

SUMMARY
Here we present the ascidian Ciona intestinalis as an alternative invertebrate system to study Alzheimer’s disease (AD) pathogenesis. Through the use of AD animal models, researchers often attempt to reproduce various aspects of the disease, particularly the coordinated processing of the amyloid precursor protein (APP) by α-, β- and γ-secretases to generate amyloid beta (Aβ)-containing plaques. Recently, Drosophila and C. elegans AD models have been developed, exploiting the relative simplicity of these invertebrate systems, but they lack a functional Aβ sequence and a β-secretase ortholog, thus complicating efforts to examine APP processing in vivo. We propose that the ascidian is a more appropriate invertebrate AD model owing to their phylogenetic relationship with humans. This is supported by bioinformatic analyses, which indicate that the ascidian genome contains orthologs of all AD-relevant genes. We report that transgenic ascidian larvae can properly process human APP695 to generate Aβ peptides. Furthermore, Aβ can rapidly aggregate to form amyloid-like plaques, and plaque deposition is significantly increased in larvae expressing a human APP695 variant associated with familial Alzheimer’s disease. We also demonstrate that nervous system-specific Aβ expression alters normal larval behavior during attachment. Importantly, plaque formation and alterations in behavior are not only observed within 24 hours post-fertilization, but anti-amyloid drug treatment improves these AD-like pathologies. This ascidian model for AD provides a powerful and rapid system to study APP processing, Aβ plaque formation and behavioral alterations, and could aid in identifying factors that modulate amyloid deposition and the associated disruption of normal cellular function and behaviors.
INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disorder, and can lead to a complete loss of memory and the inability to function independently. An estimated 27 million people worldwide are diagnosed with some form of AD, with a new diagnosis made every 71 seconds (Maslow, 2008). Despite the US$248 billion spent annually as a result of the disease, relatively little is available with regards to AD treatment. The majority of the currently available treatments for AD are drugs that focus on the symptoms of the disease process and not the cause of the disease. Furthermore, with the aging global population, the worldwide prevalence of AD is projected to quadruple by the middle of the century if no new treatments are developed (Brookmeyer et al., 2007).
One pathological hallmark of the disease process is the formation of extracellular plaques in the AD brain. These plaques consist primarily of amyloid beta (Aβ) aggregates that are believed to result from abnormal proteolytic cleavage of the type I integral membrane amyloid precursor protein (APP) (reviewed in Hardy and Selkoe, 2002; Wilquet and De Strooper, 2004; Marambaud and Robakis, 2005; Zhang and Xu, 2007). Briefly, proteolytic processing of APP can occur through either an amyloidogenic or non-amyloidogenic pathway depending on the proteinases that sequentially cleave full-length APP (Fig. 1). The majority of APP is processed down the non-amyloidogenic pathway as a result of cleavage at the cell surface by alpha-secretase (α-secretase). Cleavage by α-secretase precludes the formation of full-length Aβ and is considered neuroprotective. Alternatively, primary cleavage of APP by the beta-secretase (β-secretase) activity of the membrane-bound β-site APP-cleaving enzyme (BACE) occurs at the N-terminus of the Aβ domain and is required for Aβ peptide formation. The extracellular cleavage event by either α- or β-secretase results in the release of the APP ectodomain as soluble APP (sAPPα or sAPPβ) and a membrane-bound carboxy-terminal fragment (α-CTF or β-CTF). The gamma-secretase (γ-secretase) complex (APH-1, PEN-2, presenilin and nicastrin) subsequently processes the carboxy-terminal fragment within the transmembrane domain at the C-terminus of the Aβ sequence. This causes the release of the APP intracellular domain (AICD) and either the p3 or Aβ peptide (in combination with α-secretase or β-secretase cleavage, respectively). The two most commonly observed forms of Aβ are the soluble, 40-amino-acid Aβ (Aβ1–40) and the insoluble, 42-amino-acid Aβ (Aβ1-42) forms, the latter Aβ species being the primary component found in amyloid plaques.
Fig. 1.
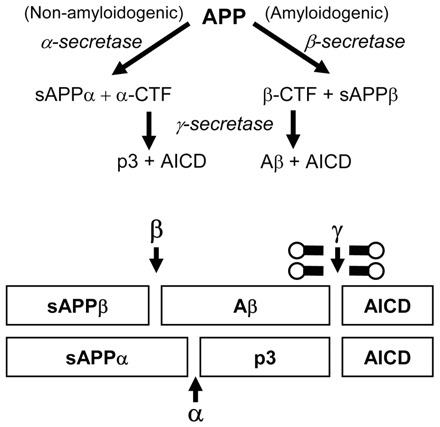
APP processing and Aβ plaque formation. Amyloidogenic processing of APP occurs as a result of the β-secretase activity of BACE. This results in the production of the sAPPβ and the β-CTF. Intramembranous cleavage of the β-CTF by the γ-secretase enzymatic complex results in the production of Aβ which can subsequently aggregate to form plaques. Non-amyloidogenic processing is a result of APP cleavage by α-secretase which takes place within the Aβ sequence, precluding its formation, and is considered to be neuroprotective.
Numerous alternative model systems have been used to study Alzheimer’s progression and treatment. A variety of neuronal and non-neuronal human cell lines are being employed to examine several aspects associated with the disease process, and for screening novel therapeutic treatments. Although these cell line-based assays can offer useful insights into the specific molecular mechanisms associated with the disease, cultured cells fail to provide the complex interactions between cell types that can only be obtained by using whole animal model systems. For these reasons, researchers have looked to use a variety of animal models, with the common assumption that the fundamental pathological mechanisms behind human diseases are also present. Various transgenic mouse models have been generated that reproduce aspects of human AD histopathology. In addition, a correlation between the associated pathology and cognitive performance has been established in many transgenic AD mouse models (Gotz et al., 2004). Although mouse model systems have been valuable in understanding certain aspects of the disease process, there are a variety of limitations regarding their use, including the high cost and length of time associated with generating a transgenic mouse displaying pathological characteristics of AD.
A growing trend in AD research has been the use of invertebrate model systems, particularly Drosophila and C. elegans, owing to their highly characterized genetics, rapid development and expansive collection of experimental procedures. Although invertebrates may lack the neuronal and behavioral complexity of mammals, their use has been significant in directly investigating the molecular and cellular processes that underlie the disease process, and have allowed for rapid and clear observation of the phenotypes commonly associated with AD (Gotz et al., 2004; Link, 2005; Wu and Luo, 2005; Crowther et al., 2006). Importantly, these rapid disease models can potentially be exploited for use in high-throughput genetic and drug screening. However, because of their distant evolutionary relationship to vertebrates, findings in these non-chordate invertebrates can be difficult to extrapolate to human neurological diseases. With regard to AD experiments, the lack of BACE exhibiting β-secretase activity in both flies and worms has complicated efforts to examine APP processing in these systems.
Recent molecular phylogenic analyses suggest that urochordates, including ascidians, are the true sister group of vertebrates (Graham, 2004; Delsuc et al., 2006) and will probably provide an excellent genomic background for modeling human diseases. In contrast to other invertebrate models, ascidians, like humans, are chordates, share a larval notochord, and undergo neurulation to form a dorsal hollow neural tube. Within 18 hours post-fertilization (hpf), free-swimming tadpole larvae contain functional central and peripheral nervous systems that are composed of approximately 350 cells, including an anterior sensory vesicle and a visceral ganglion containing motor neurons. This simple chordate nervous system is important in coordinating several aspects of tadpole behavior, including larval swimming and their ability to respond to the environmental cues that are necessary for settlement (Zega et al., 2006; Imai and Meinertzhagen, 2007).
Ascidians are amenable to extensive embryological manipulation and a number of tools have been developed to alter gene activity. Our lab has developed an efficient electroporation-based method for generating hundreds of transgenic ascidian embryos (Zeller, 2004; Zeller et al., 2006a). Fertilized ascidian eggs are mixed with supercoiled plasmid DNA, subjected to an electrical pulse and allowed to develop. A rapid period of embryogenesis allows for analysis of transgene expression within 18 hpf. Tissue-specific misexpression of wild-type or mutant proteins can be achieved by using promoter elements to drive the expression of any protein of interest. We have carefully analyzed and optimized the experimental conditions, and have shown that co-electroporated transgenes are expressed in the same cells (in >95% of embryos examined) and that the electroporation conditions can be varied to adjust the level of transgene expression in a predictable manner (Zeller et al., 2006a). However, to date, there has been no concerted effort to exploit ascidian embryos as a model for a specific human disease.
To determine whether the ascidian Ciona intestinalis could be utilized as a model for AD, transgenic larvae were generated that express wild-type and mutant forms of human APP (hAPP695). We show that expression of hAPP695 appears to be processed in a similar manner to the well-characterized Aβ cascade. Furthermore, hAPP695 expression can lead to the formation of Aβ-containing deposits as early as 23 hpf, as assessed by thioflavin S, a dye commonly used to stain for amyloid plaques (Guntern et al., 1992). Increased plaque formation can be achieved by introducing point mutations associated with familial AD into hAPP695. Transgenic ascidians expressing Aβ1–42 in the larval nervous system display severe deficiencies with their ability to settle, a behavioral response that is important for metamorphosis. Importantly, treatment of transgenic larvae with an anti-amyloid therapeutic leads to both a decrease in plaque formation, in a dose-dependent manner, and an improvement in larval attachment. This initial analysis demonstrates that the ascidian provides a rapid invertebrate model system, in a more appropriate chordate context, to study AD pathogenesis.
RESULTS
Alzheimer’s disease – relevant gene conservation
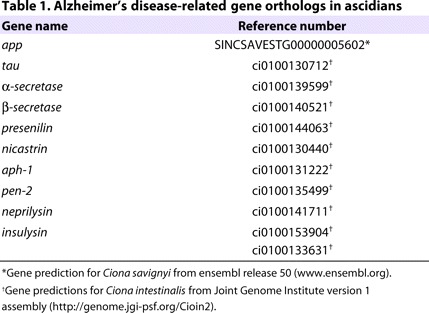
The close phylogenetic relationship between ascidians and vertebrates suggests that a large number of AD-relevant genes are present in the Ciona genome, and that they probably display a higher degree of conservation when compared with other invertebrates. Bioinformatic approaches were utilized to identify candidate ascidian homologs of genes implicated in AD (Table 1). Although we could not identify a complete APP-like sequence in the EST database of C. intestinalis, the single APP homolog believed to be present in the C. savignyi genome contains a short peptide sequence within its predicted transmembrane domain that is conserved with the Aβ region of APP homologous proteins found in vertebrates. We have also identified a putative ascidian ortholog to the protein tau, the major component of the neurofibrillary tangles associated with AD pathogenesis. In addition to APP, single candidate ascidian orthologs to proteins involved in APP processing, or the Aβ cascade, have been identified; these include α-secretase and all components of the γ-secretase enzymatic complex (Zhang and Xu, 2007). Importantly, the identification of a putative β-secretase ortholog is of particular interest owing to its absence in other invertebrate models (Fossgreen et al., 1998; Greeve et al., 2004). This identified BACE-like protein, CiBace, has 31.5% and 29.9% identity to human BACE1 and BACE2, respectively (supplementary material Fig. S1). Importantly, the regions containing the active-site aspartases Asp62 and Asp254 in CiBace are conserved and located within the active-site motif D(T/S)G, characteristic for aspartic proteases. In addition to the proteins involved in APP processing, we also identified orthologs to neprilysin and insulysin, which are enzymes that are thought to be involved in the degradation or clearance of the processed Aβ peptide (Nalivaeva et al., 2008).
Table 1.
Alzheimer’s disease-related gene orthologs in ascidians
Transgenic ascidians: a rapid in vivo model for Alzheimer’s disease
The high degree of genetic conservation among key players in AD pathogenesis led us to hypothesize that the coordinated mechanisms involved in the Aβ cascade may also be conserved in ascidians. To examine APP processing in ascidian embryos and larvae, the human APP isoform hAPP695 was expressed transiently and ubiquitously throughout the developing ascidian embryo using cis-regulatory elements from the Ciona elongation factor 1a (ef1a) gene. By fusing a codon-optimized cyan fluorescent protein (CFP) to the C-terminal end of hAPP695, transgenic ascidian larvae could easily be identified (Fig. 2A). At 23 hpf, embryos expressing hAPP695 were harvested and whole lysates were probed using the monoclonal anti-Aβ antibody (6E10) that recognizes a six-aminoacid epitope (FRHDSG) located within the Aβ sequence (McLaurin et al., 2002). In hAPP695-expressing larvae, the 6E10 antibody detected full-length hAPP695 at ∼76 kDa and a ∼4.6 kDa peptide, probably corresponding to the processed Aβ peptide (Fig. 2B). Processed peptides corresponding to single cleavage events at either the α-secretase site (∼70 kDa) or β-secretase site (∼11 kDa) were also present in hAPP695-expressing larvae, suggesting that hAPP695 can be processed in the ascidian through a similar Aβ cascade. Transgenic embryos expressing only CFP did not have any of these bands, although the antibody did react non-specifically to some proteins in ascidian embryos.
Fig. 2.
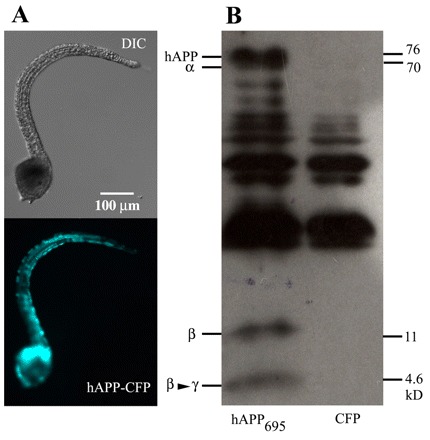
Human APP695 is properly processed in transgenic ascidian embryos. (A) Representative transgenic ascidian embryo ubiquitously expressing hAPP695 fused, by the C-terminal end, to CFP. The cis-regulatory region of the ascidian ef1a gene was used to drive ubiquitous expression of hAPP695. Images for DIC (differential interference contrast, top) and CFP (bottom) are shown. (B) Immunoblot of whole transgenic embryo lysates expressing hAPP695 or CFP alone. Full-length hAPP695 and fragments of hAPP695 processed by α-secretase (α), β-secretase (β), or β- and γ-secretase (β→γ) were detected using the Aβ-specific monoclonal antibody 6E10. APP-specific bands are only observed in transgenic embryos expressing hAPP695, suggesting that ascidians possess functional APP processing enzyme complexes. The bands that are common to both lanes are ascidian proteins cross-reacting to the human Aβ antibody.
Aggregation of the Aβ peptide into amyloid plaques is a characteristic pathological hallmark for AD pathogenesis. However, plaque generation in the current AD transgenic models is limited owing to the length of time required for formation. To determine whether Aβ-containing plaques can form in ascidians in vivo, thioflavin S staining was performed on transgenic larvae that ubiquitously expressed hAPP695 or hAPP variants (Fig. 3). PCR mutagenesis was used to generate three hAPP variants that contained alterations in sequences located near to either the β-and/or γ-secretase cleavage sites of APP that are known to be associated with early-onset familial forms of AD (FAD). The mutant constructs are: (1) hAPPmtβ, containing two point mutations (K670N/M671L) near to the β-secretase cleavage site (Mullan et al., 1992); (2) hAPPmtγ, containing three point mutations (T714I/I716V/V717I) located near to the γ-secretase site (Yoshioka et al., 1991; Eckman et al., 1997; De Jonghe et al., 2001); and (3) hAPPmtβγ, containing a combination of five point mutations at the β- and γ-secretase cleavage sites (Fig. 3A) (Oakley et al., 2006). Ubiquitous expression was driven by cis-regulatory elements of the ascidian ef1a gene. After 23 hpf, transgenic larvae that had been electroporated with equal masses of constructs were fixed and subsequently stained using both the 6E10 antibody and thioflavin S. In transgenic embryos expressing CFP alone, no thioflavin S or anti-Aβ staining was apparent (Fig. 3B). Transgenic embryos expressing hAPP695 had thioflavin S-reactive deposits that colocalized with the anti-Aβ antibody (Fig. 3C). This suggests that hAPP695 can undergo processing to generate Aβ peptides, which may subsequently aggregate to form plaques. Expression of hAPPmtβ (Fig. 3D) resulted in a significant increase in the number of thioflavin S-reactive plaques compared with wild-type hAPP695, with the average number of plaques per embryo increasing from two to 12 (Fig. 3F). However, no difference in the level of thioflavin S-reactive deposits was observed when an equal amount of hAPPmtγ was expressed (Fig. 3F). This suggested that low levels of β-cleavage might interfere with γ-secretase activity. To test this hypothesis, we generated a mutant hAPP expression construct in which both β- and γ-cleavage sites contained mutations (hAPPmtβγ). Transgenic embryos expressing this construct had significantly increased numbers of Aβ-containing plaques (Fig. 3E,F), with the average number of plaques per embryo increasing to 50. These experiments suggest that the sequence specificity at the β-secretase cleavage site appears to be conserved and recognized by the endogenous, ascidian β-secretase homolog.
Fig. 3.
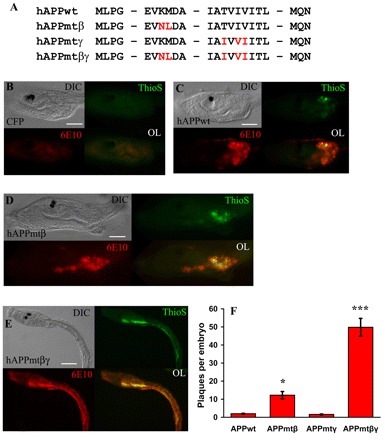
APP mutations synergize to greatly increase Aβ plaque formation in transgenic ascidian embryos. (A) Partial amino acid sequences of the wild-type (wt) and mutant hAPP695 constructs. The specific point mutations introduced by PCR are indicated in red. (B–E) Representative transgenic embryos (∼23 hpf) ubiquitously expressing CFP or hAPP. All transgenes were driven by the ef1a enhancer. Images for DIC, thioflavin S staining, mAb 6E10 staining and fluorescent overlays (OL) are shown. (B) Control transgenic embryos expressing CFP. Note the lack of thioflavin S and mAb 6E10 staining. (C) Transgenic embryo expressing hAPPwt. Note that there are some thioflavin S positive plaques and there is some 6E10 staining. (D) Transgenic embryo expressing APPmtβ. There is an increase in both thioflavin S and mAb 6E10 staining compared with either CFP-expressing or hAPP-expressing embryos. (E) Transgenic embryo expressing APPmtβγ. These transgenic embryos show the most thioflavin S and mAb 6E10 staining. (F) Quantitation of the thioflavin S-positive plaques of the embryos shown in B–E. Data are plotted as the average number of plaques per embryo ± standard error (S.E.) versus the transgene, n=50 embryos per construct. A one-way ANOVA analysis was performed on the four categories shown; F (3,196)=72.87, P=0.000. A Tukey post-hoc comparison of the four groups indicates that APPmtβ-expressing embryos had a significantly greater number of plaques compared with larvae expressing APPwt or APPmtγ, P<0.05 (*). In addition, the number of plaques in APPmtβγ-expressing larvae is much greater than for all other transgenic embryos, suggesting that mutations at both cleavage sites synergize to produce a significant increase in Aβ plaques, P<0.001 (***). Data are representative of three separate experiments. Bars, 50 μm (B–D); 100 μm (E).
Anti-amyloid drug treatment reduces the plaque load in transgenic ascidians
We next wanted to determine whether this rapid (<24 hours) and aggressive plaque phenotype generated in hAPP695-expressing ascidians would be suitable to validate therapeutic drugs that are designed to reduce Aβ aggregation and plaque formation. Transgenic larvae expressing hAPPmtβγ were allowed to develop in seawater containing varying concentrations of 3-amino-1-propanesulfonic acid (3-APS). As an anti-amyloid therapeutic, 3-APS had previously undergone clinical trials and is currently being developed as a food supplement for AD patients under the name Alzhemed (Bellus Health, Laval, Quebec). Thought to act as an Aβ mimetic, this drug has, in neuronal cultures, been shown to protect against Aβ-induced cytotoxicity and to reduce plaque burden by 24% in an early-onset AD mouse model (Geerts, 2004; Gervais et al., 2007). Treatment of hAPPmtβγ-expressing larvae with 3-APS at clinically relevant concentrations, ranging from 0.1 mM to 10 mM, reduced the number of thioflavin S-reactive deposits in a dose-dependent manner, with a 35% and 70% reduction in the presence of 1 mM and 10 mM of 3-APS, respectively (Fig. 4). There was no apparent effect on larval development (data not shown).
Fig. 4.
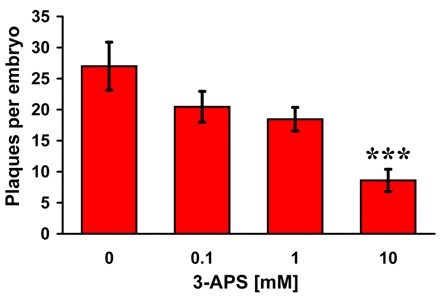
Treating transgenic embryos that express mutant human APP with an anti-amyloid drug reduces the number of thioflavin S-positive plaques in a dose-dependent manner. At the two-cell stage (1 hpf), transgenic embryos expressing APPmtβγ were grown in the presence of varying concentrations of the anti-amyloid drug 3-APS. At 23 hpf, embryos were fixed and stained for plaque formation with thioflavin S. The number of plaques per embryo ± S.E. is plotted versus drug treatment. Embryos grown in 10 mM 3-APS had significantly reduced numbers of plaques compared with other concentrations of the drug [P=0.001, 10 mM vs 0 mM (***); P=0.01, 10 mM vs 0.1 mM; P=0.01, 10 mM vs 1 mM].
Transgenic expression of human Aβ1–42 alters larval behavior
Recent studies examining ascidian larval behavior offer insights into neural function and may also provide an assay to monitor neural dysfunction (Tsuda et al., 2003a; Meinertzhagen et al., 2004; Zega et al., 2006). One well-characterized behavior controlled by the nervous system of the ascidian tadpole larvae involves the coordinated actions guiding attachment, an event necessary for metamorphosis (Cloney, 1982). To examine whether larval attachment can be perturbed, transgenic ascidian larvae were generated that express the 42-amino-acid species of Aβ (Aβ1–42), specifically in the nervous system, using cis-regulatory elements of the pan-neural synaptotagmin gene (Katsuyama et al., 2002). Aβ1–42 has long been considered to be the neurotoxic species associated with Alzheimer’s, and its accumulation in transgenic C. elegans and Drosophila has been shown to be particularly damaging (Chiang et al., 2008). Plaque staining of Aβ1–42-expressing ascidian larvae revealed thioflavin S-reactive deposits similar to those formed using full-length hAPP695, suggesting that Aβ1–42 can aggregate in vivo (Fig. 5A). When transgenic embryos expressed Aβ1–42 that was fused to CFP, the number of plaques was significantly reduced by 50%, suggesting that steric hindrance of the CFP molecule may be inhibiting Aβ aggregation (Fig. 5B,C). A similar level of reduction in the number of thioflavin S-reactive deposits was observed in Aβ1–42-expressing larvae grown in the presence of 1 mM 3-APS (Fig. 5C).
Fig. 5.
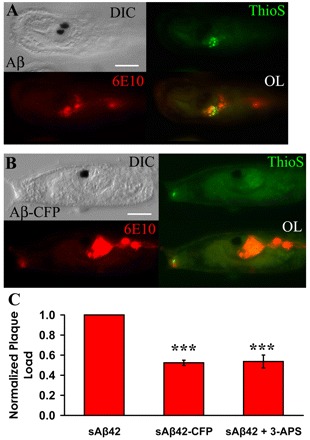
Reduction of plaque formation in transgenic ascidian embryos expressing Aβ1-42-CFP or following 3-APS treatment. (A,B) Representative transgenic ascidian tadpoles (23 hpf) expressing Aβ1–42 (A) or Aβ1–42-CFP (B) in the nervous system. In both cases, transgene expression is driven by cis-regulatory elements of the pan-neural synaptotagmin gene. Larvae were stained using both 6E10 and thioflavin S to reveal plaque deposits. Bars, 50 μm. (C) Relative quantification of thioflavin S-reactive, Aβ-containing amyloid plaques in larvae expressing Aβ1–42, Aβ1–42-CFP, or Aβ1–42 in the presence of 1 mM 3-APS. The plaque load was normalized to the number of plaques in the Aβ1–42-expressing larvae. Data are graphed as the relative plaque load per embryo ± S.E.; n=3 independent experiments, and at least 20 embryos per condition were analyzed per trial. A one-way ANOVA analysis was performed on the three separate conditions; F (2,6)=52.75, P=0.000. A Tukey post-hoc comparison indicates that fusion of CFP to Aβ1–42 and 1 mM 3-APS treatment both significantly reduced plaque deposition in vivo, P<0.001 (***).
To determine whether the expression of Aβ1–42 could affect attachment behavior, transgenic tadpoles were placed in a device that would assay for attachment to the underside of a floating dish (Fig. 6A). This apparatus was then placed in the dark for 24 hours to allow larvae to settle (Jiang et al., 2005). During the electroporation process, chorions are removed from eggs, which later negatively impacts larval settlement, and only about 35% of dechorionated larvae can attach properly (data not shown). Transgenic larvae expressing Aβ1–42 exhibited a 50% reduction in attachment compared with embryos expressing CFP alone (Fig. 6B). However, when an equal mass of Aβ1–42-CFP was expressed, the rate of attachment was significantly improved, by approximately 27%, compared with larvae expressing Aβ1–42. In addition, 1 mM 3-APS treatment significantly improved the level of attachment in Aβ1–42-expressing larvae by 35%. These results suggest that Aβ1–42 expression, and the subsequent plaque formation, hinders one or more of the behaviors that coordinate larval attachment and is, therefore, indicative of neural dysfunction.
Fig. 6.
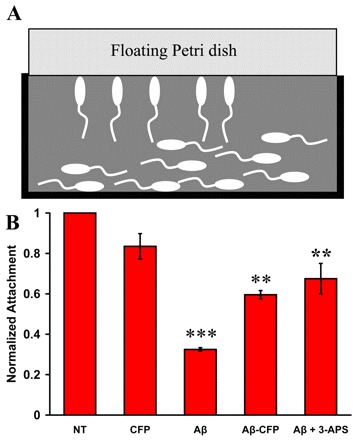
Transgenic ascidian embryos expressing human Aβ1–42 fail to undergo normal settlement behaviors. (A) Schematic of the attachment assay. At 23 hpf, non-transgenic (NT) or transgenic larvae expressing Aβ1–42, Aβ1–42-CFP, or CFP in the nervous system, were transferred to an apparatus that monitors attachment to the underside of a floating 100×15 mm Petri plate. Embryos were scored for attachment at 24 hours following the transfer. (B) Relative quantification of attachment, normalized to dechorionated nontransgenic larvae. Embryos expressing Aβ1–42 exhibit reduced attachment compared with controls (NT and CFP). To assess the effectiveness of an anti-amyloid inhibitor, transgenic embryos expressing Aβ1–42 were grown in the presence of 1 mM 3-APS. Data are plotted as the average percentage attachment ± S.E.; n=3 and at least 50 embryos per construct were analyzed per trial. A one-way ANOVA analysis was performed; F (4,10)=32.09, P=0.000. A Tukey post-hoc comparison of the five groups indicates that larvae expressing Aβ1–42 displayed a significant reduction in the levels of attachment compared with larvae expressing CFP alone, P<0.001 (***). The attachment rate is significantly improved with larvae expressing Aβ1–42-CFP or upon 3-APS treatment, P<0.01 (**).
DISCUSSION
Here we describe a rapid in vivo system to study APP processing and Aβ plaque formation using the ascidian. In transgenic ascidian larvae that express hAPP695 alone, Aβ peptides are produced and can aggregate to form plaques within 23 hpf. Mutations in hAPP695 that are associated with FAD resulted in a significant increase in plaque formation in vivo. In addition, proper neurological behavior was hindered as a result of the expression of Aβ1–42 in the ascidian nervous system. Lastly, a reduction in Aβ aggregation, either genetically by expressing an elongated form of Aβ1–42 (Aβ1–42-CFP) or chemically through the use of an anti-amyloid therapeutic, not only reduced plaque load but also improved defects in larval attachment.
In developing an animal model system for human disease, an appropriate model should have a close evolutionary relationship to humans. Consequently, mice have been used extensively as AD animal models owing to the similarity with the human brain anatomy and the existence of numerous behavioral tests to examine neural dysfunction (Eriksen and Janus, 2007). However, generating transgenic mice is not only time intensive but also costly. For these reasons, researchers have turned to invertebrate animal models that have provided much insight into some of the molecular mechanisms involved in AD pathogenesis. However, unlike both flies and worms, ascidians provide a more appropriate context to study human diseases; a fully functional chordate nervous system at 18 hpf, combined with simple behavioral assays, allows for rapid determination of neural functioning. Furthermore, because of their evolutionary history, ascidians are likely to share a larger number of genes with humans that are not present in other invertebrates. To our knowledge, this is the first invertebrate model system in which orthologs for all the genes implicated in APP processing have been identified. For these reasons, the ascidian offers a more suitable animal system to examine many of the cellular and molecular aspects associated with AD.
In addition to having AD-related genes, the Aβ cascade appears to be conserved in the ascidian. Both fly and worm models have relied on co-expression of human APP and human BACE to reconstruct the APP processing machinery. We have demonstrated that, when expressed alone, hAPP695 can be processed to produce peptide fragments consistent with the Aβ cascade. Although we cannot distinguish between the exact Aβ-containing species produced, the detection of Aβ-reactive fragments of comparable sizes to those expected following cleavage at the α-, β- or γ-secretase sites suggests that the processing pathway is conserved. The western blot also revealed additional 6E10 immunoreactive bands that were present only in the embryos expressing hAPP695 that could either be degradative products or multimeric forms of the Aβ peptide (Kayed et al., 2003; Wilquet and De Strooper, 2004). We have generated several mutant constructs that force the amyloidogenic processing of hAPP695, consistent with previous observations in a mutant hAPP695 mouse model (Oakley et al., 2006). A tenfold increase in the average number of plaques per embryo was observed when two point mutations were introduced at the β-secretase cleavage site of hAPP695 (hAPPmtβ). This double mutation, identified in a Swedish family, has been shown to result in the increased production and secretion of Aβ (Haass et al., 1995). An additional three point mutations, also known as the Austrian, Florida and London mutations, at the γ-secretase site (hAPPmtβγ) resulted in a fivefold increase in plaque formation compared with hAPPmtβ. These findings are consistent with the importance of β- and γ-site cleavage for Aβ production. However, mutations solely at the γ-secretase site failed to increase the number of plaques suggesting that the β-site cleavage acts as a rate-limiting step for plaque formation. These are important findings because all five of the mutations we studied are associated with early-onset FAD, supporting our hypothesis that the ascidian can be utilized to study the human APP processing pathway.
The ascidian system provides a much more rapid animal model to examine Aβ plaque formation in vivo. Transgenic mice AD models take, on average, eight months to display any observable plaque phenotype, with more aggressive mice AD models demonstrating plaque formation at two months of age (Shah et al., 2005; Oakley et al., 2006). Because of their rapid development, the use of invertebrate AD model systems, such as worms and flies, has reduced the time for plaque formation down to several weeks (Link, 1995; Link et al., 2003; Greeve et al., 2004). Our results in the ascidian demonstrate that the time to observable plaque formation can be reduced to less than a day. This can be attributed to the rapid development of the transient free-swimming tadpole larva. Furthermore, the promoters that were utilized activate gene transcription early in development, which allows for higher expression levels of both hAPP695 and Aβ1–42. As a result, we believe that our very aggressive plaque-forming ascidian model (APPmtβγ) can be developed for use in rapid in vivo screening to examine the effectiveness of a variety of anti-amyloid therapeutics.
In addition to this rapid plaque pathology, we have shown that, by utilizing a relatively simple behavioral assay based on levels of attachment, the expression of Aβ1–42 in ascidians causes neurological-associated changes in behavior. The expression of Aβ1–42 specifically in the nervous system significantly decreases the levels of larval attachment compared with wild-type larvae or those expressing CFP alone. Neurons located in the anterior sensory vesicle of the tadpole are thought to be involved in the mechanisms guiding larval attachment. Two pigmented cells located within the sensory vesicle, the otolith and ocellus, act as part of the sensory organ to coordinate the characteristic swimming behavior that is necessary for attachment. After hatching, swimming tadpole larvae are both photopositive and geonegative to allow for larval dispersal. This swimming behavior later switches to being photonegative and geopositive to allow for larval attachment (Svane and Young, 1989). Furthermore, resting larvae can be stimulated to swim when passing from light to dark conditions (Tsuda et al., 2003b). Previous studies have shown that laser ablation of either the otolith or the ocellus can affect this geotactic or phototactic response, respectively (Tsuda et al., 2003a). Because the ability of larval swimming appeared to not be affected in Aβ1–42-expressing embryos, we hypothesize that the reduced level of attachment observed is probably the result of a dysfunction in one or more of these behaviors.
Through our studies we also observed that, when expressed at lower levels, Aβ1–42 expression caused defects in attachment but failed to generate observable amyloid deposits. This lack of plaque deposition in larvae exhibiting defects in attachment provides additional support to recent theories that alternative Aβ-containing species may act as causative factors for Aβ-induced pathogenesis. It has recently been reported that intracellular Aβ accumulation can take on several forms and that this structural polymorphism of the Aβ species can mediate a variety of pathogenic effects prior to plaque formation in vivo (Iijima et al., 2004; Klein et al., 2004; Lesne et al., 2006). We believe that the Aβ1–42-mediated behavioral effects in our ascidian AD model are not a result of the Aβ1–42 peptide monomer itself because 3-APS treatment and fusion of CFP to Aβ1–42, which are both believed to inhibit the aggregation of Aβ, significantly improved attachment, while probably retaining the normal function, if any, of the Aβ1–42 peptide monomer. For these reasons, soluble amyloid oligomers may be a likely candidate for the Aβ-containing species mediating the observed effects on larval attachment. Oligomeric Aβ is thought to cause not only defects in locomotor functioning in a transgenic Drosophila AD model (Crowther et al., 2005; Chiang et al., 2009), but also spatial memory impairment in mice (Lesne et al., 2006).
To establish the ascidian as a more comprehensive model for AD, we have begun to examine other characteristics associated with the complex pathology of the disease. To determine whether neurodegeneration was taking place, TUNEL labeling was performed on larvae expressing human Aβ1–42. Although the mechanisms coordinating apoptotic cell death have been shown to be conserved in the ascidian (Chambon et al., 2002), we did not observe any TUNEL-positive cells in the nervous system of Aβ1–42-expressing larvae. This lack of apoptotic cell death is not altogether surprising given the previous studies in similar invertebrate AD models. For example, in studies of Drosophila expressing human Aβ it was shown that both the formation of amyloid deposits and the onset of learning defects occur well before any visible signs of neuronal cell death, with the onset of degeneration visible only in a 30-day-old Aβ42-expressing fly brain (Iijima et al., 2004). This suggests that Aβ-mediated toxicity in the ascidian may act independently of programmed cell death. Studies supported by previous research, showing that the neurons of AD patients contain increased numbers of autophagosomes and lysosomes, have also been initiated to examine whether autophagy is mediating these pathological effects (Nixon et al., 2000). Additionally, in transgenic mice expressing human mutant APP and mutant presenilin-1, Aβ was shown to partially co-localize within neuronal lysosomes (Langui et al., 2004). Interestingly, preliminary analyses using a lysosomal marker suggest that Aβ-mediated toxicity in the ascidian, resulting from the expression of human APP, may result from triggering autophagic cell death (data not shown) and will be explored further.
Our results suggest that the ascidian will be a useful, rapid, invertebrate chordate model system to study the early stages of Alzheimer’s disease. Through its use, we believe that fundamental questions regarding the molecular mechanisms coordinating AD and its pathogenesis will be answered. Furthermore, owing to its small size and experimental tractability, the ascidian may permit cost-effective and rapid screening of candidate therapeutic compounds for AD early in the drug development process. Overall, this study introduces the ascidian as a model for AD and provides the framework for understanding, and possibly treating, this disease and other debilitating neurological disorders.
METHODS
Human APP695 and Aβ1–42 transgene construction
All transgenes were constructed using the parent pSP72 vector (Promega, Madison, WI) containing the SV40 polyadenylation signal sequence, as described previously (Zeller et al., 2006b). Primers were designed to clone the cis-regulatory domain for either synaptotagmin or ef1a to drive expression of our transgenes. The cDNA clone encoding a 695-amino acid isoform of human APP (hAPP695) was kindly provided by Dr Edward Koo (Buxbaum et al., 1993) and was cloned downstream of the ef1a enhancer element. To monitor expression in vivo, hAPP695 was fused, at the C-terminus, in-frame with a codon-optimized cyan fluorescent protein (CFP) (Zeller et al., 2006b). Alternatively, hAPP695 wild-type and mutant constructs were also cloned without the CFP tag. For the generation of hAPP695 mutants, various primers were designed that would introduce the following familial AD (FAD)-associated point mutations in APP695: K670N, M671L, T714I, I716V and V717I. For the expression of Aβ1–42, primers were designed to amplify both the first 21 amino acids of hAPP695, a region previously described as the hAPP695 signal sequence, and the 42 amino acids corresponding to the Aβ1–42 peptide (Buckig et al., 2002). The two DNA fragments were assembled using overlap PCR. All expression constructs were sequence verified by the San Diego State University Microchemical Core (San Diego, CA), and the primers used for cloning are listed in supplementary material Fig. S2.
Embryo culturing and electroporation
Culturing and electroporation of Ciona intestinalis embryos has been described elsewhere (Zeller, 2004; Zeller et al., 2006a). Adult ascidians were collected under permit from several marinas in San Diego, California and maintained in a closed, recirculating seawater system under constant light to induce gamete production. Fertilized eggs were dechorionated in the presence of 1% thioglycolic acid (Sigma-Aldrich, St Louis, MO) and 0.05% protease (Type XIV; Sigma-Aldrich) in pH 9.5 seawater, and subsequently washed four times using 0.45 μm-filtered seawater. Approximately 300 μl of seawater containing eggs was added to 40-90 μg of plasmid DNA in 500 μl of 0.77 M mannitol. The egg/DNA mixture was then transferred to a 0.4-cm gap electroporation cuvette, electroporated at 3000 μF and 10 Ω (resistance), and then placed in a 60-mm gelatin-coated dish containing seawater supplemented with 0.1 mM EDTA, 10 U/ml penicillin and 10 μg/ml of streptomycin, and cultured at 18°C.
Immunohistochemistry and histology
Transgenic larvae were fixed in 2% paraformaldehyde in seawater for 10 minutes and washed twice in phosphate-buffered tris containing 0.1% Tween-20 (PBT). Embryos were then rinsed in 100% methanol and subsequently washed three times in PBT. Embryos were blocked in PBT containing 1% donkey serum for 10 minutes, and then incubated with a 1:1000 dilution of the monoclonal antibody (mAb) 6E10 (α-amyloid beta; Covance, Trenton, NJ) in PBT/1% donkey serum overnight. After washing in PBT, embryos were incubated for 1 hour with the secondary antibody, Alexa Fluor 546 goat anti-mouse IgG (Invitrogen, Carlsbad, CA), which was diluted 1:200 in PBT/1% donkey serum containing 10 μg/ml of thioflavin S (Sigma-Aldrich) for plaque staining. After washing three times in PBT, embryos were stored in 50% glycerol. Imaging was performed using a Zeiss Axioplan 2e imaging microscope.
Immunoblot analyses
For western blotting, whole embryo lysates were homogenized and equal amounts of protein were separated by 12% SDS-PAGE. Proteins were transferred onto a nitrocellulose membrane, which was then blocked overnight with 5% non-fat milk in Tris-buffered saline with Triton X-100 (TBST) overnight. The membrane was then incubated with mAb 6E10 (1:500) in TBST containing 2.5% non-fat milk. Bound antibody was detected with a goat anti-mouse peroxidase-conjugated secondary antibody (Sigma-Aldrich).
Attachment assay
At approximately 14 hpf, 3-aminobenzoic acid ethyl ester (Sigma-Aldrich) was added at a final concentration of 0.02% to paralyze the ascidian larvae and prevent non-specific attachment. At ∼22 hpf, transgenic larvae expressing either Aβ1-42 or Aβ 1–42-CFP were transferred to a gelatin-coated 15-cm Petri dish lid containing seawater supplemented with 10 U/ml penicillin and 10 μg/ml of streptomycin. A second 15-cm dish was carefully suspended on top of the first dish to sandwich the larvae between the two surfaces. The apparatus was kept in the dark and cultured at 18°C. After 24 hours, embryos were scored based on the ability to adhere to the bottom of the floating dish, and began to undergo metamorphosis.
Treatment with anti-amyloid therapeutic
3-amino-1-propanesulfonic acid (3-APS; Sigma-Aldrich) was added directly to transgenic ascidian larvae at final concentrations of 0.1, 1 and 10 mM after the 2-cell stage; the larvae were then allowed to develop at 18°C. After 23 hpf, embryos were fixed and stained for both Aβ expression, using the mAb 6E10, and plaque formation using thioflavin S, as described above. Experiments were repeated more than three times and a representative result was shown. Analysis of variance (ANOVA) analyses were calculated with SPSS software with Tukey post-hoc analysis.
Supplementary Material
Acknowledgments
The human APP695 clone was kindly provided by Barbara Cottrell and Dr Edward Koo from the University of California, San Diego School of Medicine. This work was funded by the National Science Foundation (IBN-0347937 to R.W.Z.), the SDSU Minority Biomedical Research Support Initiative for Maximizing Student Diversity (MBRS/IMSD) Program #1 R25 GM58906-08 (to M.J.V.), and the Invitrogen Corporation Fellowship Award (to M.J.V.).
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
R.W.Z. and M.J.V. developed and designed research concepts. M.J.V. conducted the experiments. The manuscript was prepared and edited by R.W.Z. and M.J.V.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.003434/-/DC1
REFERENCES
- Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. (2007). Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 3, 186–191 [DOI] [PubMed] [Google Scholar]
- Buckig A, Tikkanen R, Herzog V, Schmitz A. (2002). Cytosolic and nuclear aggregation of the amyloid beta-peptide following its expression in the endoplasmic reticulum. Histochem Cell Biol. 118, 353–360 [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Koo EH, Greengard P. (1993). Protein phosphorylation inhibits production of Alzheimer amyloid beta/A4 peptide. Proc Natl Acad Sci USA 90, 9195–9198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambon JP, Soule J, Pomies P, Fort P, Sahuquet A, Alexandre D, Mangeat PH, Baghdiguian S. (2002). Tail regression in Ciona intestinalis (prochordate) involves a caspase-dependent apoptosis event associated with ERK activation. Development 129, 3105–3114 [DOI] [PubMed] [Google Scholar]
- Chiang HC, Iijima K, Hakker I, Zhong Y. (2009). Distinctive roles of different {beta}-amyloid 42 aggregates in modulation of synaptic functions. FASEB J. 23, 1969–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PK, Lam MA, Luo Y. (2008). The many faces of amyloid beta in Alzheimer’s disease. Curr Mol Med. 8, 580–584 [DOI] [PubMed] [Google Scholar]
- Cloney R. (1982). Ascidian larvae and the events of metamorphosis. Am Zool. 22, 817–826 [Google Scholar]
- Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA, Gubb DC, Lomas DA. (2005). Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 132, 123–135 [DOI] [PubMed] [Google Scholar]
- Crowther DC, Page R, Chandraratna D, Lomas DA. (2006). A Drosophila model of Alzheimer’s disease. Methods Enzymol. 412, 234–255 [DOI] [PubMed] [Google Scholar]
- De Jonghe C, Esselens C, Kumar-Singh S, Craessaerts K, Serneels S, Checler F, Annaert W, Van Broeckhoven C, De Strooper B. (2001). Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum Mol Genet. 10, 1665–1671 [DOI] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Chourrout D, Philippe H. (2006). Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 439, 965–968 [DOI] [PubMed] [Google Scholar]
- Eckman CB, Mehta ND, Crook R, Perez-tur J, Prihar G, Pfeiffer E, Graff-Radford N, Hinder P, Yager D, Zenk B, et al. (1997). A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of A beta 42(43). Hum Mol Genet. 6, 2087–2089 [DOI] [PubMed] [Google Scholar]
- Eriksen JL, Janus CG. (2007). Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav Genet. 37, 79–100 [DOI] [PubMed] [Google Scholar]
- Fossgreen A, Bruckner B, Czech C, Masters CL, Beyreuther K, Paro R. (1998). Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc Natl Acad Sci USA 95, 13703–13708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerts H. (2004). NC-531 (Neurochem). Curr Opin Investig Drugs 5, 95–100 [PubMed] [Google Scholar]
- Gervais F, Paquette J, Morissette C, Krzywkowski P, Yu M, Azzi M, Lacombe D, Kong X, Aman A, Laurin J, et al. (2007). Targeting soluble Abeta peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol Aging 28, 537–547 [DOI] [PubMed] [Google Scholar]
- Gotz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L, Kurosinski P, Chen F. (2004). Transgenic animal models of Alzheimer’s disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry 9, 664–683 [DOI] [PubMed] [Google Scholar]
- Graham A. (2004). Evolution and development: rise of the little squirts. Curr Biol. 14, R956–958 [DOI] [PubMed] [Google Scholar]
- Greeve I, Kretzschmar D, Tschape JA, Beyn A, Brellinger C, Schweizer M, Nitsch RM, Reifegerste R. (2004). Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci. 24, 3899–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntern R, Bouras C, Hof PR, Vallet PG. (1992). An improved thioflavine S method for staining neurofibrillary tangles and senile plaques in Alzheimer’s disease. Experientia 48, 8–10 [DOI] [PubMed] [Google Scholar]
- Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ. (1995). The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1, 1291–1296 [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. (2004). Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer’s disease. Proc Natl Acad Sci USA 101, 6623–6628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai JH, Meinertzhagen IA. (2007). Neurons of the ascidian larval nervous system in Ciona intestinalis: II. Peripheral nervous system. J Comp Neurol. 501, 335–352 [DOI] [PubMed] [Google Scholar]
- Jiang D, Tresser JW, Horie T, Tsuda M, Smith WC. (2005). Pigmentation in the sensory organs of the ascidian larva is essential for normal behavior. J Exp Biol. 208, 433–438 [DOI] [PubMed] [Google Scholar]
- Katsuyama Y, Matsumoto J, Okada T, Ohtsuka Y, Chen L, Okado H, Okamura Y. (2002). Regulation of synaptotagmin gene expression during ascidian embryogenesis. Dev Biol. 244, 293–304 [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- Klein WL, Stine WB, Jr, Teplow DB. (2004). Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol Aging 25, 569–580 [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- Link CD. (1995). Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci USA 92, 9368–9372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD. (2005). Invertebrate models of Alzheimer’s disease. Genes Brain Behav. 4, 147–156 [DOI] [PubMed] [Google Scholar]
- Link CD, Taft A, Kapulkin V, Duke K, Kim S, Fei Q, Wood DE, Sahagan BG. (2003). Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol Aging 24, 397–413 [DOI] [PubMed] [Google Scholar]
- Marambaud P, Robakis NK. (2005). Genetic and molecular aspects of Alzheimer’s disease shed light on new mechanisms of transcriptional regulation. Genes Brain Behav. 4, 134–146 [DOI] [PubMed] [Google Scholar]
- Maslow K. (2008). 2008 Alzheimer’s disease facts and figures. Alzheimers Dement. 4, 110–133 [DOI] [PubMed] [Google Scholar]
- McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M, French JE, Lambermon MH, Darabie AA, Brown ME, et al. (2002). Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat Med. 8, 1263–1269 [DOI] [PubMed] [Google Scholar]
- Meinertzhagen IA, Lemaire P, Okamura Y. (2004). The neurobiology of the ascidian tadpole larva: recent developments in an ancient chordate. Annu Rev Neurosci. 27, 453–485 [DOI] [PubMed] [Google Scholar]
- Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. (1992). A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1, 345–347 [DOI] [PubMed] [Google Scholar]
- Nalivaeva NN, Fisk LR, Belyaev ND, Turner AJ. (2008). Amyloiddegrading enzymes as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res. 5, 212–224 [DOI] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM, Mathews PM. (2000). The endosomal-lysosomal system of neurons in Alzheimer’s disease pathogenesis: a review. Neurochem Res. 25, 1161–1172 [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 26, 10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, 3rd, Sudhof T, Yu G. (2005). Nicastrin functions as a gamma-secretasesubstrate receptor. Cell 122, 435–447 [DOI] [PubMed] [Google Scholar]
- Svane I, Young CM. (1989). The ecology and behavior of ascidian larvae. Oceanography and Marine Biology: An Annual Review 27, 45–90 [Google Scholar]
- Tsuda M, Sakurai D, Goda M. (2003a). Direct evidence for the role of pigment cells in the brain of ascidian larvae by laser ablation. J Exp Biol. 206, 1409–1417 [DOI] [PubMed] [Google Scholar]
- Tsuda M, Kawakami I, Shiraishi S. (2003b). Sensitization and habituation of the swimming behavior in ascidian larvae to light. Zoolog Sci. 20, 13–22 [DOI] [PubMed] [Google Scholar]
- Wilquet V, De Strooper B. (2004). Amyloid-beta precursor protein processing in neurodegeneration. Curr Opin Neurobiol. 14, 582–588 [DOI] [PubMed] [Google Scholar]
- Wu Y, Luo Y. (2005). Transgenic C. elegans as a model in Alzheimer’s research. Curr Alzheimer Res. 2, 37–45 [DOI] [PubMed] [Google Scholar]
- Yoshioka K, Miki T, Katsuya T, Ogihara T, Sakaki Y. (1991). The 717Val—Ile substitution in amyloid precursor protein is associated with familial Alzheimer’s disease regardless of ethnic groups. Biochem Biophys Res Commun. 178, 1141–1146 [DOI] [PubMed] [Google Scholar]
- Zega G, Thorndyke MC, Brown ER. (2006). Development of swimming behaviour in the larva of the ascidian Ciona intestinalis. J Exp Biol. 209, 3405–3412 [DOI] [PubMed] [Google Scholar]
- Zeller RW. (2004). Generation and use of transgenic ascidian embryos. Methods Cell Biol. 74, 713–730 [DOI] [PubMed] [Google Scholar]
- Zeller RW, Virata MJ, Cone AC. (2006a). Predictable mosaic transgene expression in ascidian embryos produced with a simple electroporation device. Dev Dyn. 235, 1921–1932 [DOI] [PubMed] [Google Scholar]
- Zeller RW, Weldon DS, Pellatiro MA, Cone AC. (2006b). Optimized green fluorescent protein variants provide improved single cell resolution of transgene expression in ascidian embryos. Dev Dyn. 235, 456–467 [DOI] [PubMed] [Google Scholar]
- Zhang YW, Xu H. (2007). Molecular and cellular mechanisms for Alzheimer’s disease: understanding APP metabolism. Curr Mol Med. 7, 687–696 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.