the enzyme and cell-based assays also indicate that BMS-791325 is a potent inhibitor of GTs 1a, 1b, 3a, 4a, and 5a with variability observed in potency against GT 6 sequences.
Selection of resistance in vitro.
To gain insight into the mechanism of BMS-791325 resistance, the HCV replicon system was used to select resistance-associated substitutions in NS5B that may emerge during treatment. Passage of replicon cells in the presence of BMS-791325 resulted in significant decreases in susceptibility to BMS-791325 versus cocultured controls treated with DMSO. Selection at 10× wild-type EC50 (40 nM and 70 nM for GTs 1a and 1b, respectively) generated GT 1a replicon cells with EC50s 34- to 46-fold greater (136 to 184 nM) than that of control and GT 1b cells with EC50s 25- to 28-fold greater (175 to 196 nM). After selection at 20× wild-type EC50 (80 nM and 140 nM for GTs 1a and 1b, respectively), GT 1a elevations (34- to 35-fold; 136 to 140 nM) were similar to those observed at 10×, whereas GT 1b elevations were somewhat higher (34- to 40-fold; 238 to 280 nM) than that at 10×.
In this study, genotypic analysis of the selected cells identified substitutions conferring resistance to BMS-791325 only at a single residue of NS5B (P495) for both GT 1a and GT 1b. The substitutions P495A, P495S, and P495L were common to the two genotypes, with an additional P495T substitution observed in GT 1a only. Each change, introduced to a wild-type replicon backbone by site-directed mutagenesis, resulted in various degrees of BMS-791325 resistance in transient replication assays (Table 5). All substitutions conferred in excess of a 10-fold loss of susceptibility versus the wild type. In general, the relative replication efficiency of the substituted replicons compared with the wild type was inversely correlated with the degree of resistance (Table 5). No change in susceptibility to asunaprevir, daclatasvir, HCV-796, or NM-283 was observed for any P495 substitution in transient assays of the GT 1a replicon (data not shown).
TABLE 5.
Transient replication assays of BMS-791325 resistance-associated NS5B variants selected in vitro
| Variant | Genotype 1a |
Genotype 1b |
||||
|---|---|---|---|---|---|---|
| EC50 (nM)a | Fold changeb | Replication efficiencyb | EC50 (nM)a | Fold changeb | Replication efficiencyb | |
| Wild type | 5.0 ± 0.6 | 1.0 | 8 ± 0.5 | 1.0 | ||
| P495A | 59 ± 5 | 12 | 0.7 | 115 ± 29 | 14 | 0.4 |
| P495S | 336 ± 36 | 67 | 0.2 | 350 ± 57 | 44 | 0.1 |
| P495L | 154 ± 25 | 31 | 0.3 | 290 ± 111 | 36 | 0.1 |
| P495T | 293 ± 124 | 59 | 0.1 | |||
| L392Ic | 5–7 | 0.1–0.4 | ||||
Mean ± standard deviation from two or more independent experiments.
Versus wild type.
Data from work of Pelosi et al. (38).
Naturally occurring variations at three positions in NS5B that are potentially associated with BMS-791325 resistance were examined in HCV GTs 1 to 6 in sequences from the European HCV database (http://euhcvdb.ibcp.fr/euHCVdb/). The three loci were as follows: P495 substitutions, as observed in these experiments; L392I, observed previously in GT 1a under in vitro selection and conferring low-level (5- to 7-fold) loss of susceptibility (38); and V494A, also observed in currently unpublished data to be associated with low-level (2- to 3-fold)-reduced GT 1a susceptibility to BMS-791325.
As shown in Table 6, the predominant amino acid associated with BMS-791325 resistance, residue P495, was conserved in all sequences for GTs 1 to 5 and in 95% of GT 6a sequences. Likewise, L392 was highly conserved (>92%) across all genotypes except GT 2, where isoleucine was present at this position in 87% and 96% of GT 2a and GT 2b sequences, respectively. An L392I substitution in GT 1a confers approximately 6-fold resistance to BMS-791325, suggesting that the naturally occurring I392 residue in GT 2 may contribute to the reduced BMS-791325 susceptibility observed. Another contributor is likely to be residue 494; V494 was 100% conserved across GT 1, GT 4, and GT 5 sequences, but more than 95% of GT 2 sequences in the database carried A494. Interestingly, although V494 was not present in GT 3a sequences at all, the C494 variant in this genotype did not affect BMS-791325 susceptibility (EC50s, 4 to 10 nM across four GT 3a patient isolates). In GT 6a sequences, the A494 variant was present in a significant minority of database sequences (21%) with V494 conserved in the remaining 79%. It is of note, therefore, that A494 was present in the NS5B sequence from the GT 6a patient isolate pt-hn001, discussed above, which demonstrated lower susceptibility to BMS-791325 in a hybrid replicon culture than did the other two GT 6a patient sequences evaluated.
TABLE 6.
Prevalence in the European HCV database (http://euhcvdb.ibcp.fr/euHCVdb/) of polymorphisms at BMS-791325 resistance-associated loci
| Genotype (n) | Residue/polymorphism at NS5B position (% of sequences): |
||
|---|---|---|---|
| 392 | 494 | 495 | |
| 1a (215) | L (97) | V (100) | P (100) |
| F (2.5) | |||
| P (0.5) | |||
| 1b (368) | L (92) | V (100) | P (100) |
| I (6) | |||
| F (2) | |||
| 2a (23) | I (87) | A (100) | P (100) |
| L (9) | |||
| X (4) | |||
| 2b (27) | I (96) | A (96) | P (100) |
| L (4) | V (4) | ||
| 3a (25) | L (100) | C (100) | P (100) |
| 4a (15) | L (100) | V (100) | P (100) |
| 5a (4) | L (100) | V (100) | P (100) |
| 6a (19) | L (100) | V (79) | P (95) |
| A (21) | L (5) | ||
BMS-791325 cross-resistance with other HCV inhibitor classes.
BMS-791325 retained full activity against mutations conferring resistance to other HCV inhibitors (Table 7). BMS-791325 was as active against HCV GT 1b replicons carrying mutations known to convey drug resistance to active-site (nucleoside) NS5B inhibitors (S282T) (43), NS3 protease inhibitors (R155Q, A156V, and D168V) (44), or NS5A replication complex inhibitors (Y93H ± L31V) (30, 45) as it was against the wild-type replicon. BMS-791325 activity was also unaffected by resistance mutations to nonnucleoside NS5B inhibitors targeting thumb site 2 (M423T) (29), palm site 1 (M414T) (46), or palm site 2 (C316Y) (47) but was reduced by known mutations to other thumb site 1 inhibitors (L392I and P495S) (38, 48, 49). The lack of cross-resistance to inhibitors targeting other HCV proteins, or different sites on NS5B, supports the use of BMS-791325 in DAA combination regimens, including those using multiple polymerase inhibitors.
TABLE 7.
BMS-791325 activity against drug-resistant genotype 1b replicons
| Protein | Mutation | BMS-791325 EC50 (nM)a | Fold change (vs wild-type Con1) |
|---|---|---|---|
| NS5B polymerase | Wild-type Con1 | 5.0 ± 1.0 | |
| S282T | 4.0 ± 1.4 | 0.8 | |
| L392I | 35 ± 12 | 7 | |
| P495S | 192 ± 21 | 38 | |
| M423T | 5.0 ± 1.0 | 1 | |
| M414T | 7.0 ± 3.0 | 1.4 | |
| C316Y | 2.0 ± 1.0 | 0.4 | |
| NS3 protease | R155Q | 5.3 ± 2.2 | 1 |
| A156V | 2.4 ± 0.4 | 0.5 | |
| D168V | 3.0 ± 0.2 | 0.6 | |
| NS5A | Y93H | 3.8 ± 0.9 | 0.8 |
| L31V + Y93H | 2.9 ± 0.6 | 0.6 |
Mean (± standard deviation) from two or more independent experiments.
In vitro drug combination studies.
Monotherapy treatment of a highly mutable, rapidly replicating pathogen can result in the rapid emergence of drug resistance (50), which has led to the development of combination regimens as a successful strategy to prevent resistance-associated therapeutic failure. Since HCV therapy with the newer agents under development is likely to benefit from the combination approach, the antiviral activity of BMS-791325 in combination with representative agents against the major HCV drug targets was examined in GT 1a replicon cultures. BMS-791325 was evaluated in dual- and triple-drug combinations with ASV (NS3 protease inhibitor), DCV (NS5A replication complex inhibitor), HCV-796 (a palm site 2 nonnucleoside NS5B inhibitor), NM-283 (a nucleoside analog NS5B inhibitor), and (unpegylated) alfa-2a interferon plus RBV (Table 8; see also Tables S1 to S6 in the supplemental material). In two-drug combinations, the compound displayed at least additive interactions with all drugs and combinations tested and was synergistic with HCV-796 across all drug ratios. Three-drug combinations of BMS-791325 with DCV and ASV, or alfa-2a interferon and RBV, also yielded additive to synergistic interactions. No antagonism was observed in any of the combinations evaluated.
TABLE 8.
Summary of BMS-791325 activity in drug combination studies
| Drug(s) in combination | Activity |
|---|---|
| IFN-α + RBV | Additive/synergistic |
| ASV | Additive |
| DCV | Synergistic/additive |
| HCV-796 | Synergistic |
| NM-283 | Synergistic/additive |
| DCV + ASV | Additive |
In vivo plasma and tissue exposures across species.
Plasma and liver concentrations of BMS-791325 were assessed in rats, dogs, and monkeys following oral dosing (Table 9). Rats dosed with BMS-791325 were monitored over 48 h, dogs were monitored over 72 h, and monkeys were monitored over 24 h. Liver contained the highest concentrations of compound across all species tested, although the liver-to-plasma ratios in dog, as determined either by concentration at the 24-h time point or using 24-h AUC values, ranged from 1.6- to 2.0-fold. Over the study duration, monkeys showed the highest liver-to-plasma ratios across the species, measured both by 24-h concentration (60:1) and by AUC (24:1). In all species, liver BMS-791325 concentration at 24 h postdose was more than 10-fold above GT 1, 3, and 5 replicon EC50s; between 6.7- and 40-fold above GT 4 replicon EC50s; and 1.5- to 14-fold above GT 6 replicon EC50s.
TABLE 9.
Liver and plasma 24-h concentrations and AUC values of BMS-791325 in nonclinical animal modelsa
| Animal (dose) and pharmacokinetic parameter | Liver | Plasma | Liver/plasma ratio |
|---|---|---|---|
| Rat (10 mg/kg) | |||
| C24 (μM) | 0.63 | 0.05 | 12.2 |
| AUCinf (μM · h) | 645.82 | 42.99 | 15 |
| Dog (3 mg/kg) | |||
| C24 (μM) | 2.13 | 1.33 | 1.6 |
| AUCinf (μM · h) | 152.92 | 78.16 | 2 |
| Monkey (3 mg/kg) | |||
| C24 (μM) | 0.12 | 0.002 | 60 |
| AUC24 (μM · h) | 51.32 | 2.13 | 24.1 |
Values were derived from one animal (dog and monkey) or two animals (rat) per time point.
DISCUSSION
BMS-791325 showed robust antiviral activity in an early single-ascending-dose study, with mean HCV RNA declines of approximately 1.5 and 2.5 log10 IU/ml at 24 h after administration of a single 100-mg or 300-mg dose, respectively, as described in the accompanying paper (51). Here, we report the preclinical profile of BMS-791325 that supported its selection for clinical development and which demonstrates the correlation between in vitro and in vivo potency and resistance.
In these studies, BMS-791325 displayed highly selective, nanomolar inhibition of recombinant HCV NS5B polymerase in vitro for all major HCV genotypes, with much weaker activity observed against GT 2. This inhibition resulted in potent antiviral activity in cell-based subgenomic replicons of HCV expressing consensus and/or clinical NS5B sequences for GTs 1a, 1b, 3a, 4a, 5a, and 6a. The antiviral activity observed against multiple clones for GTs 1, 3, 4, and 5 suggests that baseline heterogeneity within these genotypes is likely to have minimal impact on response to BMS-791325. BMS-791325 genotype specificity appears to be typical for NS5B thumb site 1 inhibitors, as reduced GT 2 activity has also been observed for other compounds active at this site (37, 49, 52, 53). This loss of activity in GT 2 apparently results from specific differences in amino acid sequence (L392I, V494A, and V499A) and protein conformation within the binding pocket (49, 53). The distribution of GT 2 is global, accounting for 10 to 20% of HCV infections overall and 15% of infections in the United States, while GT 1, which is susceptible to inhibition by BMS-791325, accounts for more than 80% of all HCV infections.
For GT 6a, although the three patient-derived NS5B sequences were 97 to 98% identical at the amino acid level, two showed susceptibilities to BMS-791325 in hybrid replicons (9 to 10 nM) similar to those of other non-GT 2 sequences, while the third was approximately 9-fold less susceptible (80 nM). Reduced susceptibility was most likely associated with an NS5B-V494A polymorphism present in this patient's virus and in approximately one-fifth of GT 6a sequences in the European HCV database. Based on mean plasma trough concentrations of approximately 380 to 400 nM following 14 days or 12 weeks of treatment with 75 mg BMS-791325 twice daily in phase 2 studies (54)—which are above the GT 6a V494A replicon EC90 values—HCV GT 6a variants with baseline V494A would likely be suppressed at this dose.
In vitro selection of BMS-791325 resistance variants at NS5B amino acid 495 (proline) is also typical of thumb site 1 inhibitors (27, 48, 54–56). The substitutions observed (P495A/S/L/T) conferred variable levels of resistance in GT 1a and 1b replicons, with the lowest (12- and 14-fold, respectively) observed for P495A and the highest (67- and 44-fold, respectively) observed for P495S. In previously described replicon experiments by Pelosi et al. (38), an L392I substitution in NS5B—conferring 6- to 16-fold elevations in BMS-791325 EC50s—was observed in GT 1a under coselection with BMS-791325 and DCV. This substitution occurred in combination with DCV-resistance-associated substitutions in NS5A and was not seen under joint selection by BMS-791325 and the NS3 protease inhibitor ASV (38) or under selection by BMS-791325 alone either by Pelosi et al. or in these experiments. Substitutions at NS5B codon 495 were also seen by Pelosi et al. (38) in combination with the same NS5A resistance substitutions but not in combination with L392I. The L392I substitution may therefore represent an alternative pathway to thumb site 1 inhibitor resistance that is more readily selected under joint selective pressure with other DAAs such as an NS5A inhibitor.
The observation that the EC50 fold change and relative replication capacity associated with proline 495 variants were inversely correlated suggests that the genetic barrier associated with the development of higher levels of drug resistance at this key position may support reversion to the wild type when inhibitor selection is removed. Although amino acid 495 was the sole locus of NS5B resistance identified in these replicon studies, in which selection was examined at 10- to 20-fold EC50 levels, in vivo selection in virologic breakthroughs from an ongoing phase 2a study of BMS-791325 with pegylated alfa-2a interferon and RBV has shown treatment-emergent linked substitutions at positions 495 and 421 (A421V) (57). The A421V substitution elevated P495-associated phenotypic resistance in GT 1a (H77c) replicons by a factor of 3 without further reducing the replication capacity but conferred only minimal (≈3-fold) levels of resistance itself (57). Although BMS-791325 did not select for A421V in these studies, this substitution was seen by Pelosi et al. in GT 1a replicons under selection with BMS-791325 either alone or in combination with DCV, but it was not linked with P495 variants (38). Further data will be needed to establish the significance of A421V as a possible compensatory mutation for P495 resistance and whether selection of the linked variants in vivo but not in vitro represents an effect of combination treatment or of in vivo selective pressure on whole-virion replication.
In vitro combination experiments showed that BMS-791325 has at least additive antiviral activity with representative agents from the major classes of oral direct-acting HCV antivirals, as well as with alfa-2a interferon and RBV. No antagonism was seen with any agent at any concentration or drug/drug ratio. These data are supportive of BMS-791325 in all-oral DAA regimens for GT 1 and also as the third agent in a mixed oral/parenteral combination with IFN-α/RBV. The synergistic antiviral activity seen with combinations of BMS-791325 and the palm site 2 NS5B inhibitor, HCV-796, demonstrates the potential value of approaching a single target with two inhibitors that bind to different sites and share no cross-resistance. The presence of three nonoverlapping allosteric inhibitor-binding sites on NS5B offers an opportunity for the sequential or combined use of nonnucleoside inhibitors in HCV infection, contrasting with the experience for HIV-1. Although nonnucleoside HIV-1 reverse transcriptase inhibitors have been important components of antiretroviral combination regimens since the introduction of nevirapine—the first inhibitor—in 1996, the presence of only a single allosteric site on the polymerase results in significant cross-resistance between individual agents that severely limits their sequential use after virologic failure (58). The potential for synergistic HCV suppression with combinations of nonnucleoside NS5B inhibitors further emphasizes the flexibility of this class of agent, although such combinations have not been clinically studied and it remains unknown how such a convergent approach would impact the emergence of multidrug NS5B resistance.
A hepatotropic disposition in vivo is a potentially beneficial property for drugs active in liver-specific disease, simultaneously maximizing target organ levels and reducing peripheral exposure and the associated potential for off-target adverse events. The liver-to-plasma ratios observed for BMS-791325 in rats, dogs, and monkeys were similar, although lower in magnitude, to the highly hepatotropic ratios seen for the NS3 protease inhibitor ASV in these species (34). In the case of ASV, accumulation occurs chiefly through active transport by liver organic anion-transporting polypeptide (OATP) 1B1 and OATP 2B1 at physiologically relevant concentrations (59). For BMS-791325, the mechanism of liver sequestration and the relative contributions of active and passive uptake remain to be elucidated. Hepatic exposures in all three animal species at 24 h postdose were greater than the GT 1 replicon protein-adjusted EC90 values, indicating that effective oral dosing would be achievable in humans.
In conclusion, these preclinical data show strong and selective nanomolar inhibition of HCV GTs 1, 3, 4, 5, and 6 by BMS-791325, with a large therapeutic window between antiviral and cytotoxic concentrations, and a very low potential for interaction with human polymerases at therapeutic levels. The antiviral properties of this compound indicate its suitability for use with a variety of other HCV therapeutics in combination regimens. Phase 2 studies of BMS-791325 included its use with DCV and ASV in an all-oral regimen in GT 1 and GT 4 treatment-naive patients and GT 1 patients with prior null response to IFN-α/RBV (study AI443-014; clinicaltrials.gov ID NCT01455090) (60), and phase 3 studies of this 3-DAA regimen are ongoing.
Supplementary Material
ACKNOWLEDGMENTS
We thank Huy Trinh for his gift of GT 6 patient sera and Mark Cockett for his support. Editorial assistance with the preparation of the manuscript was provided by Nick Fitch of Articulate Science (London, United Kingdom) and funded by Bristol-Myers Squibb.
All authors are, or were at the time that the work described was carried out, employees of Bristol-Myers Squibb and may be stockholders thereof. In addition, J. A. Bender, M. Ding, and R. G. Gentles are named in U.S. patent 7,456,166 assigned to Bristol-Myers Squibb.
Footnotes
Published ahead of print 14 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02495-13.
REFERENCES
- 1.Lavanchy D. 2009. The global burden of hepatitis C. Liver Int. 29(Suppl 1):74–81. 10.1111/j.1478-3231.2008.01934.x [DOI] [PubMed] [Google Scholar]
- 2.Simmonds P. 2004. Genetic diversity and evolution of hepatitis C virus—15 years on. J. Gen. Virol. 85:3173–3188. 10.1099/vir.0.80401-0 [DOI] [PubMed] [Google Scholar]
- 3.Di Bisceglie AM, Order SE, Klein JL, Waggoner JG, Sjogren MH, Kuo G, Houghton M, Choo QL, Hoofnagle JH. 1991. The role of chronic viral hepatitis in hepatocellular carcinoma in the United States. Am. J. Gastroenterol. 86:335–338 [PubMed] [Google Scholar]
- 4.Fattovich G, Giustina G, Degos F, Tremolada F, Diodati G, Almasio P, Nevens F, Solinas A, Mura D, Brouwer JT, Thomas H, Njapoum C, Casarin C, Bonetti P, Fuschi P, Basho J, Tocco A, Bhalla A, Galassini R, Noventa F, Schalm SW, Realdi G. 1997. Morbidity and mortality in compensated cirrhosis type C: a retrospective follow-up study of 384 patients. Gastroenterology 112:463–472. 10.1053/gast.1997.v112.pm9024300 [DOI] [PubMed] [Google Scholar]
- 5.Kiyosawa K, Sodeyama T, Tanaka E, Gibo Y, Yoshizawa K, Nakano Y, Furuta S, Akahane Y, Nishioka K, Purcell RH. 1990. Interrelationship of blood transfusion, non-A, non-B hepatitis and hepatocellular carcinoma: analysis by detection of antibody to hepatitis C virus. Hepatology 12:671–675. 10.1002/hep.1840120409 [DOI] [PubMed] [Google Scholar]
- 6.Seeff LB, Buskell-Bales Z, Wright EC, Durako SJ, Alter HJ, Iber FL, Hollinger FB, Gitnick G, Knodell RG, Perrillo RP. 1992. Long-term mortality after transfusion-associated non-A, non-B hepatitis. The National Heart, Lung, and Blood Institute Study Group. N. Engl. J. Med. 327:1906–1911 [DOI] [PubMed] [Google Scholar]
- 7.Global Burden Of Hepatitis C Working Group. 2004. Global burden of disease (GBD) for hepatitis C. J. Clin. Pharmacol. 44:20–29. 10.1177/0091270003258669 [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization. 2012. Prevention and control of viral hepatitis infection: framework for global action. World Health Organization, Geneva, Switzerland: http://www.who.int/csr/disease/hepatitis/GHP_framework.pdf Accessed 27 February 2014 [Google Scholar]
- 9.European Association for the Study of the Liver. 2014. EASL clinical practice guidelines: management of hepatitis C virus infection. J. Hepatol. 60:392–420. 10.1016/j.jhep.2013.11.003 [DOI] [PubMed] [Google Scholar]
- 10.Ghany MG, Strader DB, Thomas DL, Seeff LB, American Association for the Study of Liver Diseases 2009. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 49:1335–1374. 10.1002/hep.22759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghany M, Nelson DR, Strader DB, Thomas DL, Seeff LB. 2011. An update on treatment of genotype 1 chronic hepatitis c virus infection: 2011 practice guidelines by the American Association for the Study of Liver Diseases. Hepatology 54:1433–1444. 10.1002/hep.24641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pawlotsky JM. 2011. The results of phase III clinical trial with telaprevir and boceprevir presented at the Liver Meeting 2010: a new standard of care for hepatitis C virus genotype 1 infection, but with issues still pending. Gastroenterology 140:746–754. 10.1053/j.gastro.2011.01.028 [DOI] [PubMed] [Google Scholar]
- 13.Manns M, Marcellin P, Fred Poordad FP, Stanislau Affonso de Araujo E, Buti M, Horsmans Y, Ewa Janczewska EJ, Villamil F, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, Kalmeijer R, Beumont-Mauviel M. 2013. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype-1 infection in treatment-naive patients: results from QUEST-2, a phase III trial. J. Hepatol. 58(Suppl 1):S568 (Abstract.) 10.1016/S0168-8278(13)61412-9 [DOI] [Google Scholar]
- 14.Jacobson I, Dore GJ, Foster GR, Fried MW, Radu M, Rafalskiy VV, Moroz L, Craxi A, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, Kalmeijer R, Beumont-Mauviel M. 2013. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from QUEST-1, a phase III trial. J. Hepatol. 58(Suppl 1):S574 (Abstract.) 10.1016/S0168-8278(13)61424-5 [DOI] [Google Scholar]
- 15.Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, Jacobson IM, Kowdley KV, Nyberg L, Subramanian GM, Hyland RH, Arterburn S, Jiang D, McNally J, Brainard D, Symonds WT, McHutchison JG, Sheikh AM, Younossi Z, Gane EJ. 2013. Sofosbuvir for previously untreated chronic hepatitis C infection. N. Engl. J. Med. 368:1878–1887. 10.1056/NEJMoa1214853 [DOI] [PubMed] [Google Scholar]
- 16.Anonymous. 2002. National Institutes of Health consensus development conference statement: management of hepatitis C 2002 (June 10 to 12, 2002). Gastroenterology 123:2082–2099. 10.1053/gast.2002.1232082 [DOI] [PubMed] [Google Scholar]
- 17.Talal AH, Lafleur J, Hoop R, Pandya P, Martin P, Jacobson I, Han J, Korner EJ. 2013. Absolute and relative contraindications to pegylated-interferon or ribavirin in the US general patient population with chronic hepatitis C: results from a US database of over 45 000 HCV-infected, evaluated patients. Aliment. Pharmacol. Ther. 37:473–481. 10.1111/apt.12200 [DOI] [PubMed] [Google Scholar]
- 18.Kolykhalov AA, Mihalik K, Feinstone SM, Rice CM. 2000. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 74:2046–2051. 10.1128/JVI.74.4.2046-2051.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chinnaswamy S, Yarbrough I, Palaninathan S, Kumar CT, Vijayaraghavan V, Demeler B, Lemon SM, Sacchettini JC, Kao CC. 2008. A locking mechanism regulates RNA synthesis and host protein interaction by the hepatitis C virus polymerase. J. Biol. Chem. 283:20535–20546. 10.1074/jbc.M801490200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrus D, Ahmed-El-Sayed N, Simister PC, Miller S, Triconnet M, Hagedorn CH, Mahias K, Rey FA, Astier-Gin T, Bressanelli S. 2010. Further insights into the roles of GTP and the C terminus of the hepatitis C virus polymerase in the initiation of RNA synthesis. J. Biol. Chem. 285:32906–32918. 10.1074/jbc.M110.151316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. 1999. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 96:13034–13039. 10.1073/pnas.96.23.13034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beaulieu PL. 2009. Recent advances in the development of NS5B polymerase inhibitors for the treatment of hepatitis C virus infection. Expert Opin. Ther. Pat. 19:145–164. 10.1517/13543770802672598 [DOI] [PubMed] [Google Scholar]
- 23.Watkins WJ, Ray AS, Chong LS. 2010. HCV NS5B polymerase inhibitors. Curr. Opin. Drug Discov. Dev. 13:441–465 [PubMed] [Google Scholar]
- 24.Sofia MJ, Chang W, Furman PA, Mosley RT, Ross BS. 2012. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J. Med. Chem. 55:2481–2531. 10.1021/jm201384j [DOI] [PubMed] [Google Scholar]
- 25.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA. 2010. Discovery of a β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 53:7202–7218. 10.1021/jm100863x [DOI] [PubMed] [Google Scholar]
- 26.Poordad F, Lawitz E, Kowdley KV, Cohen DE, Podsadecki T, Siggelkow S, Heckaman M, Larsen L, Menon R, Koev G, Tripathi R, Pilot-Matias T, Bernstein B. 2013. Exploratory study of oral combination antiviral therapy for hepatitis C. N. Engl. J. Med. 368:45–53. 10.1056/NEJMoa1208809 [DOI] [PubMed] [Google Scholar]
- 27.Larrey D, Lohse AW, de Ledinghen V, Trepo C, Gerlach T, Zarski JP, Tran A, Mathurin P, Thimme R, Arastéh K, Trautwein C, Cerny A, Dikopoulos N, Schuchmann M, Heim MH, Gerken G, Stern JO, Wu K, Abdallah N, Girlich B, Scherer J, Berger F, Marquis M, Kukolj G, Böcher W, Steffgen J. 2012. Rapid and strong antiviral activity of the non-nucleosidic NS5B polymerase inhibitor BI 207127 in combination with peginterferon alfa 2a and ribavirin. J. Hepatol. 57:39–46. 10.1016/j.jhep.2012.02.015 [DOI] [PubMed] [Google Scholar]
- 28.Biswal BK, Cherney MM, Wang M, Chan L, Yannopoulos CG, Bilimoria D, Nicolas O, Bedard J, James MN. 2005. Crystal structures of the RNA-dependent RNA polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J. Biol. Chem. 280:18202–18210. 10.1074/jbc.M413410200 [DOI] [PubMed] [Google Scholar]
- 29.Rigat K, Wang Y, Hudyma TW, Ding M, Zheng X, Gentles RG, Beno BR, Gao M, Roberts SB. 2010. Ligand-induced changes in hepatitis C virus NS5B polymerase structure. Antiviral Res. 88:197–206. 10.1016/j.antiviral.2010.08.014 [DOI] [PubMed] [Google Scholar]
- 30.Lemm JA, O'Boyle D, II, Liu M, Nower PT, Colonno R, Deshpande MS, Snyder LB, Martin SW, St. Laurent DR, Serrano-Wu MH, Romine JL, Meanwell NA, Gao M. 2010. Identification of hepatitis C virus NS5A inhibitors. J. Virol. 84:482–491. 10.1128/JVI.01360-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Boyle DR, II, Nower PT, Lemm JA, Valera L, Sun JH, Rigat K, Colonno R, Gao M. 2005. Development of a cell-based high-throughput specificity screen using a hepatitis C virus-bovine viral diarrhea virus dual replicon assay. Antimicrob. Agents Chemother. 49:1346–1353. 10.1128/AAC.49.4.1346-1353.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796. 10.1038/nm1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100. 10.1038/nature08960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, Levine S, Chaniewski S, Yu F, Barry D, Chen C, Lee MS, Mosure K, Sun LQ, Sinz M, Meanwell NA, Colonno RJ, Knipe J, Scola P. 2012. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob. Agents Chemother. 56:5387–5396. 10.1128/AAC.01186-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kneteman NM, Howe AY, Gao T, Lewis J, Pevear D, Lund G, Douglas D, Mercer DF, Tyrrell DL, Immermann F, Chaudhary I, Speth J, Villano SA, O'Connell J, Collett M. 2009. HCV796: a selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus. Hepatology 49:745–752. 10.1002/hep.22717 [DOI] [PubMed] [Google Scholar]
- 36.Pierra C, Amador A, Benzaria S, Cretton-Scott E, D'Amours M, Mao J, Mathieu S, Moussa A, Bridges EG, Standring DN, Sommadossi JP, Storer R, Gosselin G. 2006. Synthesis and pharmacokinetics of valopicitabine (NM283), an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. J. Med. Chem. 49:6614–6620. 10.1021/jm0603623 [DOI] [PubMed] [Google Scholar]
- 37.Herlihy KJ, Graham JP, Kumpf R, Patick AK, Duggal R, Shi ST. 2008. Development of intergenotypic chimeric replicons to determine the broad-spectrum antiviral activities of hepatitis C virus polymerase inhibitors. Antimicrob. Agents Chemother. 52:3523–3531. 10.1128/AAC.00533-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pelosi LA, Voss S, Liu M, Gao M, Lemm JA. 2012. Effect on hepatitis C virus replication of combinations of direct-acting antivirals, including NS5A inhibitor daclatasvir. Antimicrob. Agents Chemother. 56:5230–5239. 10.1128/AAC.01209-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chou TC. 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58:621–681. 10.1124/pr.58.3.10 [DOI] [PubMed] [Google Scholar]
- 40.Wang YK, Rigat KL, Roberts SB, Gao M. 2006. A homogeneous, solid-phase assay for hepatitis C virus RNA-dependent RNA polymerase. Anal. Biochem. 359:106–111. 10.1016/j.ab.2006.09.013 [DOI] [PubMed] [Google Scholar]
- 41.Longley MJ, Ropp PA, Lim SE, Copeland WC. 1998. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry 37:10529–10539. 10.1021/bi980772w [DOI] [PubMed] [Google Scholar]
- 42.Dicker IB, Samanta HK, Li Z, Hong Y, Tian Y, Banville J, Remillard RR, Walker MA, Langley DR, Krystal M. 2007. Changes to the HIV long terminal repeat and to HIV integrase differentially impact HIV integrase assembly, activity, and the binding of strand transfer inhibitors. J. Biol. Chem. 282:31186–31196. 10.1074/jbc.M704935200 [DOI] [PubMed] [Google Scholar]
- 43.Migliaccio G, Tomassini JE, Carroll SS, Tomei L, Altamura S, Bhat B, Bartholomew L, Bosserman MR, Ceccacci A, Colwell LF, Cortese R, De Francesco R, Eldrup AB, Getty KL, Hou XS, LaFemina RL, Ludmerer SW, MacCoss M, McMasters DR, Stahlhut MW, Olsen DB, Hazuda DJ, Flores OA. 2003. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 278:49164–49170. 10.1074/jbc.M305041200 [DOI] [PubMed] [Google Scholar]
- 44.Lin C, Gates CA, Rao BG, Brennan DL, Fulghum JR, Luong YP, Frantz JD, Lin K, Ma S, Wei YY, Perni RB, Kwong AD. 2005. In vitro studies of cross-resistance mutations against two hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061. J. Biol. Chem. 280:36784–36791. 10.1074/jbc.M506462200 [DOI] [PubMed] [Google Scholar]
- 45.Fridell RA, Qiu D, Wang C, Valera L, Gao M. 2010. Resistance analysis of the hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro replicon system. Antimicrob. Agents Chemother. 54:3641–3650. 10.1128/AAC.00556-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomei L, Altamura S, Bartholomew L, Bisbocci M, Bailey C, Bosserman M, Cellucci A, Forte E, Incitti I, Orsatti L, Koch U, De Francesco R, Olsen DB, Carroll SS, Migliaccio G. 2004. Characterization of the inhibition of hepatitis C virus RNA replication by nonnucleosides. J. Virol. 78:938–946. 10.1128/JVI.78.2.938-946.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Howe AY, Cheng H, Johann S, Mullen S, Chunduru SK, Young DC, Bard J, Chopra R, Krishnamurthy G, Mansour T, O'Connell J. 2008. Molecular mechanism of hepatitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, HCV-796. Antimicrob. Agents Chemother. 52:3327–3338. 10.1128/AAC.00238-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tomei L, Altamura S, Bartholomew L, Biroccio A, Ceccacci A, Pacini L, Narjes F, Gennari N, Bisbocci M, Incitti I, Orsatti L, Harper S, Stansfield I, Rowley M, De Francesco R, Migliaccio G. 2003. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 77:13225–13231. 10.1128/JVI.77.24.13225-13231.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rydberg EH, Cellucci A, Bartholomew L, Mattu M, Barbato G, Ludmerer SW, Graham DJ, Altamura S, Paonessa G, De Francesco R, Migliaccio G, Carfi A. 2009. Structural basis for resistance of the genotype 2b hepatitis C virus NS5B polymerase to site A non-nucleoside inhibitors. J. Mol. Biol. 390:1048–1059. 10.1016/j.jmb.2009.06.012 [DOI] [PubMed] [Google Scholar]
- 50.Coffin JM. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267:483–489. 10.1126/science.7824947 [DOI] [PubMed] [Google Scholar]
- 51.Sims KD, Lemm J, Eley T, Liu M, Berglind A, Sherman D, Lawitz E, Vutikullird AB, Tebas P, Gao M, Pasquinelli C, Grasela DM. 2014. Randomized, placebo-controlled, single-ascending-dose study of BMS-791325, a hepatitis C virus (HCV) NS5B polymerase inhibitor, in HCV genotype 1 infection. Antimicrob. Agents Chemother. 58:3496–3503. 10.1128/AAC.02579-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beaulieu PL. 2006. Finger loop inhibitors of the HCV NS5B polymerase: discovery and prospects for new HCV therapy. Curr. Opin. Drug Discov. Dev. 9:618–626 [PubMed] [Google Scholar]
- 53.Pauwels F, Mostmans W, Quirynen LM, van der Helm L, Boutton CW, Rueff AS, Cleiren E, Raboisson P, Surleraux D, Nyanguile O, Simmen KA. 2007. Binding-site identification and genotypic profiling of hepatitis C virus polymerase inhibitors. J. Virol. 81:6909–6919. 10.1128/JVI.01543-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X, Li W, Huang S, He B, Chung E, Griffies A, Cooney E, Hughes EA, Kandoussi H, Sims K, Gardiner D, Bertz RJ, Eley T. 2013. Evaluation of pharmacokinetic drug-drug interaction (DDI) between BMS-791325, an NS5B non-nucleoside polymerase inhibitor, daclatasvir and asunaprevir in triple combination in HCV genotype 1-infected patients. J. Hepatol. 58(Suppl 1):S184 (Abstract.) 10.1016/S0168-8278(13)60453-5 [DOI] [Google Scholar]
- 55.Kukolj G, McGibbon GA, McKercher G, Marquis M, Lefebvre S, Thauvette L, Gauthier J, Goulet S, Poupart MA, Beaulieu PL. 2005. Binding site characterization and resistance to a class of non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase. J. Biol. Chem. 280:39260–39267. 10.1074/jbc.M506407200 [DOI] [PubMed] [Google Scholar]
- 56.Tomei L, Altamura S, Paonessa G, De Francesco R, Migliaccio G. 2005. HCV antiviral resistance: the impact of in vitro studies on the development of antiviral agents targeting the viral NS5B polymerase. Antivir. Chem. Chemother. 16:225–245 [DOI] [PubMed] [Google Scholar]
- 57.McPhee F, Falk P, Lemm J, Liu M, Kirk M, Hernandez D, Cooney E, Hughes EA, Gao M. 2012. Characterization of viral escape in HCV genotype 1-infected patients treated with BMS-791325 and pegylated interferon-alfa and ribavirin. J. Hepatol. 56(Suppl 2):S473. 10.1016/S0168-8278(12)61206-9 [DOI] [Google Scholar]
- 58.de Bethune MP. 2010. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009). Antiviral Res. 85:75–90. 10.1016/j.antiviral.2009.09.008 [DOI] [PubMed] [Google Scholar]
- 59.Eley T, Han YH, Huang SP, He B, Li W, Bedford B, Stonier M, Gardiner D, Sims K, Balimane P, Rodrigues D, Bertz RJ. 2012. In vivo and in vitro assessment of asunaprevir (BMS-650032) as an inhibitor and substrate of organic anion polypeptide (OATP) transporters in healthy volunteers. Rev. Antivir. Ther. Infect. Dis. 6:7.(Abstract.) [Google Scholar]
- 60.Everson GT, Sims KD, Rodriguez-Torres M, Hézode C, Lawitz E, Bourlière M, Loustaud-Ratti V, Rustgi V, Schwartz H, Tatum H, Marcellin P, Pol S, Thuluvath PJ, Eley T, Wang X, Huang SP, McPhee F, Wind-Rotolo M, Chung E, Pasquinelli C, Grasela DM, Gardiner DF. 2014. Efficacy of an interferon- and ribavirin-free regimen of daclatasvir, asunaprevir, and BMS-791325 in treatment-naive patients with HCV genotype 1 infection. Gastroenterology 146:420–429. 10.1053/j.gastro.2013.10.057 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.