

Abstract
Oxazolidinones represent a new class of antituberculosis drugs that exert their function by inhibiting protein synthesis. Here, we compared the activities of three oxazolidinones, linezolid, PNU-100480, and AZD5847, against latent tuberculosis using a simple model employing the streptomycin-starved Mycobacterium tuberculosis strain 18b. The in vitro drug susceptibility results showed that the three oxazolidinones had a bacteriostatic effect against actively growing bacilli but potent bactericidal activity against nonreplicating cells. In the murine model of latent infection with M. tuberculosis 18b, the efficacy of the three compounds varied greatly. Indeed, AZD5847 or its prodrug exhibited no activity or only modest activity, respectively, after 2 months of treatment, whereas both linezolid and PNU-100480 were effective against latent bacilli in mice and showed promising outcomes in combination therapy with rifampin. Moreover, the potency of PNU-100480 was significantly greater than that of linezolid, making it an attractive drug candidate in the development of new combination therapies for latent tuberculosis.
INTRODUCTION
Despite the availability of effective chemotherapy, tuberculosis (TB) is still a major global health problem, and >8.6 million cases of active disease and 1.4 million deaths were reported in 2012 (1). Moreover, approximately one-third of the world's population is latently infected with Mycobacterium tuberculosis and at risk of reactivation (1). The slow growth and dormancy of M. tuberculosis contribute to the chronic nature of the infection, and effective treatment of active disease requires lengthy multidrug regimens. This situation is now worsened by the increasing emergence of multidrug-resistant (MDR), extensively drug-resistant (XDR), and even totally drug-resistant M. tuberculosis strains (1), thus underscoring the urgent need for new molecules with potent activity against latent M. tuberculosis and no cross-resistance with existing therapeutic agents.
Oxazolidinones are one of the only three new classes of antibiotics that have been approved by the Food and Drug Administration (FDA) in the last 35 years (2). Oxazolidinones inhibit bacterial protein synthesis via competitive binding to the 23S rRNA in the catalytic site of the bacterial 50S ribosomes (3, 4). Linezolid (LZD), the first oxazolidinone that entered clinical use, exhibits bacteriostatic activity against M. tuberculosis with an MIC of less than 1 μg/ml (5) and has been used against MDR/XDR TB cases (4, 6, 7). However, long-term administration of LZD may cause severe side effects such as anemia, thrombocytopenia, and peripheral neuropathy (8). PNU-100480 (sutezolid; PNU) is an oxazolidinone analogue that has been reported to have an improved safety profile compared to LZD (2). PNU has an MIC similar to that of LZD for M. tuberculosis in vitro but has more potent bactericidal activity in ex vivo whole-blood cultures and in a murine model of TB (9–11). In mice, addition of PNU not only enhanced the bactericidal activity during the initial phase of treatment (9) but also increased the sterilizing activity of the standard first-line regimen in long-term relapse-based combination studies (12). In whole-blood assays, PNU showed cumulative activity with TMC207 and SQ109 (13). LZD and PNU were both developed by companies subsequently acquired by Pfizer, and PNU has more recently been transferred to Sequella. Another antitubercular oxazolidinone in clinical development is AZD5847 (AZD), a candidate drug from AstraZeneca (14). AZD has an MIC that equals the MICs of LZD and PNU, and its bactericidal activity has been demonstrated by 14-day daily oral dosing in the murine model of TB (2). Novel oxazolidinones currently in clinical development, such as tedizolid (15) and radezolid (16), are being optimized for the treatment of infections caused by Gram-positive bacteria.
The results of a 14-day phase II clinical trial showed a modest early bactericidal activity of LZD but no effect during the subsequent 5 days (17), whereas a recent clinical trial of LZD in patients with chronic XDR TB suggested that LZD had sterilizing ability and was effective for treating MDR/XDR TB (18). However, the study had limitations due to the small number of patients enrolled. Therefore, the in vivo potency of LZD against latent TB remains subject to question. As PNU and AZD have now advanced to phase II trials in pulmonary TB patients, they deserve further investigation to evaluate their efficacy and to determine whether they are superior to LZD in the case of latent TB.
As a novel class of antituberculosis agents, oxazolidinones are gaining considerable interest, and more derivatives will be developed because of their great potential for bactericidal activity against latent TB. A simple, rapid, and reliable model is needed for evaluating the sterilizing activities of this class of compounds for future development of new combination regimens. We recently developed such a model for latent TB that uses the streptomycin (STR)-dependent M. tuberculosis 18b strain; in the absence of streptomycin, 18b enters a nonreplicating state hypothesized to be that of the latent tubercle bacillus (19, 20). A panel of antitubercular compounds was compared using this model, and LZD was one of the drugs capable of killing streptomycin-starved 18b (SS18b) in vitro (20). In this study, we compared the activities of some previously established protein synthesis inhibitors and the three oxazolidinones against latent TB using the SS18b model both in vitro and in vivo.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
M. tuberculosis strains H37Rv and 18b were grown at 37°C with shaking in Middlebrook 7H9 (Difco) broth supplemented with 10% albumin-dextrose-catalase (ADC) enrichment, 0.2% glycerol, 0.05% Tween 80, and, in the case of 18b, 50 μg/ml streptomycin (STR), or on solid Middlebrook 7H10 medium (Difco) supplemented with 0.5% glycerol, 10% oleic acid-albumin-dextrose-catalase (OADC), and 50 μg/ml STR.
Nonreplicating SS18b was generated as follows. Strain 18b was grown to mid-logarithmic phase in STR-containing medium and washed three times in phosphate-buffered saline (PBS) containing 0.05% Tween 80 (PBST). The bacterial pellets were resuspended in medium without STR. SS18b cultures were maintained at an optical density at 600 nm (OD600) between 0.2 and 0.5 for 2 weeks (with the addition of fresh medium if necessary), by which time cells had stopped replicating.
Antimicrobials.
STR, isoniazid (INH), rifampin (RIF), kanamycin, amikacin, hygromycin, chloramphenicol, clarithromycin, and fusidic acid were purchased from Sigma-Aldrich. Other drugs were from AstraZeneca (LZD, AZD5847, AZD5847, and phosphate prodrug [pAZD]), Janssen Pharmaceuticals (TMC207), and the NIAID (PNU). Drugs for in vivo experiments were either dissolved or suspended before gavage as follows: RIF in water (21), LZD and PNU in 5% polyethylene glycol 200 and 95% methylcellulose (0.5%) (12), AZD in 0.5% hydroxypropyl methylcellulose (HPMC) plus 0.1% Tween 80, and pAZD in dextrose-NaCl solution. Drug solutions for mouse treatment were prepared weekly and stored at 4°C.
REMA.
To determine the in vitro efficacy of compounds, a 2-week-old SS18b culture (OD600 of 0.1) and an H37Rv culture (OD600 of 0.0002) were used in resazurin reduction microplate assays (REMA). Two- or three-fold serial dilutions of each test compound were prepared in 96-well plates containing tubercle bacilli in a total volume of 100 μl and then incubated for 7 days at 37°C before addition of 10 μl of 0.025% resazurin. After overnight incubation, fluorescence of the resazurin metabolite resorufin was determined (excitation at 560 nm and emission at 590 nm; gain, 80) by using a Tecan Infinite M200 microplate reader.
Drug susceptibility testing.
Drug treatment was performed in both actively growing M. tuberculosis H37Rv cells and dormant SS18b cells at 37°C with shaking. Mid-logarithmic-phase M. tuberculosis cultures were diluted to an OD of 0.05 and split into 10-ml samples, and drugs were added at the concentrations indicated in the legend to Fig. 2, with an untreated sample serving as a control. Serial dilutions of the cultures were plated on 7H10 medium supplemented with glycerol, OADC, and STR (in the case of SS18b) at day 7 after addition of drugs. The STR-dependent phenotype was checked by plating the same dilutions on 7H10 medium without STR. CFU were counted after 4 to 5 weeks of incubation at 37°C.
FIG 2.
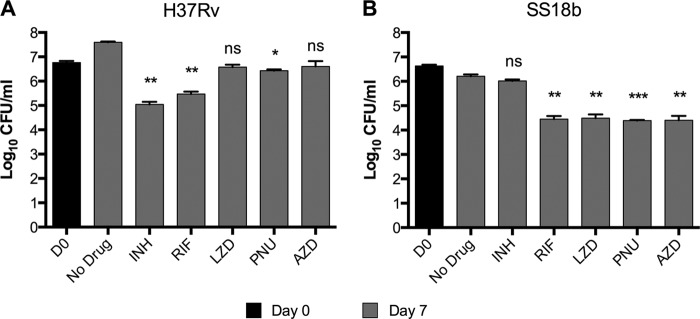
In vitro drug susceptibility test against actively growing M. tuberculosis H37Rv and nongrowing SS18b. (A) M. tuberculosis H37Rv culture was split into several samples and incubated with different drugs, as indicated. Serial dilutions were made and plated 7 days after the addition of drugs. (B) An SS18b culture was divided into several samples and processed as described for the experiment shown in panel A. Bars represent the means ± standard deviations of two experiments. Statistically significant differences relative to the untreated control (No Drug) were calculated using a Student t test. *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ns, no significant difference. Drug concentrations were as follows: INH, 0.25 μg/ml; RIF, 0.5 μg/ml; LZD, 2.5 μg/ml; PNU, 2.5 μg/ml; AZD, 2.5 μg/ml.
Hypoxia model.
Antimicrobial activity against hypoxic nonreplicating M. tuberculosis H37Rv cultures was measured as described earlier (22) with minor modifications. Briefly, H37Rv cells were grown in Dubos Tween broth in McCartney bottles with a magnetic bead using a defined headspace ratio (HSR) of 0.5. Methylene blue was added as a redox indicator (final concentration of 1.5 μg/ml) to all bottles to monitor oxygen depletion. The McCartney bottles were placed on a magnetic stirrer set at 180 rpm, inside a 37°C incubator. The methylene blue indicator started to fade by day 8 and was completely decolorized by 12 days. The antimicrobial activity of various compounds against nonreplicating cells was determined in 96-well microtiter plates using a 14-day-old hypoxia-adapted culture. The entire assay was performed in a hypoxic chamber (DuPoy) by exposing hypoxic cells to various concentrations of compounds for 7 days at 37°C. An anaerobic indicator strip was placed inside the chamber to visually confirm the removal of oxygen during the entire process. Bacterial enumeration was performed on Middlebrook 7H11 agar plates after incubation of plates for 21 to 28 days at 37°C in 5% CO2 in a humidified atmosphere. INH was used as a control to check for hypoxic activity.
Mice and infection.
Female BALB/c mice, aged 5 to 6 weeks, were obtained from Charles River Laboratories. For infections, logarithmic-phase 18b cells were suspended in PBST with 50 μg/ml STR and delivered intravenously (i.v.) in the lateral tail vein at 107 CFU per mouse. STR was given subcutaneously (s.c.) at a dose of 100 mg/kg of body weight once daily, five times/week for 3 weeks. Experiments were approved by the Swiss Cantonal Veterinary Authority (authorization number 2218).
Treatment.
Drug treatment was initiated 4 weeks after infection. Drugs were administered by gavage at the following doses (mg/kg): RIF, 10; LZD, 100; PNU, 100; AZD, 125; and pAZD, 500. AZD and pAZD were given six times a week for 8 weeks. When combined with RIF, oxazolidinones were administered 1 h after RIF. Treatment efficacy was assessed by counting the number of CFU in lungs at the end of treatment. Control and treated mice were sacrificed, the lungs were homogenized, and dilutions were plated on 7H10 agar enriched with 10% OADC and supplemented with STR (50 μg/ml), cycloheximide (10 μg/ml), and ampicillin (50 μg/ml). The plates were incubated for 28 days at 37°C before CFU were enumerated. In a different mouse study, LZD and PNU were administered five times a week for 4 weeks. Treatment efficacy was assessed by lung and spleen CFU counts at the end of treatment.
Statistical analysis.
CFU counts were log10 transformed before analysis, expressed as mean log10 CFU ± standard deviation (SD), and compared using unpaired Student's t tests in GraphPad Prism, version 6.0.
RESULTS
In vitro activities against SS18b determined by REMA.
In our initial work, we showed that fusidic acid, a weak protein synthesis inhibitor, was slightly active against SS18b (19, 20), and this prompted us to assess the behavior of other translation inhibitors in this model. We therefore tested the old drugs, amikacin, chloramphenicol, clarithromycin, fusidic acid, and hygromycin, using the REMA and CFU assays. The results showed that these compounds, especially amikacin and hygromycin, were active against nonreplicating 18b cells (see Fig. S1 in the supplemental material). Encouraged by these findings, we then tested the activities of three oxazolidinones (LZD, PNU, and AZD) and control drugs against SS18b using the REMA.
Figure 1 shows the results obtained after 7 days of incubation of SS18b cultures with the compounds. The positive-control drugs, RIF and TMC207, whose activity against nonreplicating bacilli has been well demonstrated (19, 20), consistently caused a concentration-dependent decrease in fluorescence. INH was used as a negative control, showing no activity against dormant cells but, rather, an increase in fluorescence, a common signature of cell wall inhibitors (20). All three oxazolidinones were effective against SS18b. For both LZD and PNU, the activity of the drug was already seen by fluorescence readout at 50 ng/ml with maximum activity at 400 ng/ml, whereas for AZD activity was observed only at 150 ng/ml or at higher concentrations. In all cases, M. tuberculosis H37Rv served as a reference strain to determine the MICs of all compounds, and the results indicated that all three oxazolidinones were active against growing bacteria with similar MICs of about 400 ng/ml (Fig. 1).
FIG 1.

Resazurin reduction microplate assays (REMA). Serial 3-fold dilutions of each drug were prepared in a 96-well plate, and their activities against M. tuberculosis strains H37Rv and SS18b were evaluated by REMA. Graphs show the correlations between drug concentrations on the x axes and fluorescence levels on the y axes. Bars represent the means ± standard deviations of two experiments.
Validation of in vitro activities against SS18b by CFU levels.
To validate the drug susceptibility results obtained by REMA, we then measured the bactericidal activities of the three oxazolidinones by CFU determination. Figure 2 shows the results of the experiments carried out on actively growing M. tuberculosis H37Rv (Fig. 2A) and nonreplicating SS18b bacilli (Fig. 2B). The untreated H37Rv culture showed more than a 1-log increase in the number of CFU over 7 days of incubation. Both INH and RIF were active, leading to decreases in CFU of 1.7 log (P = 0.0026) and 1.3 log (P = 0.0041), respectively. All three oxazolidinones were able to prevent the growth of the bacteria at the concentrations tested. However, LZD and AZD did not cause a significant decrease in terms of CFU numbers, indicating that they are bacteriostatic drugs. On the other hand, PNU was the only bactericidal oxazolidinone compound although it caused only a 0.3-log (P = 0.03) reduction in the number of CFU.
The above-mentioned drugs were then tested against SS18b. No growth was seen in the absence of STR in the untreated sample, confirming that 18b cells were arrested in the nongrowing state. Addition of INH did not cause a significant decrease in CFU level (P = 0.1012), whereas RIF had potent activity against nonreplicating 18b cells, with a 1.8-log decrease (P = 0.0034) in the number of CFU, in agreement with previous reports (19, 20). In contrast to what was observed when cultures were actively multiplying, the three oxazolidinones exhibited strong killing activity against dormant cells and caused a 1.8-log CFU reduction (P < 0.01), similar to that obtained with RIF. Taken together, these data confirmed the results obtained by REMA.
Efficacy of the oxazolidinones against nonreplicating bacteria was also assessed using the Wayne and Haynes model of hypoxia (22). Interestingly, when tested against hypoxic M. tuberculosis H37Rv cells, both PNU and AZD displayed good bactericidal activity, mirroring the results obtained with SS18b, whereas LZD performed less well (see Fig. S2 in the supplemental material).
In vivo activities of AZD5847.
Interestingly, oxazolidinones displayed greater bactericidal activity against nonreplicating than growing cells in vitro, which prompted us to investigate their efficacy against SS18b in vivo. After infection with M. tuberculosis 18b, BALB/c mice received STR for 3 weeks to allow bacterial replication in the lungs. Treatment began 10 days after STR withdrawal, when the mice had established a chronic infection. In the first experiment, AZD alone and in combination with RIF was tested. After 2 months of treatment (Fig. 3), a reduction of more than 3 logs in the number of CFU (P < 0.0001) was observed in the lungs of mice treated with RIF in comparison with the lungs of untreated mice. In contrast, AZD treatment did not cause any decrease in lung CFU numbers, suggesting that it had no bactericidal activity against nonreplicating bacilli in mice. Furthermore, the combination of AZD and RIF was as potent as RIF alone (P = 0.7533), indicating no synergistic, additive, or antagonistic effects.
FIG 3.
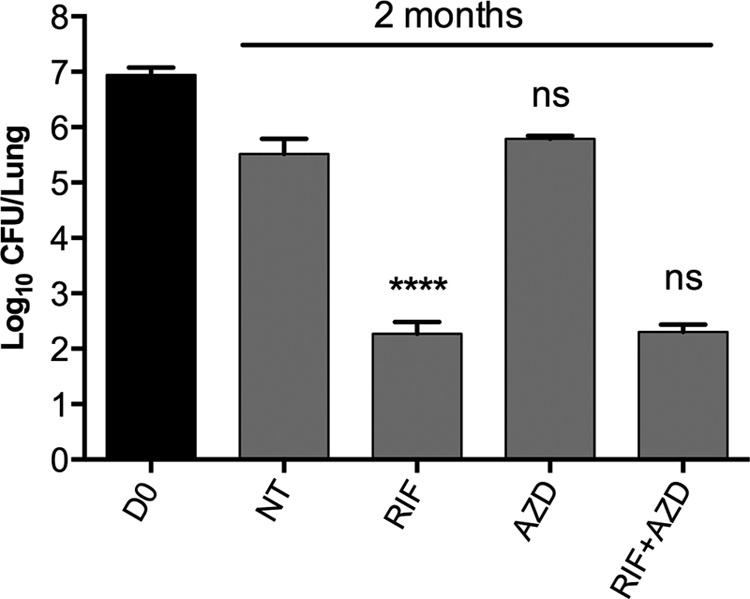
In vivo efficacy of AZD5847 against nonreplicating M. tuberculosis 18b. M. tuberculosis 18b-infected mice were treated for 2 months with RIF, AZD, or their combination, starting at 1 month postinfection. Untreated controls (NT) and treated animals were sacrificed at the end of treatment, and CFU counts were determined by plating lung homogenates on 7H10 plates with STR. Bars represent the means ± standard deviations of groups of four mice. Statistically significant differences relative to the untreated controls (for AZD and RIF) or to RIF (for RIF plus AZD) were calculated using a Student t test. ****, P < 0.0001; ns, no significant difference. D0, day 0.
The discrepancy between the in vivo and in vitro potency of AZD was further investigated in another mouse study that examined the efficacy of pAZD, the phosphate prodrug of AZD5847 that provides better pharmacokinetic and pharmacodynamic parameters (AstraZeneca, unpublished data). pAZD alone and in combination with RIF was tested in the same SS18b mouse model of chronic infection. After 1 month of treatment (Fig. 4A), RIF caused a 2-log reduction in the bacterial burden in the lung, while pAZD showed no activity against SS18b in vivo. In addition, the potency of RIF combined with pAZD was equivalent to that of RIF alone. At the end of the experiment, after 2 months of treatment (Fig. 4B), pAZD reduced the bacterial burden by only 0.6 log (P = 0.0027) in mouse lungs, a considerably poorer performance than that of RIF (3-log killing; P < 0.0001). Moreover, combination of pAZD and RIF showed neither synergistic nor additive effects. Overall, these results indicated that AZD was not effective at killing latent SS18b in vivo or that, in the case of its prodrug, the activity was minimal.
FIG 4.
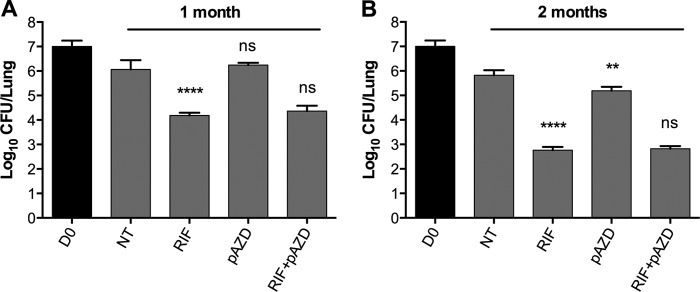
In vivo efficacy of pAZD against nonreplicating M. tuberculosis 18b. M. tuberculosis 18b-infected mice were treated for 2 months with RIF, pAZD, or their combination, starting at 1 month postinfection. Untreated controls (NT) and treated animals were sacrificed after 1 month (A) and 2 months (B) of treatment, and CFU counts were determined by plating lung homogenates on 7H10 plates with STR. Bars represent the means ± standard deviations of groups of 4 mice. Statistically significant differences relative to untreated controls (for pAZD and RIF) or to RIF (for RIF plus pAZD) were calculated using a Student t test. **, P < 0.005; ****, P < 0.0001; ns, no significant difference.
In vivo potency of linezolid and PNU-100480.
The efficacy of LZD and PNU was tested in the same mouse model of SS18b chronic infection. Figure 5 shows the CFU results from mouse lungs (Fig. 5A) and spleens (Fig. 5B) after 1 month of treatment. RIF reduced the bacterial burden by 2.1 logs (P < 0.0001) and 1.6 logs (P < 0.0001) in the lungs and spleens, respectively. LZD was effective against dormant bacilli and had better activity in the spleens (1-log CFU reduction; P = 0.0002) than in the lungs (0.6-log CFU reduction; P = 0.0026). In contrast, PNU exhibited greater efficacy than LZD as it decreased the infection level by 2.5 logs (P < 0.0001) and 1.7 logs (P < 0.0001) in the lungs and spleens, respectively. When combined with RIF, LZD caused an additional 0.6-log reduction in both mouse lungs (P = 0.0095) and spleens (P = 0.029), while PNU further reduced the number of CFU by 2.1 logs in both lungs (P = 0.0011) and spleens (P = 0.0014). These data confirmed that PNU has stronger bactericidal activity against nonreplicating M. tuberculosis than LZD at the doses tested, in accordance with the findings from other in vivo studies (9, 12). Moreover, no antagonism was found between RIF and LZD or PNU, suggesting that the two oxazolidinones could be included in the development of a more potent combination therapy with rifamycins.
FIG 5.
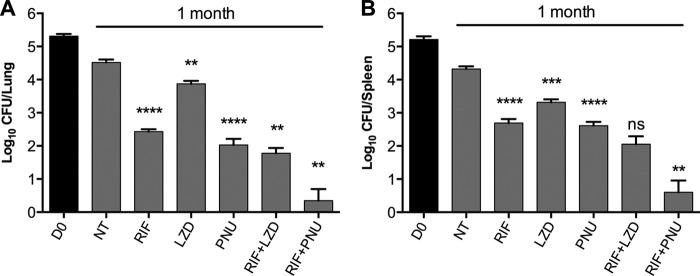
In vivo efficacy of LZD and PNU against nonreplicating M. tuberculosis 18b. Mice infected with M. tuberculosis 18b were treated for 1 month with the indicated drugs or drug combinations, starting at 1 month postinfection. Untreated controls (NT) and treated animals were sacrificed at the end of treatment, and CFU counts were determined by plating lung (A) and spleen (B) homogenates on 7H10 plates with STR. Bars represent the means ± standard deviations of groups of 4 mice. Statistically significant differences relative to untreated controls (for RIF, LZD, or PNU alone) or to RIF (for combinations) were calculated using a Student t test. **, P < 0.005; ***, P < 0.0005; ****, P < 0.0001.
DISCUSSION
We compared the bactericidal activity of various old and new protein synthesis inhibitors, including three oxazolidinones, in the nonreplicating SS18b model of latent TB. Of the previously examined drugs, amikacin and hygromycin showed modest activity and were able to reduce CFU levels in vitro by about 40% compared to the activity of RIF. In contrast, all three oxazolidinones were equally as potent as RIF against nongrowing bacilli. However, their in vivo efficacy varied extensively. PNU had significantly greater activity than LZD at a dose of 100 mg/kg. The enhanced activity of PNU compared to that of LZD is not consistent with their similar MICs, nor can it be explained by a more favorable pharmacokinetic profile since the steady-state area under the concentration-time curve (AUC) for PNU and its metabolites is three times lower than the AUC for LZD (9). Other factors must therefore play a role, including permeability through the eukaryotic cell membrane and intracellular accumulation. Indeed, in a whole-blood assay it has been shown that PNU accumulated in macrophages, which could partially explain the superior activity of PNU over LZD (10, 11). Importantly, our study demonstrated that PNU has the strongest bactericidal activity against nonreplicating SS18b in the murine model and, in combination with RIF, resulted in at least an additive effect that caused significant reduction of the bacterial burden in the animals. It is worth pointing out that the results obtained here in the SS18b model, in terms of CFU log reduction over time upon treatment with PNU, match what has been reported in BALB/c mice chronically infected with H37Rv (9, 12). This validated potent activity of PNU in both chronic and nonreplicating mouse models of TB shows promise for the compound in the clinic. Together with its low-toxicity profile in humans (10, 11), these results indicate that PNU deserves further attention in the context of developing new combination therapies for active and latent tuberculosis.
The most recent of the three oxazolidinones, AZD, unfortunately showed no detectable activity against 18b in vivo. However, the phosphorylated prodrug form, pAZD, which has improved pharmacokinetic properties, did display modest activity at the highest concentration tested after 2 months of treatment. In contrast to the findings obtained with all the other drugs tested here and in previous studies (19, 20), the in vitro potency of AZD was not mirrored by in vivo efficacy in the SS18b model. Previous investigations from AstraZeneca have shown that AZD is active in an ex vivo model (14) and in an in vivo model using H37Rv (23), indicating that, like PNU, this oxazolidinone can cross the eukaryotic cell membrane and thus reach the bacteria engulfed in the macrophages. PNU, the active metabolite of PNU, and LZD are 52%, 89%, and 69% free in human plasma, respectively, in contrast to AZD, which is 20% free (24, 25). Therefore, the relatively poor efficacy of AZD in the SS18b model compared to LZD or PNU could be due to differences in exposure to free drug at the target site. Nevertheless, further studies have to be undertaken in order to explain these differences.
Taken together, our results showed that oxazolidinones and some other ribosome inhibitors, especially amikacin and hygromycin, were active against SS18b cells but to different extents. Notably, oxazolidinones were more active on nonmultiplying cells than on rapidly growing bacteria. This suggests that translation might still occur at a very low level in the absence of streptomycin and that de novo protein synthesis is required during latency. Thus, in our experiments, the oxazolidinones probably shut down the residual protein synthesis, thereby killing the nonreplicating bacilli. Alternatively, oxazolidinones may irreversibly damage ribosomes, leading to insufficient translational capacity remaining to support growth when the SS18b cells reactivate.
One also cannot exclude the possibility that there may be a second oxazolidinone target in latent M. tuberculosis and therefore that SS18b is effectively killed via inhibition of both this target and residual translation. Genetic studies confirmed that mutations in the peptidyl transferase center of 23S rRNA or in genes encoding ribosomal proteins (rplV, rplD, and rplC) are associated with oxazolidinone resistance, thereby defining the main target (26). In addition, Sander and coworkers identified two classes of LZD-resistant mutants and described a nonribosomal mechanism of resistance in Mycobacterium smegmatis, which was verified by an in vitro peptidyl transferase assay (27). This finding was supported by another study where mutations leading to low-level LZD resistance occurred in unidentified but nonribosomal genes in M. tuberculosis (28).
Overall, the data reported here indicate that the translation machinery is a vulnerable target during latent tuberculosis infection, similar to transcription, ATP biosynthesis, and DNA metabolism (19, 20). Due to their great potency, oxazolidinones represent a class of translation inhibitors that offers tremendous potential for developing more powerful antituberculosis regimens in general as well as better regimens for the treatment of MDR/XDR cases.
Supplementary Material
ACKNOWLEDGMENTS
We thank Koen Andries and C. E. Barry III for kindly providing drugs.
The research leading to these results received funding from the European Community's Seventh Framework Programme (grant 260872) and the Swiss National Science Foundation (grant 31003A-140778). This research was supported in part by Intramural Research Program of the NIH, NIAID.
Footnotes
Published ahead of print 24 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02410-14.
REFERENCES
- 1.World Health Organization. 2012. Global tuberculosis report 2012. World Health Organization; Geneva, Switzerland: (http://www.who.int/tb/publications/global_report/en/) [Google Scholar]
- 2.Shaw KJ, Barbachyn MR. 2011. The oxazolidinones: past, present, and future. Ann. N. Y. Acad. Sci. 1241:48–70. 10.1111/j.1749-6632.2011.06330.x [DOI] [PubMed] [Google Scholar]
- 3.Ippolito JA, Kanyo ZF, Wang D, Franceschi FJ, Moore PB, Steitz TA, Duffy EM. 2008. Crystal structure of the oxazolidinone antibiotic linezolid bound to the 50S ribosomal subunit. J. Med. Chem. 51:3353–3356. 10.1021/jm800379d [DOI] [PubMed] [Google Scholar]
- 4.Patel U, Yan YP, Hobbs FW, Jr, Kaczmarczyk J, Slee AM, Pompliano DL, Kurilla MG, Bobkova EV. 2001. Oxazolidinones mechanism of action: inhibition of the first peptide bond formation. J. Biol. Chem. 276:37199–37205. 10.1074/jbc.M102966200 [DOI] [PubMed] [Google Scholar]
- 5.Huang TS, Liu YC, Sy CL, Chen YS, Tu HZ, Chen BC. 2008. In vitro activities of linezolid against clinical isolates of Mycobacterium tuberculosis complex isolated in Taiwan over 10 years. Antimicrob. Agents Chemother. 52:2226–2227. 10.1128/AAC.00414-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fortun J, Martin-Davila P, Navas E, Perez-Elias MJ, Cobo J, Tato M, De la Pedrosa EG, Gomez-Mampaso E, Moreno S. 2005. Linezolid for the treatment of multidrug-resistant tuberculosis. J. Antimicrob. Chemother. 56:180–185. 10.1093/jac/dki148 [DOI] [PubMed] [Google Scholar]
- 7.Park IN, Hong SB, Oh YM, Kim MN, Lim CM, Lee SD, Koh Y, Kim WS, Kim DS, Kim WD, Shim TS. 2006. Efficacy and tolerability of daily-half dose linezolid in patients with intractable multidrug-resistant tuberculosis. J. Antimicrob. Chemother. 58:701–704. 10.1093/jac/dkl298 [DOI] [PubMed] [Google Scholar]
- 8.Migliori GB, Eker B, Richardson MD, Sotgiu G, Zellweger JP, Skrahina A, Ortmann J, Girardi E, Hoffmann H, Besozzi G, Bevilacqua N, Kirsten D, Centis R, Lange C, TBNET Study Group. 2009. A retrospective TBNET assessment of linezolid safety, tolerability and efficacy in multidrug-resistant tuberculosis. Eur. Respir. J. 34:387–393. 10.1183/09031936.00009509 [DOI] [PubMed] [Google Scholar]
- 9.Williams KN, Stover CK, Zhu T, Tasneen R, Tyagi S, Grosset JH, Nuermberger E. 2009. Promising antituberculosis activity of the oxazolidinone PNU-100480 relative to that of linezolid in a murine model. Antimicrob. Agents Chemother. 53:1314–1319. 10.1128/AAC.01182-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallis RS, Jakubiec W, Kumar V, Bedarida G, Silvia A, Paige D, Zhu T, Mitton-Fry M, Ladutko L, Campbell S, Miller PF. 2011. Biomarker-assisted dose selection for safety and efficacy in early development of PNU-100480 for tuberculosis. Antimicrob. Agents Chemother. 55:567–574. 10.1128/AAC.01179-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallis RS, Jakubiec WM, Kumar V, Silvia AM, Paige D, Dimitrova D, Li X, Ladutko L, Campbell S, Friedland G, Mitton-Fry M, Miller PF. 2010. Pharmacokinetics and whole-blood bactericidal activity against Mycobacterium tuberculosis of single doses of PNU-100480 in healthy volunteers. J. Infect. Dis. 202:745–751. 10.1086/655471 [DOI] [PubMed] [Google Scholar]
- 12.Williams KN, Brickner SJ, Stover CK, Zhu T, Ogden A, Tasneen R, Tyagi S, Grosset JH, Nuermberger EL. 2009. Addition of PNU-100480 to first-line drugs shortens the time needed to cure murine tuberculosis. Am. J. Respir. Crit. Care Med. 180:371–376. 10.1164/rccm.200904-0611OC [DOI] [PubMed] [Google Scholar]
- 13.Wallis RS, Jakubiec W, Mitton-Fry M, Ladutko L, Campbell S, Paige D, Silvia A, Miller PF. 2012. Rapid evaluation in whole blood culture of regimens for XDR-TB containing PNU-100480 (sutezolid), TMC207, PA-824, SQ109, and pyrazinamide. PLoS One 7:e30479. 10.1371/journal.pone.0030479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balasubramanian V, Solapure S, Iyer H, Ghosh A, Sharma S, Kaur P, Deepthi R, Subbulakshmi V, Ramya V, Ramachandran V, Balganesh M, Wright L, Melnick D, Butler SL, Sambandamurthy VK. 2014. Bactericidal activity and mechanism of action of AZD5847, a novel oxazolidinone for treatment of tuberculosis. Antimicrob. Agents Chemother. 58:495–502. 10.1128/AAC.01903-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prokocimer P, Bien P, Surber J, Mehra P, DeAnda C, Bulitta JB, Corey GR. 2011. Phase 2, randomized, double-blind, dose-ranging study evaluating the safety, tolerability, population pharmacokinetics, and efficacy of oral torezolid phosphate in patients with complicated skin and skin structure infections. Antimicrob. Agents Chemother. 55:583–592. 10.1128/AAC.00076-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaadt R, Sweeney D, Shinabarger D, Zurenko G. 2009. In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent. Antimicrob. Agents Chemother. 53:3236–3239. 10.1128/AAC.00228-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dietze R, Hadad DJ, McGee B, Molino LP, Maciel EL, Peloquin CA, Johnson DF, Debanne SM, Eisenach K, Boom WH, Palaci M, Johnson JL. 2008. Early and extended early bactericidal activity of linezolid in pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 178:1180–1185. 10.1164/rccm.200806-892OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee M, Lee J, Carroll MW, Choi H, Min S, Song T, Via LE, Goldfeder LC, Kang E, Jin B, Park H, Kwak H, Kim H, Jeon H-S, Jeong I, Joh JS, Chen RY, Olivier KN, Shaw PA, Follmann D, Song SD, Lee J-K, Lee D, Kim CT, Dartois V, Park S-K, Cho S-N, Barry CE. 2012. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N. Engl. J. Med. 367:1508–1518. 10.1056/NEJMoa1201964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sala C, Dhar N, Hartkoorn RC, Zhang M, Ha YH, Schneider P, Cole ST. 2010. Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 54:4150–4158. 10.1128/AAC.00821-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang M, Sala C, Hartkoorn RC, Dhar N, Mendoza-Losana A, Cole ST. 2012. Streptomycin-starved Mycobacterium tuberculosis 18b, a drug discovery tool for latent tuberculosis. Antimicrob. Agents Chemother. 56:5782–5789. 10.1128/AAC.01125-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang M, Li SY, Rosenthal IM, Almeida DV, Ahmad Z, Converse PJ, Peloquin CA, Nuermberger EL, Grosset JH. 2011. Treatment of tuberculosis with rifamycin-containing regimens in immune-deficient mice. Am. J. Respir. Crit. Care Med. 183:1254–1261. 10.1164/rccm.201012-1949OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wayne LG, Hayes LG. 1996. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64:2062–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balasubramanian V, Gaonkar S, Solapure S, Sambandamurthy V, Shandil R, Mahesh K, Sharma S, Kaur P, Deepthi R, Subbulakshmi V, Ramya V, Ramachandran V, Reddy J, Giridhar J, Deshpande A, Bharath S, Kumar N, Balganesh M, Nandi V, Wright I, Melnick D. 2011. AZD5847, an oxazolidinone for the treatment of tuberculosis: pre-clinical studies, poster F1-1364 Abstr. 51st Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC [Google Scholar]
- 24.Louie A, Brown D, Files K, Swift M, Fikes S, Drusano G. 2012. Pharmacodynamics of PNU-100480 (U, Sutezolid), a new oxazolidinone, in combination with its active metabolite in the killing of Mycobacterium tuberculosis (Mtb) in an in vitro hollow fiber infection model (HFIM), abstr A-414, p 1265 Abstr 52nd Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC [Google Scholar]
- 25.Gravestock MB, Acton DG, Betts MJ, Dennis M, Hatter G, McGregor A, Swain ML, Wilson RG, Woods L, Wookey A. 2003. New classes of antibacterial oxazolidinones with C-5, methylene O-linked heterocyclic side chains. Bioorg. Med. Chem. Lett. 13:4179–4186. 10.1016/j.bmcl.2003.07.033 [DOI] [PubMed] [Google Scholar]
- 26.Beckert P, Hillemann D, Kohl TA, Kalinowski J, Richter E, Niemann S, Feuerriegel S. 2012. rplC T460C identified as a dominant mutation in linezolid-resistant Mycobacterium tuberculosis strains. Antimicrob. Agents Chemother. 56:2743–2745. 10.1128/AAC.06227-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sander P, Belova L, Kidan YG, Pfister P, Mankin AS, Bottger EC. 2002. Ribosomal and non-ribosomal resistance to oxazolidinones: species-specific idiosyncrasy of ribosomal alterations. Mol. Microbiol. 46:1295–1304. 10.1046/j.1365-2958.2002.03242.x [DOI] [PubMed] [Google Scholar]
- 28.Hillemann D, Rusch-Gerdes S, Richter E. 2008. In vitro-selected linezolid-resistant Mycobacterium tuberculosis mutants. Antimicrob. Agents Chemother. 52:800–801. 10.1128/AAC.01189-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.