

Abstract
Corynebacterium glutamicum is an industrially important producer of amino acids and organic acids, as well as an emerging model system for aromatic assimilation. An IclR-type regulator GenR has been characterized to activate the transcription of genDFM and genKH operons for 3-hydroxybenzoate and gentisate catabolism and represses its own expression. On the other hand, GlxR, a global regulator of the cyclic AMP (cAMP) receptor protein-fumarate nitrate reductase regulator (CRP-FNR) type, was also predicted to be involved in this pathway. In this study, electrophoretic mobility shift assays and footprinting analyses demonstrated that GlxR bound to three sites in the promoter regions of three gen operons. A combination of site-directed mutagenesis of the biding sites, promoter activity assay, and GlxR overexpression demonstrated that GlxR repressed their expression by binding these sites. One GlxR binding site (DFMx) was found to be located −13 to +8 bp upstream of the genDFM promoter, which was involved in negative regulation of genDFM transcription. The GlxR binding site R-KHx01 (located between positions −11 to +5) was upstream of the genKH promoter sequence and involved in negative regulation of its transcription. The binding site R-KHx02, at which GlxR binds to genR promoter to repress its expression, was found within a footprint extending from positions −71 to −91 bp. These results reveal that GlxR represses the transcription of all three gen operons and then contributes to the synchronization of their expression for 3-hydroxybenzoate and gentisate catabolism in collaboration with the specific regulator GenR.
INTRODUCTION
The gentisate (2,5-dihydroxybenzoate [GEN]) pathway is one of the important ring cleavage pathways in the catabolism of various aromatic compounds, including 3-hydroxybenzoate (3-HBA) (1, 2), naphthalene (3, 4), salicylate (5, 6), 3,6-dichloro-2-methoxybenzoate (7), and xylenol (8). The initial reaction of this pathway is the ring cleavage oxidation of gentisate, catalyzed by gentisate 1,2-dioxygenase (9). Subsequently, the ring fission product maleylpyruvate is degraded via either direct hydrolysis to maleate and pyruvate (10, 11) or isomerization to fumarylpyruvate before hydrolysis to fumarate and pyruvate (1, 4, 9, 12). Two LysR-type transcriptional regulators, NagR in Ralstonia sp. strain U2 (13) and MhbR in Klebsiella pneumoniae M5a1 (14), have been reported to activate the expression of enzymes involved in naphthalene and 3-HBA catabolism via gentisate pathways.
Corynebacterium glutamicum, a Gram-positive bacterium with a high G+C content, plays a prominent role for the microbial production of amino acids, organic acids, and alcohols (15–20). Derivative strains of Corynebacterium were used to produce 2.5 million tons of glutamate as monosodium glutamate per year as well as several thousand tons of other amino acids such as lysine, isoleucine, tryptophan, and threonine (16, 21). Recently, C. glutamicum ATCC 13032 has been found to utilize a number of aromatic compounds as its sole carbon and energy source, including 3-HBA and GEN (22). Its catabolic pathway of 3-HBA degradation via GEN was catalyzed by genD-, genF-, genM-, and genH-encoded enzymes (1, 23–25). In addition to these catabolic genes, GenK was identified to actively transport gentisate to facilitate GEN utilization (23). More recently, IclR-type regulator GenR has been characterized to be a dual-function protein that acts as an activator of the transcription of genDFM and genKH operons for 3-HBA and GEN catabolism in response to 3-HBA and GEN and as a repressor of its own expression (25).
It has been recognized that expression of genes in C. glutamicum, including those involved in aromatic catabolism, is generally regulated by pathway-specific regulators (22, 26). However, the presence of global regulators has also been observed when GlxR binding sites were found in the potential operator regions involved in aromatic degradation (27). GlxR is a major global transcriptional regulator of C. glutamicum, belonging to the family of cyclic AMP (cAMP) receptor protein-fumarate nitrate reductase regulators (CRP-FNR). It was first characterized as a factor repressing the promoter activity of a gene coding for an enzyme in the glyoxylate pathway and presenting as dimers (28). Subsequently, a total of 215 potential GlxR binding sites were predicted bioinformatically and 77 sites were experimentally confirmed (29). Recently, in silico detection and chromatin immunoprecipitation in conjunction with microarray (ChIP-chip) analyses detected 209 GlxR binding sites in the C. glutamicum genome, and 84 regions were previously described (30). Among these 209 binding sites, 72 of them were verified by in vitro binding assays to be associated with regulons consisting of genes for carbon metabolism, nitrogen metabolism, respiration, resuscitation, cell wall formation, and cell division (27, 29, 30). More recently, GlxR was demonstrated to positively regulate expression of genes for aerobic respiration, ATP synthesis, and glycolysis and to be necessary for expression of genes for cell separation and mechanosensitive channels (30). Interestingly, negative regulation of GlxR was found in the expression of a citrate uptake gene (30). Even though in vitro binding assays indicated its possible involvement in regulating the expression of the paa gene cluster for phenylacetic acid catabolism, whether or not the GlxR was involved in aromatic metabolism in this strain still remains unknown (31). The study of the global regulatory factor GlxR will help to improve the ability of C. glutamicum in industrial production as well as aromatic degradation. In this article, we show that GlxR represses the transcription of gen operons and is involved in 3-HBA and GEN catabolism in C. glutamicum by assays of mobility shift electrophoresis, footprinting, overexpression, and promoter activity.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture media.
The bacterial strains and plasmids used are listed in Table 1 and primers in Table 2. Escherichia coli was grown aerobically on a rotary shaker (200 rpm) at 37°C in lysogeny broth (LB) or on an LB plate with 1.5% (wt/vol) agar. C. glutamicum strains were grown in LB medium or in mineral salt medium (MM) supplemented with 2 g liter−1 of glucose on a rotary shaker (200 rpm) at 30°C (32). Aromatic compounds were added at a final concentration of 2 mM as carbon and energy sources in MM and supplemented with 0.05 g liter−1 of yeast extract to meet the requirement of vitamins for the strains. According to a previous report (1), the overnight cultures of C. glutamicum strains in MM with 2 mM 3-HBA are usually controlled within 10 h of growth for enzyme assays and gene transcription measurements. Brain heart infusion broth medium (BHIBM) was used for generation of mutants and maintenance of C. glutamicum (33). When required for selection, antibiotics were added at the following concentrations: kanamycin (Kan), 50 μg ml−1 for E. coli and 25 μg ml−1 for C. glutamicum; chloramphenicol (Cam), 30 μg ml−1 for E. coli and 10 μg ml−1 for C. glutamicum; ampicillin, 100 μg ml−1 for E. coli; and nalidixic acid (Nx), 50 μg ml−1 for C. glutamicum.
TABLE 1.
Bacteria and plasmids involved in this study
| Strain or plasmid | Relevant characteristicsa | Reference or source |
|---|---|---|
| Bacteria | ||
| Corynebacterium glutamicum | ||
| ATCC 13032 | Wild type, plasmid free, Nxr | ATCC |
| RES167 | Restriction-deficient mutant of ATCC 13032, Δ(cglIM-cglIR-cglIIR) | 55 |
| Escherichia coli | ||
| DH5α | λ− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | Gibco BRL |
| Rosetta(DE3)pLysS | Camr, F− ompT hsdSB(rB− mB−) gal dcm (DE3), pLysSRARE vector (Camr) | Novagen |
| Trans1-T1 phage resistant | F− ϕ80(lacZ)ΔM15 ΔlacX74 hsdR(rK− mK+)ΔrecA1398 endA1 tonA | TransGen Biotech |
| Plasmids | ||
| pMD18-T | Ampr, lacZα, cloning vector for sequencing | TaKaRa |
| pET-28a(+) | Expression vector, Kanr, C/N-terminal His tag/thrombin/T7 tag, T7 lac promoter, T7 transcription start, f1 origin, lacI | Novagen |
| pZWHJ024 | Kanr, 681-bp PCR fragment of glxR gene ORF in C. glutamicum ATCC 13032 genome cloned into pET-28a(+) NdeI/EcoRI site, His tag banding C-terminal GlxR | This study |
| pZWHJ002 | Camr, pBL1 oriVCg, pK18, oriVEc, trrnB of E. coli, lacZ promoterless, derivative of pXMJ19 with lacZ digested with the enzyme combination NarI/HindIII, deletion of Ptac lacq, shuttle vector | 25 |
| pZWHJ006 | Camr, derivative of pZWHJ002 with 463-bp promoter of genDFM operon for detecting promoter activation | 25 |
| pZWHJ007 | Camr, derivative of pZWHJ002 with 457-bp promoter of genR operon for detecting promoter activation | 25 |
| pZWHJ008 | Camr, derivative of pZWHJ002 with 427-bp promoter of genKH operon for detecting promoter activation | 25 |
| pZWHJ025 | Camr, pZWHJ006 with site DFMx mutated | This study |
| pZWHJ026 | Camr, pZWHJ007 with site R-KHx01 mutated | This study |
| pZWHJ027 | Camr, pZWHJ007 with site R-KHx02 mutated | This study |
| pZWHJ028 | Camr, pZWHJ007 with sites R-KHx01 and R-KHx02 mutated | This study |
| pZWHJ029 | Camr, pZWHJ008 with site R-KHx01 mutated | This study |
| pZWHJ030 | Camr, pZWHJ008 with site R-KHx02 mutated | This study |
| pZWHJ031 | Camr, pZWHJ008 with sites R-KHx01 and R-KHx02 mutated | This study |
| pZWHJ033 | Camr, pBL1 oriVCg, pK18, oriVEc, trrnB of E. coli, carrying glxR gene, derivative of pXMJ19 with glxR from RES167 chromosome, digested with the enzyme combination SalI and EcoRI, shuttle vector | This study |
oriVCg, C. glutamicum oriV; oriVEc, E. coli oriV.
TABLE 2.
Primers used in this study
| Use and published name | Plasmid or probe(s) | Sequence (5′–3′) |
|---|---|---|
| For construction of the GlxR expression plasmid | ||
| glxre01 | pZWHJ024 | GGAATTCCATATGGAAGGTGTACAGGAGATCCTG |
| glxre02 | CCGGAATTCTTATCGAGCGCGACGTGCC | |
| glxre08 | pZWHJ033 | ACGCGTCGACAAAGGAGGACAACCAAAGCAGTGGAAGGTGTACAGGAGATCCTG |
| glxre09 | CCGGAATTCTTATCGAGCGCGACGTGCC | |
| For mutated GlxR binding sites | ||
| pDFMxmut01 | DFMax | GTGACGTGGAACATATTGGGCTTTGAAAGGTGACTC |
| pDFMxmut02 | TATGTTCCACGTCACAAAGTGTTCAAGGGCTAGACGTATGC | |
| pR-KHxmut01 | R-KHax01 | CACAGCCAAGTTGTGATCAAAGGGATTCCGCTATCTG |
| pR-KHxmut02 | CACAACTTGGCTGTGCTCGATTATGTGTTTATGAC | |
| pR-KHxmut03 | R-KHax02 | CCGAGACACGACTCTGTGTGAAGTCCTCCCGATTCTGGTGCG |
| pR-KHxmut04 | AGAGTCGTGTCTCGGAGCATCGAATAGCTCCCAAAC | |
| For EMSA of His6-GlxR | ||
| pgenDFM04 | DFMan and DFMaxm | CCCAAGCTTGCGCCCATTGTTGGAGTCC |
| pgenDFM09 | CGCGGATCCATGGGGGGAATTTTCAGAGCTG | |
| pgenR04 | R-KHan | CCCAAGCTTGGCGACGTTATCCATAATC |
| pgenKH07 | CCCAAGCTTGCCAATGAGCACCGCAGCCAAG | |
| pgenR02 | R-KHax01 and R-KHax01 m | CCCAAGCTTGCACCGGAAATGGTCAAAC |
| pgenR03 | CGCGGATCCTCTGGTGCGTGTGATGTC | |
| pgenKH11 | R-KHax02 and R-KHax02 m | CGCGGATCCTATGACATCACACGCACCAG |
| pgenKH09 | CCCAAGCTTAAGTTCCACTCCTTAGCCAG |
Construction of plasmids and strains.
DNA manipulation was carried out as described previously (34). E. coli and C. glutamicum were transformed by electroporation according to a method described previously (33).
The glxR gene was amplified using primers glxre01 and glxre02 and was digested with NdeI and EcoRI before being inserted into pET28a(+) to produce pZWHJ024. N-terminal His-tagged GlxR (His6-GlxR) was expressed in E. coli Rosetta(DE3)pLysS carrying pZWHJ024. The glxR gene amplified using primers glxre08 and glxre09 was cloned into SalI and EcoRI sites of plasmid pXMJ19 to generate plasmid pZWHJ033. The glxR gene was overexpressed in C. glutamicum carrying pZWHJ033.
The GlxR binding sites DFMx, R-KHx01, and R-KHx02 were mutated using the In-Fusion Advantage PCR cloning method with the templates of pZWHJ006, pZWHJ007, and pZWHJ008, respectively, as described previously (25). To mutate site DFMx, 5-bp substitutions (GTGN8TTC to CACN8GTG) were generated using the mutagenic primers pDFMxmut01 and pDFMxmut02 to generate pZWHJ025. In the same manner, constructs pZWHJ026 and pZWHJ029 were made by replacing TGTAACTTGGCTCACC with CACAACTTGGCTGTGC at the R-KHx01 sites on pZWHJ007 and pZWHJ008, respectively (substitutions are in boldface). Constructs pZWHJ027 and pZWHJ030 were made by replacing TGTGACAGAGTCAACTCTCGG with TCACACAGAGTCGTGTCTCGG at the R-KHx02 site on pZWHJ007 and pZWHJ008, respectively. Constructs pZWHJ028 and pZWHJ031, each containing both mutated sites R-KHx01 and R-KHx02, were respectively obtained by replacing TGTGACAGAGTCAACTCTCGG with TCACACAGAGTCGTGTCTCGG at the R-KHx02 site on pZWHJ026 and pZWHJ029. The promoters with or without mutation are represented in Fig. 1.
FIG 1.

Organization of the gen cluster, physical locations of promoters for activity detection, and probes for EMSAs. The region of the DNA fragment in the genome of Corynebacterium glutamicum ATCC 13032 is indicated (54). The promoter or probes below the gen cluster contained the following mutated GlxR binding sites: mutated site DFMx (from GTGN8TTC to CACN8GTG), indicated with an asterisk; mutated site R-KHx01 (from TGTN9CAC to CACN9GTG), indicated with a rhombus (◆); mutated site R-KHx02 (from GTGN8AAC to CACN8GTG), indicated with a filled circle (•).
GlxR overexpression and purification.
N-terminal His-tagged GlxR (His6-GlxR) was expressed in E. coli Rosetta(DE3)pLysS carrying pZWHJ024. The cells were grown at 37°C to an optical density at 600 nm (OD600) of 0.4 in LB supplemented with 50 μg ml−1 kanamycin. IPTG (isopropyl-β-d-thiogalactopyranoside) was then added to a final concentration of 0.1 mM, and the cultures were incubated at 30°C for another 5 h. His6-tagged GlxR was purified using Ni-nitrilotriacetic acid (Ni-NTA) agarose chromatography (Novagen) before being stored in glycerol at 4°C, and its purity was monitored by SDS-PAGE.
EMSAs.
DNA probes containing both wild-type GlxR binding sites and their mutants (illustrated in Fig. 1) were prepared as BamHI-HindIII fragments with 5′ overhanging ends by PCR amplification using primers described in reference 25 or in Table 2. The probes were treated with calf intestine alkaline phosphatase (Promega, Beijing, China) and then labeled with [γ-32P]ATP by T4 polynucleotide kinase (Promega) to fill the 5′ recessed ends. The specific competitors were the nonradiolabeled probes. The nonspecific competitor (155 bp) from the genD gene was amplified using primers ptgenIKL01 and pgenDFM01 (25) from the C. glutamicum genome. Electrophoretic mobility shift assays (EMSAs) were performed as described previously (35). GlxR-DNA complexes were generated in 20-μl reaction mixtures, which were incubated for 30 min at 25°C. Each reaction mixture contained 2 μl 10× binding buffer (100 mM Tris, 500 mM KCl, 10 mM dithiothreitol [DTT]; pH 7.5), 2 ng (1.2 to 1.6 nM) of each probe, and various amounts of purified His6-GlxR. The EMSAs were run at 4°C on 5.0% native polyacrylamide gels for 1 h. Gels were dried and exposed in a cassette (Yuehua, Shantou, China) using a phosphor storage screen (PerkinElmer, Boston, MA, USA). Images of radioactive filters were obtained and quantified with a Cyclone Plus storage phosphor system and OptiQuant image analysis software (PerkinElmer).
DNase I footprinting.
Footprinting assays were performed as described previously (36), and the primers were the ones used in a DNase I footprinting study with GenR (25). The probe was prepared by labeling the 5′ end of the primers with [γ-32P]ATP and T4 polynucleotide kinase before PCR. The footprinting reaction mixture contained 200 ng labeled DNA probe and different concentrations of His6-GlxR in 100 μl EMSA reaction buffer. After incubation of the mixture at 25°C for 30 min, 11 μl DNase reaction buffer and different concentrations of RNase-free DNase (Promega) were added to the binding mixture and incubated at 25°C for 1 min. The reaction was stopped by the addition of 10 μl stop solution [200 mM EGTA (Sigma), pH 8.0] and100 μl nuclease-free water. An equal volume of a 1:1 mixture of Tris-saturated phenol-chloroform was then added to the above-described solution. DNA fragments in the aqueous phase were precipitated by adding DNAmate (TaKaRa, Dalian, China) and directly suspended in 10 μl formamide loading dye (80% deionized formamide, 10 mM NaOH, 1.25 mM EDTA, 0.1% xylene cyanol FF, and 0.1% bromophenol blue). Samples were then denatured at 95°C for 5 min and run on 6% polyacrylamide-urea sequencing gels next to the corresponding sequencing ladder of the AccuPower DNA sequencing kit (Bioneer, Seoul, South Korea). After electrophoresis, the gels were dried and exposed to Kodak X-ray film.
Enzyme assays.
β-Galactosidase activity was determined in Miller units (37, 38). C. glutamicum cells were grown to an OD600 of 1.2 and then harvested and washed once in 1 ml Z buffer (0.06 M Na2HPO4·12H2O, 0.04 M NaH2PO4·2H2O, 0.01 M KCl, 0.001 M MgSO4, 0.05 M β-mercaptoethanol) before being resuspended with the same buffer and treated with 2% toluene for 10 min at 37°C (38). The permeabilized cells were then incubated with o-nitrophenyl-β-d-galactopyranoside (ONPG; Sigma-Aldrich, St. Louis, MO) at 30°C, and β-galactosidase activity was determined in Miller units as previously described (37). The 3-HBA 6-hydroxylase activity (39) and maleylpyruvate isomerase activity (40) were determined as described previously. Protein concentration was determined according to the Bradford method (41).
RNA preparation and transcription analysis.
Total RNA from C. glutamicum was isolated using the hot-phenol method (42). For transcription analysis, total RNA was digested with 1 U μg−1 recombinant DNase I (TaKaRa) for 1 h at 37°C, and 1 μg RNA was reversely transcribed with PrimeScript reverse transcriptase (TaKaRa). The resulting cDNA was amplified using quantitative real-time PCR (RT-qPCR), using the primers from a previous study (25). RT-qPCR was performed in a CFX Connect real-time PCR detection system (Bio-Rad) in a 25-μl reaction volume using iQ SYBR green Supermix (Bio-Rad). All samples were run in triplicate in three independent experiments. Relative expression levels were estimated using the 2−ΔΔCT method (where CT is the threshold cycle), and the 16S rRNA gene served as a reference for normalization (43).
Statistical analysis.
Statistical analysis was performed with SPSS version 20.0.0 software. Paired-samples tests were used to calculate probability values (P) for the transcription of genDFM, genR, and genKH and promoter activity containing DFMa or DFMax01m. And one-way analysis of variance (ANOVA) was used to calculate probability values (P) for β-galactosidase activity analyses of Ra, KHa promoter containing GlxR binding sites, or their corresponding mutants. P values of <0.05 and <0.01 were considered to be statistically significant and greatly statistically significant, respectively.
RESULTS AND DISCUSSION
GlxR binds to the promoter regions of the gen operons.
Although in silico detection and ChIP-chip analyses have indicated that GlxR binds to the promoter regions of 281 operons comprising 439 genes in this strain, including those of the gen cluster for 3-HBA and GEN catabolism (30), studies on the biochemical and physiological roles of GlxR are very limited.
To investigate whether GlxR directly regulates the transcription of the gen cluster for 3-HBA and GEN catabolism, recombinant GlxR was purified for analysis of its ability to bind to the DNA fragments encompassing the promoter regions of genDFM, genR, and genKH operons (DFMan and R-KHan in Fig. 1) in EMSA. The purified His6-GlxR was incubated with fragments of DNA spanning approximately −180 to +120 bp relative to their translational start sites of target genes. The addition of His6-GlxR to reaction mixtures caused a shift in the mobility of the promoter DNA fragments at protein concentrations ranging from 7.5 to 180 nM (Fig. 2A and C). A saturating concentration of 80 nM His6-GlxR, at which a retarded and stable protein-DNA complex was formed, was used to further examine its specificity of binding to probes. After addition of excess specific cold probe, the shift was found to be abrogated (Fig. 2B and D), indicating a specific interaction between GlxR and promoter DNA fragments. The nonspecific competitor is also able to decrease GlxR binding ability to promoter regions, although this competition is relatively weak. Unlike the specific regulator GenR, the global regulator GlxR, with more than 200 DNA binding sites in C. glutamicum (27, 30), may have a lower specificity in the binding to the promoter region of gen operons.
FIG 2.

Electrophoretic mobility shift assays for determination of GlxR binding sites upstream of genDFM, genR, and genKH operons. (A and C) Effects of cAMP on the binding affinity of GlxR for the upstream regions of genDFM, genR, and genKH operons. The probes for genDFM (DFMan in panel A), genR, and genKH operons (R-KHan in panel C) (25) were incubated with increasing amounts of His6-GlxR, while the amounts of cAMP were constant. The concentrations (nM) of purified His6-GlxR and the presence of 500 nM cAMP (+) or absence of cAMP (−) are indicated above the lanes. Glucose served as a control. (B and D) Competition assays using unlabeled specific competitor (SCR) and nonspecific competitor (NSCR) DNA. Labeled probe and unlabeled competitor of 25-fold, 50-fold, and 100-fold more in amount were incubated with His6-GlxR for 30 min at 25°C. Each lane contained 0.5 ng (0.3 to 0.4 nM) of 32P-labeled DFMan and R-KHan probes. The free probes are indicated by open arrows and the retarded DNA fragments by solid arrows.
GlxR has been found to be involved in the regulation of many genes by binding to partial region of promoters in a cAMP-dependent manner in vitro (27, 29). However, recent findings indicated that its regulation is not entirely dependent on the cAMP level, and the cAMP level required for GlxR binding to DNA varies depending on the target site (30). In the current study, although GlxR was found to bind to the corresponding regions regardless of the presence or absence of cAMP (Fig. 2A and C), cAMP could enhance the affinity of GlxR for these promoters to some degrees.
It has been demonstrated that creating a glxR deletion mutant is problematic in several cases, or generated mutants showed severe growth defects (28, 30, 44). Attempts were also made in the current study, but no such mutant was obtained. The involvement of GlxR in regulating physiological function, including aromatic degradation, in C. glutamicum was generally performed by in vitro binding and promoter-reporter assays (27, 29, 30). Following in vitro binding assays in this study, specific GlxR binding sites with promoters of gen operons were subsequently determined.
Determination of the GlxR binding sites.
The transcriptional start sites (TSSs) of gen operons have been determined in previous work (25). To further determine the regulatory role of GlxR in the expression of gen operons, the DNA binding sites of each operon were identified by DNase I footprinting analyses. For genDFM, His6-GlxR was found to protect a region (designated DFMx) from −13 to +8 bp relative to TSS (Fig. 3A). The footprint on the R-KH promoter region covered two regions, one (designated R-KHx01) from −24 to −9 bp relative to the genR TSS and from −11 to +5 bp relative to the genKH TSS, and the other (designated R-KHx02) from −71 to −91 bp relative to the genR TSS and from +52 to +72 bp relative to the genKH TSS, as shown in Fig. 3B with primer pegenR01 and in Fig. 3C with primer pegenKH04, respectively. Sites DFMx and R-KHx01 were almost the same as those previously reported (27, 29, 30). A novel site, R-KH02, detected in the footprinting analysis of this study was not identified by in silico detection (27, 29) or ChIP-chip analysis (30). In addition, the probe R-KHan contained two GlxR binding sites (Fig. 1). The regulators binding two sites through higher-order complex formation have been reported (45, 46), although most of the regulator proteins bind two sites without change in the configuration as indicated in the review articles (47, 48). There is a possibility that the GlxR-DNA complex in this study is formed in a variety of formations. Unfortunately, we have not captured the significant changes by in vitro experiments, including EMSA (Fig. 2C) and DNase I footprinting (Fig. 3B and C).
FIG 3.

Identification of the GlxR binding sites upstream of genDFM (A), genR (B), and genKH (C) operons by DNase I footprinting analysis. The reaction mixtures contained approximately 200 ng of end-labeled PCR products. These were amplified with prnagRD02 primer and 32P-labeled pegenDFM01 primer (A), pgenR01 and 32P-labeled pegenR01 (B), or pgenKH02 and 32P-labeled pegenKH04 (C) (25). Before DNase I treatment, labeled DNA was preincubated with His6-GlxR for 30 min at 25°C. Standards were generated by sequencing with 32P-labeled primers pegenDFM01(A), pegenR01 (B), and pegenKH04 (C). The concentrations (μM) of purified His6-GlxR and the GlxR-protected sequences are indicated. The nucleotide sequence around the TSS (+1) (25) is shown by solid arrows, and the GlxR binding sites are represented by brackets. Arrows indicate the direction of transcription. (D and E) Organization of the upstream region of the genDFM operon (D) and the genR-genKH promoter region (E) is shown in detail. The first codon of each gene is given in italics and boldface. The sequences in the dotted boxes are site R-KHxu as proposed previously (30), and the regulator GlxR binding sites are showed in the box. The TSSs are shown by +1 (boldface) and arrows and the putative −10 and −35 promoter sequences are underlined. The regulator GenR binding sites are highlighted in gray.
In order to find a consensus sequence from the above-described three DNA regions of gen promoters interacting with GlxR, bioinformatics analysis was performed using the motif-based sequence analysis tool MEME (multiple Em for motif elicitation) (49). A consensus sequence in the form of an imperfect palindrome ([T]GTGN8T[A]T[A]C) (where bases in square brackets are occasionally present) was therefore revealed as shown in Fig. 4A. This imperfect palindrome was obtained from the following: (i) the imperfect palindrome in site DFMx is GTGN8TTC; (ii) the imperfect palindrome of the complementary sequence for site R-KHx01 (GGTGAGCCAAGTTACA) is GTGN8TAC, but the perfect palindrome GTGN9ACA of this site matches better with the reported palindrome; (iii) site R-KHx02 is TGTGN8AAC. The consensus motif obtained is almost the same as the previously reported 5′-TGTGAN6TCACA-3′ (27, 29, 44, 50–52) and 5′-TGTGN8CACA-3′ (30) for GlxR in C. glutamicum (Fig. 4B). However, site R-KHxu, identified by ChIP-chip, was not detected in footprinting analysis in this study (Fig. 3). It is worth noting that our conclusion of this consensus motif of GlxR binding sites was based on footprinting analysis rather than in silico detection or ChIP-chip analysis used in previous determinations.
FIG 4.
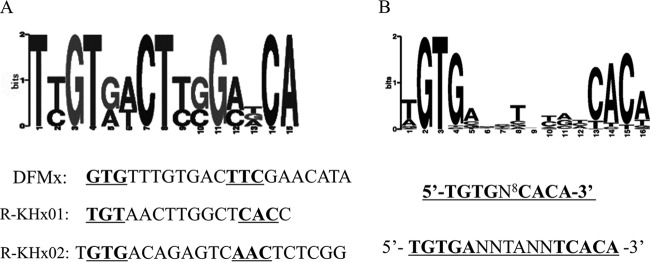
Sequence logos of motifs identified in GlxR binding sites by MEME (multiple Em for motif elicitation). (A) Motifs identified in GlxR binding sites for 3-HBA/GEN catabolic genes by MEME. (B) Data modified from references 27, 29, 30, and 44.
Role of the conserved sequence in GlxR binding and transcriptional regulation of genDFM, genR, and genKH operons.
To further confirm the GlxR binding sites identified in the upstream regions of gen operons, the effects of mutations within the three GlxR binding sites were examined in vitro. Toyoda et al. have examined effects of mutations within the GlxR binding sites in vivo and in vitro by exchanging the nucleotides corresponding to the positions of GTG and CAC in the consensus motif to CAC and GTG, respectively (5′-TGTG-N8-CACA-3′ to 5′-TCAC-N8-GTGA-3′) (30). According to this reported method, the underlined and bolded sequences in Fig. 4A have been used for further study. The nucleotides in the consensus motif of DFMx (GTGN8TTC), R-KHx01 (TGTN9CAC), and R-KHx02 (GTGN8AAC) (Fig. 4A) were changed to form DFMxm (CACN8GTG), R-KHx01m (CACN9GTG), and R-KHx02m (CACN8GTG), respectively. Probes containing both wild-type and mutated sites (Fig. 1) were used with purified His6-GlxR in EMSAs. As shown in Fig. 5, the introduced mutations abolished the GlxR binding, indicating that the consensus sequences [T]GTGN8T[A]T[A]C and GTGN9ACA were an essential determinant of GlxR binding activity.
FIG 5.
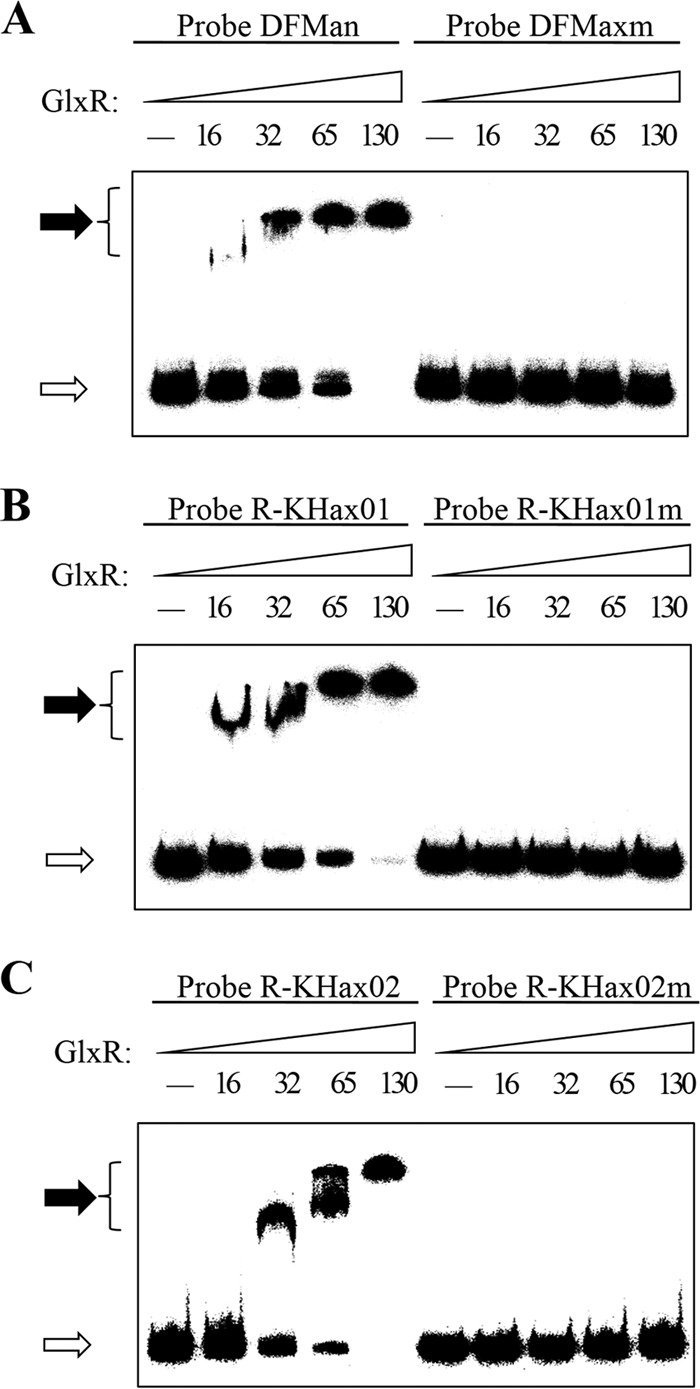
Mutational analyses of the four GlxR binding sites. EMSAs using the wild-type and mutated DNA fragments. Probes DFMan, R-KHax01, and R-KHax02 contained the intact GlxR binding sites DFMx, R-KHx01, and R-KHx02, respectively. Probes DFMaxm, R-KHax01m, and R-KHax02m contained the mutated GlxR binding sites described in Materials and Methods. The amounts (nM) of His6-GlxR used in lanes 1 to 5 are indicated. The free probes are indicated by open arrows, and the retarded DNA fragments are indicated by solid arrows.
In addition to the evidence that three GlxR binding sites in gen operons were bound by GlxR in vitro (Fig. 3), their roles in vivo were also investigated subsequently. The same mutation on consensus sequence for these binding sites used in EMSA was also introduced in the genDFM promoter of pZWHJ006, the genR promoter of pZWHJ007, and the genKH promoter of pZWHJ008 (Fig. 1). The resulting seven constructs were transformed into strain RES167 for β-galactosidase activity assays. As shown in Fig. 6B, the activity in the presence of genDFM promoter containing mutated site DFMax had an approximately 2-fold increase under the GEN-induced condition and a 1.5-fold increase under the 3-HBA condition in comparison with its wild-type promoter. For the genR promoter containing the mutated site R-KHx01m, its activity was similar to that in its wild-type counterpart. However, a 2-fold β-galactosidase activity was observed with the genR promoter containing mutated site R-KHx02m or sites R-KHx12m with a double mutation. On the other hand, the transcription of genKH was increased 1.5-fold in the presence of promoter containing mutant site R-KHx01m or double mutated site R-KHx12m, but it was not affected by the mutated site R-KHx02m. These results suggested that the transcription of genDFM, genR, and genKH was negatively regulated by the GlxR binding to sites DFMx, R-KHx02, and R-KHx01, respectively. And the consensus sequences [T]GTGN8T[A]T[A]C and GTGN9ACA were essential for the negative regulation. It is worth noting that GlxR binding site R-KHx01 overlaps each of the −10 regions of genR and genKH promoters. In the mutation procedure, the nucleotides in the consensus motif of R-KHx01 (TGTN9CAC) were changed to form R-KHx01m (CACN9GTG). The two −10 regions have been changed in the following manner: −10 region of genR, from TACAAT to TTGTAT; −10 region of genKH, from TTGATT to TTGATC. However, higher activities were detected from promoters of genR and genKH operons containing the mutated GlxR binding site. Therefore, the mutations in the GlxR binding sites have no effect on the −10 region of genR and genKH operons.
FIG 6.

GlxR attenuates upregulation of the transcription of genDFM, genR, and genKH operons. (A) qRT-PCR analyses examining the transcription of genDFM, genR, and genKH in strains RES167 and RES167/pZWHJ033. RNA samples were isolated from strains RES167 and RES167/pZWHJ033 grown on MM with 2 mM 3-HBA overnight. The levels of gene expression in each sample were calculated as the fold expression ratio after normalization to the 16S rRNA gene transcript. The values are averages of two independent RT-qPCR experiments. Error bars indicate standard deviations. These data are derived from at least three independent measurements. There was a significant difference in the transcription of genDFM, genR, and genKH between strains RES167 and RES167/pZWHJ033 (P < 0.001, paired-samples test). (B) β-Galactosidase activity driven by genDFM, genR, and genKH promoters with their corresponding GlxR binding sites in strain RES167. DFMa, Ra, and KHa containing GlxR binding sites are the GenR binding sites reported as previously (25). DFMax01m, Rax01m, Rax02m, Rax12m, KHax01m, KHax02m, and KHax12m are the constructs containing the mutated GlxR binding sites described in Materials and Methods and shown in Fig. 1. The β-galactosidase activity analyses were performed as given in the text. The data are derived from at least three independent measurements, and error bars indicate standard deviations. There was a significant difference in promoter activity between GlxR binding sites and its mutated sites from strains RES167 grown on MM with GEN or 3-HBA (P < 0.001, one-way ANOVA).
Previous work has identified three GlxR binding sites in the gen cluster by bioinformatics analysis or ChIP-chip analyses (27, 30), but one of them was not among the three sites identified in this study. In footprinting analyses for two directions of the genR-KH region (Fig. 3B and C), the sequencing results demonstrated that GlxR bound to three sites at the promoter regions of gen operons (Fig. 3). Based on the mutation and β-galactosidase assays, the GlxR binding site DFMx identified is similar to the GlxR binding site obtained by in silico detection (27) and ChIP-chip analysis (30), which is necessary for negative regulation of the transcription of genDFM. A single binding site, R-KHx01, required for repression of the transcription of genKH, matches the GlxR binding site of in silico detection (27). However, GlxR binding site R-KHx02, involved in negative regulation of the activity of the genR promoter for the expression of GenR regulator from this study, was not detected by in silico and ChIP-chip (27, 30). In contrast, site R-KHxu, identified by ChIP-chip, was not detected in footprinting analysis of this study (Fig. 3). Our study has demonstrated that the prediction of two GlxR binding sites for the gen cluster by in silico analysis and ChIP-chip was valuable for experimental studies of its regulation, although one of three predicted sites did not match the experimental data.
GlxR represses the expression of gen operons involved in the 3-HBA and GEN pathway.
It has been predicted that global regulator GlxR may be involved in the 3-hydroxybenzoate and gentisate pathways by activating the expression of genDFM genes in C. glutamicum, based on bioinformatics and ChIP-chip analyses (29, 30). In order to analyze the involvement of GlxR in the expression of genes in the gen cluster, the biochemical activity of enzymes encoded by genH in the genKH operon and genM in the genDFM operon was assayed in the glxR-overexpressed recombinant strain RES167/pZWHJ033, with 3-HBA induction. genH-encoded 3-HBA 6-hydroxylase activity of 0.31 ± 0.01 U/mg protein was present in wild-type strain RES167, but a much lower activity of 0.05 ± 0.01 U/mg protein was found in the glxR-overexpressed recombinant strain RES167/pZWHJ033. For genM-encoded maleylpyruvate isomerase, strain RES167 exhibited an activity of 1.88 ± 0.04 U/mg protein and the glxR-overexpressed recombinant strain showed a lower activity (0.80 ± 0.02 U/mg protein). To further assess the effects of GlxR on the expression of the 3-HBA and GEN catabolic operons, transcription of gen operons was also analyzed in the glxR-overexpressed recombinant strain RES167/pZWHJ033 by RT-qPCR. As shown in Fig. 6A, transcriptional levels of genDFM, genR, and genKH operons were one-fifth, one-seventh, and one-sixth, respectively, in recombinant RES167/pZWHJ033, in comparison with that in the wild-type strain under the 3-HBA-induced conditions. No enzyme activity for 3-hydroxybenzoate/gentisate catabolism and promoter activity (β-galactosidase assays) of gen operons was found to be present in the absence of 3-hydroxybenzoate and gentisate in strain RES167 (1, 25). These results indicated that the overexpression of glxR repressed all three operons to significant degrees. Recently, a unique recombineering method for constructing a mutant strain has been established in Corynebacterium glutamicum to study its biological function in vivo (21). If the mutation of GlxR binding sites in the glxR overexpression strain had been performed using this method, the relationship between the effects of the binding site mutations on gene expression and the binding of GlxR would have been made much more direct.
In C. glutamicum, a dual-function protein, GenR of the IclR family, has previously been identified to positively regulate the expression of genDFM and genKH operons for degradation of 3-HBA and GEN and repress the expression of its own encoding gene (genR) (25). We have now demonstrated that a CRP/FNR-type GlxR is an additional regulator for repressing the expression of three gen operons for 3-HBA and GEN catabolism. It is likely that GlxR contributes to the synchronization of expression of gen operons for 3-HBA and GEN catabolism in collaboration with the specific regulator GenR. Despite the fact that GlxR has been predicted to be involved in the activation or repression of considerable genes of this strain (30, 53), no thorough study on this global regulator has been conducted for its regulation of a catabolic pathway until this one.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant 31271333) and the National Key Basic Research Program of China (973 Program, grant 2012CB721003).
Footnotes
Published ahead of print 2 May 2014
REFERENCES
- 1.Shen XH, Jiang CY, Huang Y, Liu ZP, Liu SJ. 2005. Functional identification of novel genes involved in the glutathione-independent gentisate pathway in Corynebacterium glutamicum. Appl. Environ. Microbiol. 71:3442–3452. 10.1128/AEM.71.7.3442-3452.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones DCN, Cooper RA. 1990. Catabolism of 3-hydroxybenzoate by the gentisate pathway in Klebsiella pneumoniae M5a1. Arch. Microbiol. 154:489–495. 10.1007/BF00245233 [DOI] [PubMed] [Google Scholar]
- 3.Fuenmayor SL, Wild M, Boyes AL, Williams PA. 1998. A gene cluster encoding steps in conversion of naphthalene to gentisate in Pseudomonas sp. strain U2. J. Bacteriol. 180:2522–2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou NY, Fuenmayor SL, Williams PA. 2001. nag genes of Ralstonia (formerly Pseudomonas) sp. strain U2 encoding enzymes for gentisate catabolism. J. Bacteriol. 183:700–708. 10.1128/JB.183.2.700-708.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rani M, Prakash D, Sobti RC, Jain RK. 1996. Plasmid-mediated degradation of o-phthalate and salicylate by a Moraxella sp. Biochem. Biophys. Res. Commun. 220:377–381. 10.1006/bbrc.1996.0413 [DOI] [PubMed] [Google Scholar]
- 6.Ohmoto T, Sakai K, Hamada N, Ohe T. 1991. Salicylic acid metabolism through a gentisate pathway by Pseudomonas sp. TA-2. Agric. Biol. Chem. 55:1733–1737. 10.1271/bbb1961.55.1733 [DOI] [Google Scholar]
- 7.Werwath J, Arfmann HA, Pieper DH, Timmis KN, Wittich RM. 1998. Biochemical and genetic characterization of a gentisate 1,2-dioxygenase from Sphingomonas sp. strain RW5. J. Bacteriol. 180:4171–4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poh CL, Bayly RC. 1980. Evidence for isofunctional enzymes used in meta-cresol and 2,5-xylenol degradation via the gentisate pathway in Pseudomonas alcaligenes. J. Bacteriol. 143:59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lack L. 1959. The enzymic oxidation of gentisic acid. Biochim. Biophys. Acta 34:117–123. 10.1016/0006-3002(59)90239-2 [DOI] [PubMed] [Google Scholar]
- 10.Bayly RC, Chapman PJ, Dagley S, Diberardino D. 1980. Purification and some properties of maleylpyruvate hydrolase and fumarylpyruvate hydrolase from Pseudomonas alcaligenes. J. Bacteriol. 143:70–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu K, Liu TT, Zhou NY. 2013. HbzF catalyzes direct hydrolysis of maleylpyruvate in the gentisate pathway of Pseudomonas alcaligenes NCIMB 9867. Appl. Environ. Microbiol. 79:1044–1047. 10.1128/AEM.02931-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu TT, Zhou NY. 2012. Novel l-cysteine-dependent maleylpyruvate isomerase in the gentisate pathway of Paenibacillus sp. strain NyZ101. J. Bacteriol. 194:3987–3994. 10.1128/JB.00050-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones RM, Britt-Compton B, Williams PA. 2003. The naphthalene catabolic (nag) genes of Ralstonia sp. strain U2 are an operon that is regulated by NagR, a LysR-type transcriptional regulator. J. Bacteriol. 185:5847–5853. 10.1128/JB.185.19.5847-5853.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin LX, Liu H, Zhou NY. 2010. MhbR, a LysR-type regulator involved in 3-hydroxybenzoate catabolism via gentisate in Klebsiella pneumoniae M5a1. Microbiol. Res. 165:66–74. 10.1016/j.micres.2008.08.001 [DOI] [PubMed] [Google Scholar]
- 15.Kumagai H. 2000. Microbial production of amino acids in Japan. Adv. Biochem. Eng. Biotechnol. 69:71–85. 10.1007/3-540-44964-7_3 [DOI] [PubMed] [Google Scholar]
- 16.Smith KM, Cho KM, Liao JC. 2010. Engineering Corynebacterium glutamicum for isobutanol production. Appl. Microbiol. Biotechnol. 87:1045–1055. 10.1007/s00253-010-2522-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wendisch VF, Bott M, Eikmanns BJ. 2006. Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and amino acids. Curr. Opin. Microbiol. 9:268–274. 10.1016/j.mib.2006.03.001 [DOI] [PubMed] [Google Scholar]
- 18.Ikeda M. 2003. Amino acid production processes. Adv. Biochem. Eng. Biotechnol. 79:1–35. 10.1007/3-540-45989-8_1 [DOI] [PubMed] [Google Scholar]
- 19.Inui M, Kawaguchi H, Murakami S, Vertes AA, Yukawa H. 2004. Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J. Mol. Microbiol. Biotechnol. 8:243–254. 10.1159/000086705 [DOI] [PubMed] [Google Scholar]
- 20.Hermann T. 2003. Industrial production of amino acids by coryneform bacteria. J. Biotechnol. 104:155–172. 10.1016/S0168-1656(03)00149-4 [DOI] [PubMed] [Google Scholar]
- 21.Binder S, Siedler S, Marienhagen J, Bott M, Eggeling L. 2013. Recombineering in Corynebacterium glutamicum combined with optical nanosensors: a general strategy for fast producer strain generation. Nucleic Acids Res. 41:6360–6369. 10.1093/nar/gkt312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen XH, Zhou NY, Liu SJ. 2012. Degradation and assimilation of aromatic compounds by Corynebacterium glutamicum: another potential for applications for this bacterium? Appl. Microbiol. Biotechnol. 95:77–89. 10.1007/s00253-012-4139-4 [DOI] [PubMed] [Google Scholar]
- 23.Xu Y, Wang SH, Chao HJ, Liu SJ, Zhou NY. 2012. Biochemical and molecular characterization of the gentisate transporter GenK in Corynebacterium glutamicum. PLoS One 7:e38701. 10.1371/journal.pone.0038701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang YF, Zhang JJ, Wang SH, Zhou NY. 2010. Purification and characterization of the ncgl2923-encoded 3-hydroxybenzoate 6-hydroxylase from Corynebacterium glutamicum. J. Basic Microbiol. 50:599–604. 10.1002/jobm.201000053 [DOI] [PubMed] [Google Scholar]
- 25.Chao HJ, Zhou NY. 2013. GenR, an IclR-type regulator, activates and represses the transcription of gen genes involved in 3-hydroxybenzoate and gentisate catabolism in Corynebacterium glutamicum. J. Bacteriol. 195:1598–1609. 10.1128/JB.02216-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schroder J, Tauch A. 2010. Transcriptional regulation of gene expression in Corynebacterium glutamicum: the role of global, master and local regulators in the modular and hierarchical gene regulatory network. FEMS Microbiol. Rev. 34:685–737. 10.1111/j.1574-6976.2010.00228.x [DOI] [PubMed] [Google Scholar]
- 27.Kohl TA, Baumbach J, Jungwirth B, Puhler A, Tauch A. 2008. The GlxR regulon of the amino acid producer Corynebacterium glutamicum: in silico and in vitro detection of DNA binding sites of a global transcription regulator. J. Biotechnol. 135:340–350. 10.1016/j.jbiotec.2008.05.011 [DOI] [PubMed] [Google Scholar]
- 28.Kim HJ, Kim TH, Kim Y, Lee HS. 2004. Identification and characterization of glxR, a gene involved in regulation of glyoxylate bypass in Corynebacterium glutamicum. J. Bacteriol. 186:3453–3460. 10.1128/JB.186.11.3453-3460.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohl TA, Tauch A. 2009. The GlxR regulon of the amino acid producer Corynebacterium glutamicum: detection of the corynebacterial core regulon and integration into the transcriptional regulatory network model. J. Biotechnol. 143:239–246. 10.1016/j.jbiotec.2009.08.005 [DOI] [PubMed] [Google Scholar]
- 30.Toyoda K, Teramoto H, Inui M, Yukawa H. 2011. Genome-wide identification of in vivo binding sites of GlxR, a cyclic AMP receptor protein-type regulator in Corynebacterium glutamicum. J. Bacteriol. 193:4123–4133. 10.1128/JB.00384-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X, Kohl TA, Ruckert C, Rodionov DA, Li LH, Ding JY, Kalinowski J, Liu SJ. 2012. Phenylacetic acid catabolism and its transcriptional regulation in Corynebacterium glutamicum. Appl. Environ. Microbiol. 78:5796–5804. 10.1128/AEM.01588-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konopka A. 1993. Isolation and characterization of a subsurface bacterium that degrades aniline and methylanilines. FEMS Microbiol. Lett. 111:93–99. 10.1111/j.1574-6968.1993.tb06367.x [DOI] [Google Scholar]
- 33.Eggeling L, Reyes O. 2005. 23 experiments, p 535–566 In Eggeling L, Bott M. (ed), Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton, FL [Google Scholar]
- 34.Sambrook J, Fritsch EF, Maniatis T. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 35.Hellman LM, Fried MG. 2007. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2:1849–1861. 10.1038/nprot.2007.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galas DJ, Schmitz A. 1978. DNase footprinting—a simple method for detection of protein-DNA binding specificity. Nucleic Acids Res. 5:3157–3170. 10.1093/nar/5.9.3157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller JH. 1972. Experiments in molecular genetics, p 352–355 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 38.Tanaka Y, Okai N, Teramoto H, Inui M, Yukawa H. 2008. Regulation of the expression of phosphoenolpyruvate: carbohydrate phosphotransferase system (PTS) genes in Corynebacterium glutamicum R. Microbiology 154:264–274. 10.1099/mic.0.2007/008862-0 [DOI] [PubMed] [Google Scholar]
- 39.Wang LH, Hamzah RY, Yu Y, Tu SC. 1987. Pseudomonas cepacia 3-hydroxybenzoate 6-hydroxylase: induction, purification, and characterization. Biochemistry 26:1099–1104. 10.1021/bi00378a017 [DOI] [PubMed] [Google Scholar]
- 40.Feng J, Che YS, Milse J, Yin YJ, Liu L, Ruckert C, Shen XH, Qi SW, Kalinowski J, Liu SJ. 2006. The gene ncgl2918 encodes a novel maleylpyruvate isomerase that needs mycothiol as cofactor and links mycothiol biosynthesis and gentisate assimilation in Corynebacterium glutamicum. J. Biol. Chem. 281:10778–10785. 10.1074/jbc.M513192200 [DOI] [PubMed] [Google Scholar]
- 41.Bradford MM. 1976. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 72:248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 42.Lin-Chao S, Bremer H. 1986. Effect of the bacterial-growth rate on replication control of plasmid pBR322 in Escherichia coli. Mol. Gen. Genet. 203:143–149. 10.1007/BF00330395 [DOI] [PubMed] [Google Scholar]
- 43.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 44.Letek M, Valbuena N, Ramos A, Ordonez E, Gil JA, Mateos LM. 2006. Characterization and use of catabolite-repressed promoters from gluconate genes in Corynebacterium glutamicum. J. Bacteriol. 188:409–423. 10.1128/JB.188.2.409-423.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lopez-Sanchez A, Rivas-Marin E, Martinez-Perez O, Floriano B, Santero E. 2009. Co-ordinated regulation of two divergent promoters through higher-order complex formation by the LysR-type regulator ThnR. Mol. Microbiol. 73:1086–1100. 10.1111/j.1365-2958.2009.06834.x [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto K, Ishihama A. 2003. Two different modes of transcription repression of the Escherichia coli acetate operon by IclR. Mol. Microbiol. 47:183–194. 10.1046/j.1365-2958.2003.03287.x [DOI] [PubMed] [Google Scholar]
- 47.Molina-Henares AJ, Krell T, Guazzaroni ME, Segura A, Ramos JL. 2006. Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol. Rev. 30:157–186. 10.1111/j.1574-6976.2005.00008.x [DOI] [PubMed] [Google Scholar]
- 48.Tropel D, van der Meer JR. 2004. Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol. Mol. Biol. Rev. 68:474–500. 10.1128/MMBR.68.3.474-500.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2:28–36 [PubMed] [Google Scholar]
- 50.Han SO, Inui M, Yukawa H. 2008. Effect of carbon source availability and growth phase on expression of Corynebacterium glutamicum genes involved in the tricarboxylic acid cycle and glyoxylate bypass. Microbiology 154:3073–3083. 10.1099/mic.0.2008/019828-0 [DOI] [PubMed] [Google Scholar]
- 51.Han SO, Inui M, Yukawa H. 2007. Expression of Corynebacterium glutamicum glycolytic genes varies with carbon source and growth phase. Microbiology 153:2190–2202. 10.1099/mic.0.2006/004366-0 [DOI] [PubMed] [Google Scholar]
- 52.Jungwirth B, Emer D, Brune I, Hansmeier N, Puhler A, Eikmanns BJ, Tauch A. 2008. Triple transcriptional control of the resuscitation promoting factor 2 (rpf2) gene of Corynebacterium glutamicum by the regulators of acetate metabolism RamA and RamB and the cAMP-dependent regulator GlxR. FEMS Microbiol. Lett. 281:190–197. 10.1111/j.1574-6968.2008.01098.x [DOI] [PubMed] [Google Scholar]
- 53.Nishimura T, Teramoto H, Toyoda K, Inui M, Yukawa H. 2011. Regulation of the nitrate reductase operon narKGHJI by the cAMP-dependent regulator GlxR in Corynebacterium glutamicum. Microbiology 157:21–28. 10.1099/mic.0.044552-0 [DOI] [PubMed] [Google Scholar]
- 54.Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns B, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Kramer R, Linke B, McHardy A, Meyer F, Mockel B, Pfefferle W, Puhler A, Rey D, Ruckert C, Rupp O, Sahm H, Wendisch V, Wiegrabe I, Tauch A. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J. Biotechnol. 104:5–25. 10.1016/S0168-1656(03)00154-8 [DOI] [PubMed] [Google Scholar]
- 55.Tauch A, Kirchner O, Loffler B, Gotker S, Puhler A, Kalinowski J. 2002. Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr. Microbiol. 45:362–367. 10.1007/s00284-002-3728-3 [DOI] [PubMed] [Google Scholar]