

Abstract
The enzymes involved in the initial steps of anaerobic benzene catabolism are not known. To try to elucidate this critical step, a metatranscriptomic analysis was conducted to compare the genes transcribed during the metabolism of benzene and benzoate by an anaerobic benzene-degrading, nitrate-reducing enrichment culture. RNA was extracted from the mixed culture and sequenced without prior mRNA enrichment, allowing simultaneous examination of the active community composition and the differential gene expression between the two treatments. Ribosomal and mRNA sequences attributed to a member of the family Peptococcaceae from the order Clostridiales were essentially only detected in the benzene-amended culture samples, implicating this group in the initial catabolism of benzene. Genes similar to each of two subunits of a proposed benzene-carboxylating enzyme were transcribed when the culture was amended with benzene. Anaerobic benzoate degradation genes from strict anaerobes were transcribed only when the culture was amended with benzene. Genes for other benzoate catabolic enzymes and for nitrate respiration were transcribed in both samples, with those attributed to an Azoarcus species being most abundant. These findings indicate that the mineralization of benzene starts with its activation by a strict anaerobe belonging to the Peptococcaceae, involving a carboxylation step to form benzoate. These data confirm the previously hypothesized syntrophic association between a benzene-degrading Peptococcaceae strain and a benzoate-degrading denitrifying Azoarcus strain for the complete catabolism of benzene with nitrate as the terminal electron acceptor.
INTRODUCTION
Benzene is a petroleum-derived monoaromatic hydrocarbon that is present in crude oil and gasoline and is intensively used in the chemical processing industry (1). Benzene and other hydrocarbons frequently contaminate groundwater and soil, where they are readily biodegraded aerobically (2). When oxygen becomes limiting, as is often the case for contaminated sites, other electron acceptors typically enable continued biodegradation of many pollutants. Benzene biodegrades anaerobically much less readily than other monoaromatic compounds due to the absence of a substituent on the aromatic ring. Nevertheless, the past 2 decades of research have shown that benzene can be metabolized under nitrate-reducing (3, 4), sulfate-reducing (5–7), iron-reducing (8–10), and methanogenic (11, 12) conditions. Despite the interest in this process, the benzene-activating mechanism has remained elusive. Based on the metabolites detected during benzene metabolism, three initiating mechanisms have been proposed: hydroxylation to phenol, methylation to toluene, and carboxylation to benzoate (11, 13–15). Phenol, toluene, or benzoate would then be converted to the central aromatic intermediate, benzoyl-coenzyme A (CoA). The genes encoding enzymes involved in the anaerobic metabolism of these hypothetical intermediates and the downstream benzoyl-CoA are relatively well known (16). Distinct pathway variants for benzoyl-CoA catabolism are known from facultative anaerobes in the Alpha- and Betaproteobacteria (bcr/bad and bzd genes) (17–19) and from strict anaerobes in the Deltaproteobacteria (bam genes) (16, 20).
Benzoate has been nearly universally detected as a metabolite in benzene-degrading cultures (13, 15, 21). The benzoate measured may also originate from benzoyl-CoA that is hydrolyzed during extraction or analysis. In one of the most recent studies using labeled substrates, Kunapuli et al. (21) identified [13C6]benzoate in the supernatant of an iron-reducing culture supplied with [13C6]benzene and demonstrated that the carboxyl group came from bicarbonate. Subsequently, two proteins specifically expressed in the presence of benzene were identified in the same culture from a comparative metaproteomic study (22). These authors proposed, based on sequence homology to phenol carboxylases, that these proteins encoded two subunits of a putative anaerobic benzene carboxylase (Abc), which were designated AbcA and AbcD (22). Recently, Holmes et al. (23) also identified benzene-specific transcripts of a gene similar to abcA in Ferroglobus placidus when grown on benzene versus acetate. Phenol was also suggested as a metabolite in anaerobic benzene degradation (14, 15, 24), and it was also suggested to form abiotically in iron-reducing cultures upon exposure to oxygen during extraction (21). Zhang et al. (25) identified the upregulation of genes involved in phenol metabolism when Geobacter metallireducens was growing on benzene, indicating that the phenol metabolic pathway may indeed be linked to anaerobic benzene degradation.
Anaerobic benzene biodegradation has most frequently been observed in mixed cultures, although a few isolates have been described (4, 23, 26, 27). Across enrichment cultures, bacteria from the classes Deltaproteobacteria and Clostridia have been suggested as key microorganisms that activate the benzene ring (5, 7, 12, 28–30). In this study, a comparative metatranscriptomic analysis was performed on a benzene-degrading, nitrate-reducing enrichment culture. This culture has been maintained since 1997 and, through clone libraries and growth experiments, has been shown to contain operational taxonomic units (OTUs) from the families Peptococcaceae, Rhodocyclaceae (Dechloromonas, Azoarcus, and an undefined genus), and Burkholderiaceae, as well as the phyla Chlorobi and Planctomycetes (29, 31, 32). Using OTU-specific quantitative PCR, we observed increases in cell numbers for most of these taxa during benzene metabolism, but the highest increase observed was for Peptococcaceae (31), although organisms within Rhodocyclaceae were generally the most abundant. The growth of Planctomycetes spp. in the highly reducing conditions of these cultures is likely due to their ability to couple ammonium oxidation (supplied in the medium) to nitrite reduction (produced from nitrate reduction) (32). In the current metatranscriptomic study, we confirm our previous hypothesis of interactions between the strict anaerobes and the facultative, nitrate-reducing partners. We also provide compelling evidence for carboxylation carried out by Peptococcaceae as a key component of the initial steps in benzene catabolism.
MATERIALS AND METHODS
Nitrate-reducing enrichment cultures.
These cultures originated from microcosms prepared with soil and groundwater from a decommissioned gasoline station on Cartwright Avenue in Toronto, ON, Canada (3, 10); hence the name “Cartwright cultures.” Over the past 16 years, these cultures have been maintained in multiple 1- to 5-liter culture bottles with ca. monthly benzene (190 to 256 μM) and nitrate (2 to 5 mM) amendments and periodic transfers (10 to 50%) into sterile defined iron sulfide-reduced mineral medium (33). All cultures in this study were incubated statically and in the dark inside a Coy anaerobic chamber (Coy Laboratory Products, Madison, WI) supplied with an atmosphere of 80% N2, 10% H2, and 10% CO2.
Differential transcription experiments.
The goal of these experiments was to compare transcript levels between cultures amended with benzene and with benzoate. To try to minimize effects related to transferring the mixed culture, the same culture bottle was first amended with one substrate (e.g., benzoate), and once that substrate was consumed, the culture bottle was then amended with the second substrate (e.g., benzene). Between the two amendments, the culture was starved for 1 week. Two distinct experiments were conducted 1 month apart. In experiment 1, the culture was amended with benzoate first and then with benzene. RNA was extracted twice from the same culture, once as benzoate was being consumed and once as benzene was being consumed. In experiment 2, the order of the substrates was reversed, with benzene added first and then benzoate. For each experiment, 200 ml of Cartwright culture was purged of any remaining benzene with a steady gentle flow of gas (80% N2 and 20% CO2) and transferred to a 250-ml sterile, anaerobic clear Boston round glass bottle (Scientific Instrument Services, Ringoes, NJ, USA), amended with 2 mM sodium nitrate, and sealed with a screw-cap Mininert valve (Vici precision sampling, Baton Rouge, LA, USA). In experiment 1, the culture was initially amended with 128 μM benzoate. Degradation was rapid and was complete in less than 1 day. The culture was reamended with 128 μM benzoate and 2 mM nitrate, and a 50-ml culture sample was taken 2 h later, when ∼25% of the second feeding of benzoate was consumed (Fig. 1a). Once benzoate was depleted, the culture was starved for a week and then amended with 128 μM benzene (aqueous concentration) and 2 mM nitrate. A 50-ml culture sample was taken when ∼70% of benzene was depleted after a long lag time of 65 days (Fig. 1a). In experiment 2, the culture bottle was initially supplied with 128 μM benzene (aqueous concentration) and 2 mM nitrate. A 50-ml culture sample was taken after 13 days when ∼70% of benzene was consumed (Fig. 1b). Once benzene was depleted, the culture was starved for a week and then amended with 128 μM benzoate and 2 mM nitrate. The second 50-ml sample was taken after a few hours had elapsed, when ∼5% of benzoate was depleted (Fig. 1b). A total of four culture samples (2 pairs) for RNA extraction were thus obtained. Additional 200-ml culture bottles for each experiment were prepared similarly to the bottles described above to confirm the reproducibility of degradation profiles. Benzene, nitrate, and nitrite concentrations were monitored in all bottles, but RNA was only extracted from the two main experimental bottles as described above.
FIG 1.

Benzene and benzoate concentration profiles during differential transcriptomic experiments. Solid lines denote benzene concentrations (μM), while dashed lines show benzoate concentrations (μM). The circles identify RNA extraction sampling points. Note that the time scale varies from days (for benzene degradation) to hours (for benzoate degradation). (a) Experiment 1, benzoate followed by benzene amendment; the corresponding RNA samples were sequenced using pyrosequencing technology. (b) Experiment 2, benzene followed by benzoate amendment; the corresponding RNA samples were sequenced using Illumina technology.
Analytical methods.
Benzene concentrations were measured by injecting 300 μl of culture headspace into a Hewlett-Packard 6890 series gas chromatograph equipped with an HP-5 30-m by 0.32-mm-inner-diameter column and a flame ionization detector (FID). The injector, FID detector, and oven temperatures were at 200°C, 250°C, and 85°C, respectively. The carrier gas was nitrogen at a flow rate of 3 ml/min. Benzoate concentrations were measured by injecting 10-μl liquid samples withdrawn from the cultures into a Hewlett-Packard 1090 series II high-performance liquid chromatograph (HPLC). This HPLC was equipped with a Hypersil BDS C18 (5-μm particle size, 250-mm-long by 2-mm-inner-diameter) column and a UV detector at 230 nm. The mobile phase contained 25% acetonitrile and 75% KH2PO4 at a concentration of 50 mM (adjusted to pH 3 using phosphoric acid). The mobile phase flow rate was 0.3 ml/min. The retention time for benzoate was 2.8 min. Nitrate and nitrite concentrations were analyzed by injecting 20 μl of 1- to 10-times-diluted liquid samples onto a Dionex DX-100 series ion chromatograph equipped with an IonPac AS9 ion exchange column and an AG9 guard column. The mobile phase contained 5 mM sodium bicarbonate and 12.25 mM sodium carbonate. The eluent flow rate was 1 ml/min.
RNA extraction.
For all 50-ml culture aliquots, RNA extractions were conducted immediately upon sampling. RNA extractions were performed based on the protocol utilized by Waller et al. (34), with modifications. First, 50 ml of culture was dispensed into an anaerobic centrifuge tube on ice inside the anaerobic chamber. Cells were collected by centrifugation at 9,900 × g for 20 min at 4°C. The cell pellet was resuspended in 250 μl of ice-cold diethylpyrocarbonate-treated extraction buffer comprised of 1.4 M NaCl, 22 mM EDTA, and 35 mM sodium dodecyl sulfate. An amount of 0.9 ml of ice-cold acid phenol-chloroform-isoamyl alcohol (125:24:1, pH 4.5; Ambion, Austin, TX) was added to the mixture of cells and the extraction buffer. This mixture was transferred into a 1.5-ml screw-cap microcentrifuge tube containing 100 μl of sterile zirconia/silica beads (0.5 mm; BioSpec Products Inc., Bartlesville, OK). The tube was agitated horizontally in a Mini-Beadbeater-96 (Biospec Products Inc., Bartlesville, OK) for 4 min and then centrifuged at 1,4000 × g for 15 min at 4°C. The aqueous phase was transferred into a new 1.5-ml microcentrifuge tube containing 0.9 ml of ice-cold acid phenol-chloroform-isoamyl alcohol and then centrifuged at 1,4000 × g for 15 min at 4°C (this step was repeated twice). The supernatant was moved to a new microcentrifuge tube, and RNA was precipitated by adding 0.1 volume of ammonium acetate (7.5 M; Sigma-Aldrich) and 1.1 volume of isopropanol and incubating at −20°C overnight. The RNA pellet was collected by centrifugation for 15 min at 4°C and resuspended in RNase-/DNase-free water (Sigma-Aldrich). Contaminating DNA was removed using the Turbo DNA-free kit according to the manufacturer's instructions (Ambion, Austin, Texas).
cDNA library preparation and sequencing.
Sequencing was performed by the Center for Applied Genomics (The Hospital for Sick Children, MaRS Center, Toronto, ON, Canada). The two RNA samples from experiment 1 (benzoate/benzene) (Fig. 1a) were sequenced using a Roche GS FLX Titanium sequencer (Roche Applied Sciences/454 Life Sciences, Branford, CT), and the two RNA samples from experiment 2 (benzene/benzoate) (Fig. 1b) were sequenced using an Illumina Genome Analyzer II (Illumina Inc., San Diego, CA). The longer reads from pyrosequencing were selected for better assembly, while Illumina technology provided more sequencing depth. RNA samples for Roche 454 pyrosequencing were reverse transcribed using the Roche standard cDNA rapid library preparation method (Roche Applied Sciences/454 Life Sciences). The cDNA library was then quantified with an Agilent Bioanalyzer before sequencing. The average library sizes were 1,200 and 1,000 bp for samples from the benzoate-degrading and benzene-degrading time points, respectively. RNA for Illumina sequencing was reverse transcribed, and the cDNA libraries were prepared with the TrueSeq RNA sample low-throughput protocol (Illumina Inc.). The cDNA library was then quantified with the Kapa library quantification kit (Kapa Biosystems, Woburn, MA) before sequencing. The average library sizes were 203 and 216 bp for the samples from the benzene-degrading and the benzoate-degrading time points, respectively. The average quality score for each nucleotide in all the Illumina reads was calculated with FastQC (version 10.0) on the JGI Galaxy platform (35–37). Only reads with average quality scores above 20 were processed via the in silico pipeline; ambiguous nucleotides were not removed in this process.
Pyrosequencing data analysis pipeline.
The pipeline for identifying rRNA and mRNA sequences obtained by pyrosequencing (Roche/454) is depicted in Fig. 2. First, all RNA reads were compared to a small subunit and a large subunit rRNA reference database, which was constructed from publicly available databases (38), using BLASTn (39). The BLASTn output file was analyzed using MEGAN (MEtaGenome ANalyzer) software version 4 (40, 41) to generate a tree that clusters all sequences to their lowest common ancestor (LCA) in order to compare the microbial profiles of the culture when consuming benzene versus benzoate. RNA sequences that had no BLASTn matches to the ribosomal databases or that did but were not assigned to any taxonomy by MEGAN were considered non-rRNA. All of the unassigned sequences were assembled using Newbler software (454 Life Sciences, Roche, Branford, Connecticut). All contigs, isotigs, and singletons from the Newbler assembly result were further translated in six reading frames and compared to the GenBank nonredundant protein database using BLASTx.
FIG 2.

Sequence analysis pipeline. Data processing steps specific for pyrosequencing and Illumina sequencing are in separate dashed boxes. Programs used are highlighted in bold. Sequences removed from respective data pools are shown in light gray.
Illumina data analysis pipeline.
To extract phylogenic information from the RNA sequences, a random selection of 10% of the raw RNA reads was compared to the SILVA SSUrdb (small subunit reference database) (42) using BLASTn (Fig. 2). The BLASTn results were further analyzed with MEGAN version 4 (40, 41) for microbial community comparison, similar to the pyrosequencing pipeline. Coding sequences were pooled and enriched bioinformatically from the complete data set for functional analysis as follows. First, all raw reads were aligned to the SILVA SSUrdb and LSUrdb (large subunit reference database) using Bowtie (43). All reads without significant alignment to the rRNA database were pooled (benzene and benzoate degrading) and then assembled using MIRA3 (44). Reads that did not assemble (singletons) were removed (Fig. 2). Assembled sequences (contigs) were subsequently compared again with the SILVA SSUrdb and LSUrdb using BLASTn to remove the remaining rRNA sequences.
Identification of differentially transcribed genes.
To identify differentially transcribed genes, the number of raw reads in each assembled contig was normalized to reads per kilobase per million reads (RPKM) according to the following equations (45): benzene RPKM = number of benzene reads in each non-rRNA contig/(total number of benzene non-rRNA reads × contig length) × 109, and benzoate RPKM = number of benzoate reads in each non-rRNA contig/(total number of benzoate non-rRNA reads × contig length) × 109.
In the above equations and throughout the remaining text, “benzene reads” refers to cDNA sequence reads from the culture metabolizing benzene and “benzoate reads” refers to those from the culture metabolizing benzoate. To determine the relative transcription level of a specific contig, we calculated a contig specificity value, defined as the ratio of the normalized benzene and benzoate read numbers for a particular contig, as follows: contig specificity = benzene RPKM/benzoate RPKM.
When multiple contigs referred to the same gene, the specificity for a particular gene was calculated as follows: gene specificity = Σ(contig length × contig specificity)/Σcontig lengths.
Read values of zero, where a transcript was present only in one data set and not the other, were replaced with 0.5 before normalization so that a defined specificity could be calculated under all conditions (46). Since we were focusing on the relatively abundant genes, only contigs with an average coverage equal to or higher than 3 and a length exceeding 100 bp were annotated with BLASTx. The BLASTx result was then used as an input for MEGAN analysis to determine a taxonomic assignment for each contig. Each annotated contig was assigned to the lowest common taxonomic level shared by the top 10% of its BLAST hits.
PCRs for extending the contig containing the putative carboxylase gene abcA.
To determine the similarity between previously published sequences (22) and the abc gene operon retrieved in the work described herein, PCR primers (see Table S1 in the supplemental material) were designed targeting the intragenic region between the putative carboxylase genes abcD, abcA, and ubiX and the putative benzoate-coenzyme A ligase bzlA genes. The primers were designed to be complementary to the mRNA contigs identified in this study that were homologous to these genes. PCRs were conducted using a PTC-200 Peltier thermal cycler (MJ Research Inc., Waltham, MA). Each PCR mixture contained 1× ThermoPol PCR buffer, 5 U Taq polymerase (New England BioLabs Inc., Mississauga, ON, Canada), 0.3 mM deoxynucleoside triphosphates, 0.4 mM forward and reverse primers, and 30 to 100 ng genomic DNA template from the Cartwright culture. The temperature program was as follows: initial denaturation at 94°C for 5 min, followed by 29 cycles of denaturation at 94°C for 30 s, primer annealing for 1 min at 60°C, and chain extension for 45 s at 72°C, with a final extension at 72°C for 7 min. The PCR product was purified using a GeneJET PCR purification kit (Thermo Scientific, Ottawa, ON, Canada). The purified DNA samples were then submitted to the Centre for Applied Genomics (Toronto) for Sanger sequencing. Sequencing was performed using an ABL 3730 xl DNA analyzer with the same primers as were used for the PCRs.
Nucleotide sequence accession number.
Sequences for all annotated contigs are available in the supplemental material (Tables S2 and S3). The 16S rRNA sequence for the Peptococcaceae has been deposited in GenBank under accession number KJ522755.
RESULTS AND DISCUSSION
Benzene and benzoate degradation rates in Cartwright enrichment culture.
The typical benzene degradation rates in the Cartwright enrichment culture are 5 to 10 μM/day; these rates have been reasonably consistent for many years. A stoichiometry of 10 to 14 mol nitrate per mole benzene consumed has also been consistent over time (32). This stoichiometry is consistent with the oxidation of benzene to CO2 coupled to the incomplete reduction of nitrate to nitrite, as reported previously (3, 29). Benzene oxidation linked to complete reduction of nitrate to nitrogen would result in a lower ratio, below 6 mol nitrate per mole benzene (29). After benzene is depleted, nitrite is reduced to N2 in these cultures (3). The electron donors for this process are likely fatty acids or other carbohydrates released from biomass or sulfide present in the medium. In the additional culture bottles prepared similarly to those for the metatranscriptomic experiments, the consumption ratios were 10.3 ± 0.4 mol (mean ± standard deviation) of nitrate reduced/mole of benzoate oxidized and 14.3 ± 0.7 mol of nitrate reduced/mole of benzene oxidized (n = 6), consistent with prior results. Transient nitrite accumulation was also observed during both benzene and benzoate degradation (see Fig. S1 in the supplemental material), which is typical in these cultures.
The culture metabolized benzoate rapidly, at rates over 20 times higher than for benzene (Fig. 1) without any prior exposure to this substrate. Since benzoate—or its CoA derivative—is a probable intermediate of benzene degradation (13, 15, 21), one would expect that feeding the culture with benzoate would stimulate the degradation of benzene; however, this was not the case in these cultures, leading to the hypothesis that different organisms degrade benzoate and benzene in this culture. Rather, when subcultures were grown with benzoate and then subsequently amended with benzene, a lag time of at least 20 to 30 days was observed prior to the onset of benzene degradation in all replicates, similar to or longer than the typical lag times observed upon transferring the culture into fresh medium amended with benzene (31). In the experiments described here, the lag before the onset of benzene degradation after the addition of benzoate was as long as 65 days (Fig. 1a). To avoid the negative impact of feeding benzoate prior to benzene, we reversed the order of feedings in the second experiment (Fig. 1b).
RNA extraction and sequencing.
RNA extraction from these slow-growing cultures is difficult and was complicated by the presence of ferrous sulfide, a reductant in the medium, which causes macromolecules to precipitate after cell lysis. In the absence of effective rRNA removal techniques, the loss of mRNA during enrichment steps was deemed a significant risk. Therefore, once the protocol was optimized enough to obtain extractions of high-quality RNA, total RNA was sequenced without further mRNA enrichment to avoid sample loss. Fortuitously, analysis of the sequenced rRNA provided useful phylogenetic information on active members of the community. The first pair of samples sequenced using Roche 454 was successful, but the depth of coverage was not sufficient to determine enrichment levels. Illumina sequencing of a second pair of samples provided 50 times greater coverage despite the shorter read length (Table 1). Functional analysis of the sequence data as detailed below revealed the value in sequencing RNA even without a reference metagenome, because many contigs with functional genes of interest were successfully assembled and could be annotated, providing insights into the dynamics of the culture on the two substrates tested, as described below.
TABLE 1.
Sequence information summary for the RNA samples extracted from the Cartwright culture during growth on benzoate and benzene using pyrosequencing or Illumina sequencing
| Sequencing method and parameter | Benzene RNA | Benzoate RNA |
|---|---|---|
| Pyrosequencing (avg read length, 160 bp) | ||
| Total reads | 196,075 | 250,500 |
| Non-rRNA reads (% of total reads) | 2,153 (1.1) | 4,970 (2) |
| Non-rRNA contigs/isotigs | 81 | 219 |
| Annotated mRNA contigs (BLASTx) | 75 | 189 |
| Annotated mRNA singletons (BLASTx) | 490 | 1,831 |
| Illumina sequencing (read length, 75 bp) | ||
| Total reads | 26,893,212 | 28,567,030 |
| Non-rRNA reads (%) | 849,441 (3) | 1,149,965 (4) |
| Non-rRNA contigs | 366,368a | |
| Contigs that passed the abundance thresholdb | 37,353a | |
| Annotated mRNA contigs (BLASTx) | 14,038a |
The number shown is the number of contigs assembled from pooled benzene and benzoate RNA reads.
The abundance threshold is a contig length greater than 100 b and average coverage greater than 3.
Processing RNA sequence reads.
The total numbers of RNA and non-rRNA sequence reads obtained from pyrosequencing and Illumina protocols are summarized in Table 1. Illumina sequencing of the RNA samples (amended with benzene first and then with benzoate) generated 100 times more reads than the RNA samples sequenced via pyrosequencing (amended with benzoate first and then with benzene). The non-rRNA sequences accounted for 1 to 4% of the total reads, consistent with other studies indicating 1 to 5% mRNA in total cellular RNA in prokaryotic cells (47).
Active microbial community profiles from rRNA sequence reads.
The large number of rRNA sequence reads offered a unique window into the taxonomic affiliations of the active organisms in the culture during growth on each of the two substrates. The MEGAN program assigns reads into different clusters based on the LCA principle. Because Illumina reads are short and because ribosomal sequences are in general highly similar to each other, the MEGAN tree generated from the Illumina data (see Fig. S2 in the supplemental material) assigned most reads to higher taxonomical levels than in the tree generated with the pyrosequencing data (Fig. 3). However, both trees are sufficiently similar to alleviate concern that the lower sequencing depth of pyrosequencing affected the observed community. When normalized to 100,000 reads per sample, the family Peptococcaceae was the only branch with a distinctly higher number of ribosomal sequences when the culture was growing on benzene versus benzoate (Fig. 3, 3,849 versus 27; see also Fig. S2, 920 versus 252). All other dominant bacterial groups, including Rhodocyclales, Burkholderiales, Chlorobi, and Chloroflexi, had similar rRNA sequence abundance in both cases (Fig. 3; see also Fig. S2). These taxonomic results are entirely consistent with previous clone libraries (29, 32) and a previous 16S rRNA gene-based quantitative PCR investigation of the same culture that showed an increase of Peptococcaceae abundance linked to benzene depletion (31). Peptococcaceae have also been implicated in the initial attack on benzene in several other enrichment cultures, via 16S rRNA-based terminal restriction length polymorphism (7), stable isotope probing (28), and denaturing gradient gel electrophoresis (30). Our Peptococcaceae 16S rRNA sequence (GenBank accession number KJ522755) is 97% identical to clones obtained from the previously characterized iron-reducing Clostridium enrichment culture BF (28) and 98% identical to clones from a nitrate-reducing, benzene-degrading chemostat (30).
FIG 3.

Taxonomic comparison of active microbial communities from MEGAN analysis of pyrosequencing reads. Data from the benzoate-amended culture are in red, while data from the benzene-amended culture are in blue. The size of each circle at a tree node is proportional to the number of reads assigned to the corresponding taxon. The numbers in brackets are the total number of reads assigned to that node plus the number of reads in the subtree rooted at that node for each data set (benzoate; benzene). For comparison purposes, the number of reads was normalized to 100,000 reads per data set. A companion figure representing the Illumina sequence data is provided in Fig. S2 in the supplemental material.
Analysis of nonribosomal (mRNA) reads.
To identify genes that were specifically transcribed in the presence of benzene, we primarily relied on the Illumina data set because it afforded a much greater depth of sequencing. Only 3.8% of the nonribosomal contigs passed the threshold of read depth and length and had a hit from BLASTx analysis (Table 1). Contigs assigned to the order Clostridiales (mostly assigned to the family Peptococcaceae) were more abundant when the culture was degrading benzene (Fig. 4a), while contigs attributed to Rhodocyclales, Burkholderiales, and Planctomycetes were equally or slightly more abundant when the culture was degrading benzoate (Fig. 4b to d). These function-based data are entirely consistent with the rRNA community abundance analysis above. The complete set of annotated contigs and associated best hit identity scores and sequences are provided in Table S2 in the supplemental material. A complete set of annotated pyrosequencing data is also provided in Table S3. To further analyze these data, we first validated the results by mapping contigs to genes from pathways expected to be found in the culture, namely, anaerobic benzoate catabolism and nitrate respiration. These pathways were indeed well represented. Next, we tried to identify the most highly transcribed benzene-specific transcripts and associated pathways. Finally, we also searched for the presence of gene transcripts for other metabolic pathways that may or may not be present, including anaerobic toluene and phenol metabolism and aerobic aromatic pathways. The results of these analyses of the sequence data are summarized in Table 2, as well as a series of tables in the supplemental material (see Tables S4 to S10). The salient features of these analyses are further described below.
FIG 4.

Log-log plot (base 2) of normalized coverage (RPKM) for assembled contigs from dominant microbial groups assigned by MEGAN, comparing RNA samples from benzoate-amended (y axis) and benzene-amended (x axis) cultures. Clostridiales (Peptococcaceae) (a); Rhodocyclales (Azoarcus and Dechloromonas) (b); Burkholderiales (c); Planctomycetes (d). The red circles denote contigs encoding carboxylase/decarboxylase genes assigned to Clostridiales.
TABLE 2.
Summary of transcribed genes from pyrosequencing and Illumina sequencing data
| Gene(s) | Detected by pyrosequencing in culture with: |
Illumina sequencing results |
Protein annotation | Function | Pathway | |||
|---|---|---|---|---|---|---|---|---|
| Benzene | Benzoate | Gene specificitya | Taxon of closest match | % identity or range | ||||
| bzdA | No | Yes | 0.2 | Azoarcus | 64–87 | Benzoate-CoA ligase | Benzoate CoA ligase | Benzoate |
| bclA | No | No | 1.0 | Bacteria | 77.0 | Benzoate-CoA ligase | ||
| bzlA | No | No | 30 | Clostridiales | 78–93 | Putative benzoate-CoA ligase | ||
| bamB | No | No | 17 | Clostridiales | 91 | Putative aldehyde ferredoxin oxidoreductase | Benzoyl-CoA reductase (ferredoxin-dependent) | Benzoate |
| bamC | No | No | 55 | Clostridiales | 88 | Putative iron-sulfur binding protein | ||
| bamD | Yes | No | ND | Clostridiales | 85 | Putative NADH oxidoreductase | ||
| bamE | Yes | No | 101 | Clostridiales | 74–82 | Putative iron-sulfur binding protein | ||
| bcrA/badF | No | No | 3.0 | Proteobacteria | 61–86 | Benzoyl-CoA reductase, alpha subunit | Benzoyl-CoA reductase (ATP-dependent) | Benzoate |
| bcrB/badE | No | Yes | 3.0 | Proteobacteria | 67–78 | Benzoyl-CoA reductase, beta subunit | ||
| bcrC/badD | No | No | Benzoyl-CoA reductase, gamma subunit | |||||
| bcrD/badG | No | No | 3.0 | Proteobacteria | 71–78 | Benzoyl-CoA reductase, delta subunit | ||
| bzdN | No | Yes | 0.2 | Azoarcus | 89–100 | Benzoyl-CoA reductase, gamma subunit | ||
| bzdO | No | Yes | 0.3 | Azoarcus | 88–100 | Benzoyl-CoA reductase, beta subunit | ||
| bzdP | No | Yes | 0.1 | Azoarcus | 92–96 | Benzoyl-CoA reductase, delta subunit | ||
| bzdQ | No | Yes | 0.1 | Azoarcus | 98–100 | Benzoyl-CoA reductase, alpha subunit | ||
| bzdW | Yes | Yes | 0.2 | Azoarcus | 90.0 | Cyclohex-1-ene-1-carboxyl-CoA hydratase | Hydratase | Benzoate |
| bzdX | No | Yes | 0.1 | Azoarcus | 94.0 | 6-Hydroxycyclohex-1-ene-1-carboxyl-CoA dehydrogenase | Dehydrogenase | Benzoate |
| had | No | Yes | ND | Proteobacteria | 71–73 | 6-Hydroxycyclohex-1-ene-1-carboxyl-CoA dehydrogenase | ||
| bzdY | No | Yes | 0.1 | Azoarcus | 89–95 | 6-Oxocyclohex-1-ene-1-carbonyl-CoA hydrolase | Hydrolase | Benzoate |
| oah | No | No | 4.0 | Proteobacteria | 73–91 | 6-Oxocyclohex-1-ene-1-carbonyl-CoA hydrolase | ||
| abcA | Yes | No | 21 | Clostridiales | 95–100 | Putative anaerobic benzene carboxylase | Putative carboxylases | Unconfirmed pathway |
| abcD | Yes | No | 62 | Clostridiales | 94–96 | Putative anaerobic benzene carboxylase | ||
| ppsA | No | No | 6.0 | Proteobacteria | 81–83 | Phenylphosphate synthase alpha subunit | Phenyl phosphate synthase | Phenol |
| ppsB | No | No | 12 | Proteobacteria | 79.17 | Phenylphosphate synthase, beta subunit | ||
| ppsC | No | No | 4.0 | Proteobacteria | 56.14 | Protein stimulating phenylphosphate synthetase activity | ||
| ppcB | No | No | 72 | Deltaproteobacteria | 79–87 | Phenylphosphate carboxylase subunit beta | Phenylphosphate carboxylase | Phenol |
| hbcL | No | No | 2.0 | Azoarcus | 39–84 | 4-Hydroxybenzoate CoA ligase | 4-Hydroxybenzoate CoA ligase | Phenol |
| hcrA | No | No | 4.0 | Proteobacteria | 68 | 4-Hydroxybenzoyl-CoA reductase alpha subunit | Hydroxybenzoyl-CoA reductase | Phenol |
| ubiD-like | No | No | 14 | Clostridiales | 89–100 | Putative 3-polyprenyl-4-hydroxybenzoate carboxy-lyase | Decarboxylases | Ubiquinoneb |
| ubiX-like | No | No | 21 | Clostridiales | 91 | Putative 3-octaprenyl-4-hydroxybenzoate carboxy-lyase | ||
| ubiX | No | No | 1.5 | Deltaproteobacteria | 71–91 | 3-Octaprenyl-4-hydroxybenzoate carboxy-lyase (UbiX) | ||
| ubiX | No | No | 0.2 | Azoarcus | 71–93 | 3-Octaprenyl-4-hydroxybenzoate carboxy-lyase (UbiX) | ||
| ubiD | No | No | 0.2 | Azoarcus | 82–91 | 3-Polyprenyl-4-hydroxybenzoate carboxy-lyase (UbiD) | ||
Specificity was calculated from the number of reads per contig (see Materials and Methods). ND, not detected in the Illumina sequencing results. A visual representation of the majority of these genes within pathway assignments is presented in Fig. 6. The numbers of reads per gene (and thus gene specificity) were calculated by adding up the number of reads per contig associated with the same gene and also corresponding to microorganisms of the same genus (e.g., Azoarcus) as reported in Table S4 in the supplemental material. Aromatoleum aromaticum EBN1 was renamed Azoarcus, and therefore, genes from this species were considered Azoarcus genes.
Genes other than decarboxylase genes from the ubiquinone biosynthesis pathway are shown in Table S10 in the supplemental material.
Transcription of genes involved in anaerobic benzoate metabolism.
Genes coding for multiple distinct anaerobic benzoate catabolism pathway enzymes, including bzd-like genes found in Azoarcus and Aromatoleum spp., bcr/bad-like genes found in Thauera and Alphaproteobacteria spp., and bam-like genes found in the strictly anaerobic Geobacter and Peptococcaceae spp. (18–20), were all identified in the Illumina sequencing data (Fig. 5; see also Table S4 in the supplemental material). In Azoarcus genomes, the bzd genes are located in a catabolic operon (bzdNOPQMSTUVWXYZA) (17). We identified bzd-like transcripts for most of the genes in this operon, all transcribed at a relatively constant specificity and showing higher transcript abundance on benzoate than on benzene; these genes cover the entire upper pathway of anaerobic benzoate biodegradation (Fig. 5, Table 2; see also Table S4). Some of these genes have normalized read counts (reads per kilobase per million reads [RPKM]) of above 1,000, among the most abundant transcripts from the benzoate-metabolizing culture (Fig. 5, blue triangles). When the culture was metabolizing benzene, the same genes were also transcribed but at 8 to 10 times lower abundance (Table 2; see also Table S4). A second set of benzoate degradation genes that are more closely related to enzymes from obligate anaerobes (bam-like genes) were also identified in the data. These bam-like genes were almost exclusively transcribed when the culture was growing on benzene (Fig. 5, red triangles), including a putative benzoate-CoA ligase gene, bzlA (similar to bamY), and several genes encoding subunits of an ATP-independent benzoyl-CoA reductase, bamBCE. These genes were detected at normalized read counts that ranged between 40 and 100 RPKM on benzene (see Table S4) and were virtually undetected when the culture was grown on benzoate (see Table S4, pink shading). Finally, a third group of anaerobic benzoyl-CoA reductases (bcr/bad genes) and associated downstream genes that have been described in Betaproteobacteria, such as Thauera spp., and in Alphaproteobacteria, including Rhodopseudomonas palustris and Magnetospirillum spp. (16, 19, 48), were also detected (Fig. 5, green triangles). These transcripts were slightly more abundant in the presence of benzene (consistent specificity of 2 to 5). The bcr and bad genes are homologous to each other (13), and bcr genes have been found in diverse taxa. Therefore, these transcripts were not confidently assigned taxonomically with MEGAN (see Table S4). At this time, we cannot assign the bcr/bad genes to one of the known taxa within the culture.
FIG 5.

Log-log plot (base 2) of normalized coverage (RPKM) for contigs associated with benzoate catabolic genes, comparing RNA samples from benzoate-amended (y axis) and benzene-amended (x axis) cultures. Blue, bzd-type genes are related to Azoarcus or Aromatoleum spp.; green, bcr/bad-type genes are related to Thauera or Alphaproteobacteria spp.; red, bam-type genes are related to Geobacter or Peptococcaceae spp.
A similar but less sensitive transcriptional response was seen in the pyrosequencing data: all of the bzd-like benzoate catabolic genes were transcribed when the culture was amended with benzoate (see Table S5 in the supplemental material). However, these genes were not detected in samples from the benzene-amended culture. Rather, two subunits of the ATP-independent benzoyl-CoA reductase, bamD and bamE, were identified in this case (Table 2; see also Table S5), confirming the abundance of these transcripts during benzene metabolism in the culture. Finally, transcripts of several other genes associated with benzoate catabolism were also detected in the benzoate-degrading cultures in both sequencing data sets, including genes encoding the transcriptional regulator bzdR and the benzoate transporter bzdB (see Tables S4 and S5) (16, 49). The abundance and extent of pathway coverage of detected transcripts for anaerobic benzoate metabolism, consistent with the central role of benzoate in anaerobic benzene and benzoate metabolism, provides confidence in the validity of the data set.
Transcripts mapped to nitrate and nitrite reduction pathways.
The denitrifying genes (nar, nir, nor, and noz) identified in the Illumina data set were dominated by those attributed to Azoarcus and Planctomycetes (see Table S6 in the supplemental material). These genes are all components of a membrane-associated nitrate respiratory process. The contigs associated with this pathway were typically 5 to 10 times more abundant during benzoate metabolism (specificity of ≤0.2), consistent with the higher relative abundance of transcripts from Azoarcus and Planctomycetes under this condition (Fig. 4). In the pyrosequencing data, all nitrate respiration genes identified were related to Azoarcus spp. and were essentially only detected during benzoate degradation (see Table S7). These data confirm known physiological processes in the culture and establish relative transcript abundances for the dominant taxa in these RNA samples.
Annotation of contigs more highly transcribed during benzene metabolism.
We next examined all of the contigs that were more highly transcribed in the presence of benzene (i.e., with specificity greater than 10) and their corresponding best BLASTx hits (see Table S8 in the supplemental material). Many sequences from this data set matched strikingly well with metagenomic sequences from the benzene-degrading “Clostridia enrichment culture BF,” with an identity level higher than 90% on the amino acid level, including several putative carboxylase or decarboxylase genes associated with aromatic compound metabolism (Table 2; see also Table S8). Two of the highly transcribed benzene-specific genes were homologs of abcA and abcD, two subunits of a putative benzene carboxylase identified in Clostridia enrichment culture BF (22), as well as in a pure archaeal culture, Ferroglobus placidus (23). In addition, benzene-specific genes encoding proteins similar to those encoded by ubiD and ubiX, enzymes involved in the carboxylation or decarboxylation of aromatic compounds in the ubiquinone biosynthesis pathway, were highly transcribed, along with other nonspecifically annotated carboxy-lyases. Proteins similar to UbiD and UbiX were also identified in culture BF by Abu Laban et al. (22). Taken together, these results point to a significant role for carboxylation during anaerobic benzene catabolism. In our data set, ubiD and ubiX sequences from different microbial groups were identified (Table 2; see also Table S4), but only those associated with obligate anaerobes and not those associated with facultative anaerobes were specifically transcribed on benzene. Transcripts related to other genes in the ubiquinone biosynthetic pathway (such as ubiA and ubiB) did not share the pattern of exclusive transcription during benzene metabolism (see Table S9). This suggests that proteins homologous to UbiD and UbiX may be specifically involved in benzene metabolism, perhaps in addition to or instead of their normal role in the ubiquinone pathway. By far, the most highly transcribed genes on benzene were predominantly affiliated with the Firmicutes (see Table S8, green), and a large number of these genes were annotated as hypothetical proteins or transposases.
Transcription of genes involved in anaerobic toluene and phenol metabolism.
Alternative paths from benzene to benzoate or benzoyl-CoA are possible via methylation to toluene or hydroxylation to phenol. Therefore, contigs were also mapped to the pathways from toluene to benzoyl-CoA via fumarate addition and from phenol to benzoyl-CoA via phosphorylation. No genes in the toluene activation pathways were identified in any of the contigs generated from pyrosequencing or Illumina sequencing, regardless of substrate. In contrast, transcripts that map to the phenol pathway were detected (Table 2, Fig. 6; see also Table S4 in the supplemental material): transcripts for phenylphosphate synthase genes (pps) and, particularly, for phenylphosphate carboxylase genes (ppc) were detected and were more abundant when the culture was degrading benzene. Genes for subsequent steps, 4-hydroxybenzoate ligase (hbcl) and 4-hydroxybenzoyl-CoA reductase (alpha subunit; hcrA) were also transcribed. These data indicate that an anaerobic phenol metabolism pathway is possibly active in the culture, though the specificities for each step are variable (Table 2, Fig. 6).
FIG 6.

Gene transcripts identified in RNA samples corresponding to known or hypothesized enzymes involved in the anaerobic metabolism of monoaromatic compounds in different anaerobes. Three parallel yet distinct pathways are known for benzoate catabolism: one found in Azoarcus/Aromatoleum spp. (bzd genes, blue), one identified in Thauera/Alphaproteobacteria spp. (bcr/bad genes, green), and the other in obligate anaerobes (bam genes, red) (54). Sequences corresponding to each of these pathways were identified in the culture. The numbers adjacent to gene names are the values for specificity (for benzene relative to benzoate). Question marks indicate where corresponding bcr/bad and bam genes were not found in the current data set. The putative carboxylase genes proposed for the Clostridium enrichment culture BF (6) are shown in boldface black, while the genes associated with the anaerobic phenol degradation pathway are shown in black but not in boldface. All contigs associated with these genes and their associated specificities are provided in Table S4 in the supplemental material.
Transcripts mapped to oxygenase genes.
The possibility exists that benzene could be activated (oxidized to phenol) by molecular oxygen produced by nitrite dismutation during denitrification (50). Transcripts for oxygenases associated with oxidation of monoaromatic compounds were identified in samples from the culture degrading both substrates (see Table S10 in the supplemental material). The most abundant oxygenases were benzoyl-CoA oxygenases (box genes) from Betaproteobacteria (e.g., Azoarcus), which were more highly transcribed on benzoate (see Table S10, blue). Other proteobacterial oxygenase transcripts, including the genes encoding 4-hydroxybenzoate 3-monooxygenase, phenol 2-monooxygenase, and an aromatic ring-opening dioxygenase, were detected and were transcribed at similar levels on either substrate. No genes similar to toluene mono- or dioxygenases or benzene mono- or dioxygenases, known for activating the unsubstituted benzene ring aerobically (51, 52), were identified in the sequence data.
Could oxygen be activating benzene in this nitrate-reducing culture?
Although oxygenase transcripts were detected, our data indicate that strictly anaerobic conditions prevailed in the Cartwright culture and that any role of molecular oxygen was minimal. The cultures were maintained with FeS-reduced mineral medium in sealed bottles inside an anaerobic chamber. Therefore, oxygen, if any was present, did not come from outside the culture bottles. The most abundant oxygenase transcripts detected were homologous to benzoyl-CoA oxygenases (box gene cluster), which are induced by benzoyl-CoA and have been shown to be expressed regardless of aerobic or anaerobic conditions (53). In addition, we detected abundant transcripts for the benzoyl-CoA reductase genes bzdNOPQ, whose expression is strongly repressed by oxygen (54, 55). We also identified transcripts from anammox (anaerobic ammonium oxidation) bacteria within Planctomycetes, which are notoriously sensitive to oxygen (56). Moreover, none of the oxygenases were more highly transcribed with benzene than with benzoate. We conclude that oxygenases were most likely transcribed in the culture due to nonspecific induction by benzoyl-CoA, as previously reported (53), or by other intermediates during benzene or benzoate degradation.
Is carboxylation the mechanism for benzene activation?
Having identified a member of the Peptococcaceae (Clostridiales) as the only organism whose transcription level increased upon the addition of benzene, it is fair to conclude that this organism must be initiating benzene metabolism in this culture, as has been suggested previously (7, 28, 30, 31). Given the apparent enrichment of Peptococcaceae transcripts associated with carboxylases, it is clear that carboxylation plays an important role in benzene metabolism, but is it indeed the activating mechanism? We were able to rule out a mechanism via toluene because of the absence of bssA-like transcripts. As well, ethylbenzene dehydrogenase genes identified in the nitrate-reducing strain EBN1 (49) were not identified in our transcriptome data (see Tables S2 and S3 in the supplemental material), so a reaction similar to the ethylbenzene hydroxylation process was not evident. Rather, in addition to transcripts associated with COG0043 (carboxylase/lyase of aromatic compounds), we observed enrichment of COG1013/-14 (oxoacid:ferredoxin oxidoreductase) (see Table S8) during benzene metabolism, which is consistent with the important role ferredoxin plays in redox reactions and energy conservation in strict anaerobes. However, carboxylation is also one of the steps in the anaerobic activation of phenol in strict anaerobes (57, 58), and therefore, the transcription of carboxylases could be stimulated by the presence of phenol. Phenol is converted to benzoyl-CoA via several reactions catalyzed by phenylphosphate synthase, phenylphosphate carboxylase, 4-hydroxybenzoate-CoA ligase, and 4-hydroxybenzoyl-CoA reductase (Fig. 6) (49, 57, 58). The transcription of these phenol-induced genes (59) could mean that phenol was produced in the culture. Phenol has been detected in anaerobic benzene-degrading cultures (11, 13–15) and has been postulated as a metabolite of benzene degradation (25). However, in an iron-containing culture, it has been shown that phenol can be produced abiotically from benzene during the extraction procedure when the sample becomes exposed to oxygen, and thus, the detection of phenol may be an artifact of the extraction method (21). We have considered whether in fact phenol could be produced from benzene abiotically in our cultures, catalyzed by reactive oxidized iron or hydroxyl radicals generated from FeS (in the medium) by reaction with nitrate, nitrite, or oxygen resulting from the disproportionation of nitrite. While we cannot exclude an intermittent or occasional role for such reactions, it is difficult to imagine that the culture would continue to degrade benzene at consistent rates and stoichiometry for so many years with an initial reaction entirely dependent on the internal production of reactive oxidized species to drive a Fenton-type (60) reaction.
Gene neighborhood of putative abcDA anaerobic benzene carboxylase genes.
Abu Laban et al. (22) identified a specific benzene-expressed protein band of 60 kDa in the proteome of benzene-grown cells of culture BF. This protein, which they named AbcA, had similarity to a known subunit of phenylphosphate carboxylases (PpcA). In further making the case for the role of abcA and abcD as components of an anaerobic benzene carboxylase and not phenol carboxylase, the authors noted that the gene cluster containing open reading frames (ORFs) for abcA (similar to ppcA) and abcD (similar to ppcD) genes did not contain either ORFs similar to the remaining subunits ppcB and ppcC needed for phenol carboxylation or ORFs similar to the ppsABC genes encoding phenylphosphate synthase, and therefore, they concluded that this gene cluster was not involved in anaerobic phenol metabolism but, rather, contained putative anaerobic benzene carboxylase (Abc) genes (22). Given the extraordinary similarity between the Peptococcaceae sequences in our culture and those of culture BF (most of them have above 90% identity on the amino acid level), we attempted to determine whether the abcA gene neighborhood was the same in both cultures as well. Using PCR primers (see Table S1 in the supplemental material) spanning the intergenic regions between the putative carboxylase genes abcD, abcA, and ubiX and the putative benzoate-coenzyme A ligase bzlA genes (in the order found in culture BF), we determined that these PCR products successfully bridged the adjacent genes with intergenic regions smaller than 120 bp (Fig. 7). The sequenced PCR products and the published partial coding sequence of contig 11418 from culture BF (22) have greater than 80% nucleotide identity in the overlapped region, and therefore, the genes abcD, abcA, bzlA, and ubiX are found with the identical unusual gene organization in the two cultures. The presence of bzlA, encoding a benzoate-CoA ligase, next to putative carboxylases is consistent with the proposed direct carboxylation pathway. We therefore conclude that the most likely scenario in our cultures is that benzene is primarily activated via carboxylation, with the potential for a minor fraction of benzene being first biotically or abiotically oxidized to phenol owing to the presence of traces of molecular oxygen from dismutation of nitric oxide (50) or of metal oxides in the medium (61). Any phenol thus formed would subsequently be phosphorylated prior to carboxylation (Fig. 6).
FIG 7.
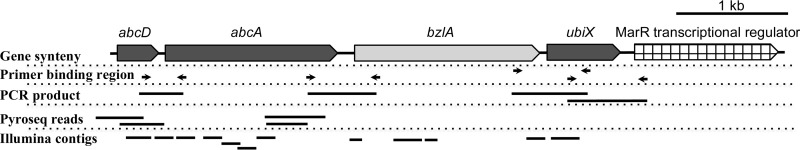
Conserved gene synteny in the neighborhood of putative abcA, abcD, bzlA, and ubiX genes (the gene synteny shown here was reported by Abu Laban et al. [22]). Primers were designed to target the intragenic regions to determine whether the operonic structure previously proposed was also observed in this study (primer sequences are included in Table S1 in the supplemental material). The PCR products were sequenced in both forward and reverse directions. Primers were designed such that each amplified region has at minimum a 70-bp overlap with the targeted flanking genes. The relevant pyrosequencing reads and Illumina contigs are also mapped to the predicted operon. The visible overlaps between Illumina contigs were all shorter than 70 bp, and thus, these contigs were not assembled.
Syntrophic relationships in the nitrate-reducing, benzene-degrading culture.
Whether benzene is directly or indirectly (via phenol) carboxylated to benzoate, it is clear that benzoate is a central metabolite in this culture. We identified three distinct parallel clusters of benzoate metabolism genes (Fig. 5), derived from at least two distinct organisms (Peptococcaceae and Azoarcus, and possibly one other). Azoarcus spp. is the main player in benzoate metabolism, as revealed by the abundance of Azoarcus-related transcripts during benzoate degradation. Moreover, it is clear that Azoarcus does not degrade benzene and that feeding the culture benzoate does not sustain benzene-degrading organisms in the culture, since no Peptococcaceae transcripts were identified on benzoate and benzene-degrading activity was negatively impacted by first feeding benzoate to the culture (Fig. 1a). Nevertheless, benzene degradation clearly does proceed via the intermediate benzoate. Benzoate metabolism genes from Peptococcaceae, as well as from an unidentified proteobacterium, were transcribed, indicating that more than one organism may be degrading benzoate in the culture. Given that the lower part of the benzoate pathway was either less well represented in the sequence data collected or less well identified by homology search, we could not determine the nature of the products of benzoate metabolism that were ultimately transferred to Azoarcus or to other Proteobacteria for nitrate reduction. The most likely products are the typical small-molecule metabolites implicated in syntrophic relationships, such as acetate, formate, and hydrogen. The 8- to 10-fold-lower transcription levels of benzoate pathway genes in Azoarcus during benzene degradation than during benzoate degradation are consistent with the much lower rates of benzene and, hence, benzoate degradation. It is possible that Azoarcus, in addition to obtaining electrons from products like acetate, hydrogen, or formate, is also scavenging electrons from benzoate or other metabolites produced by Peptococcaceae, from leaky or lysed cells. Alternatively, perhaps the presence of trace amounts of benzoate in the medium from benzene is sufficient to activate the transcription of benzoate pathway genes in Azoarcus. It is even possible that benzene itself activates the transcription of these genes, because the benzoate concentrations were previously shown to be extremely low (<10 nM) in these benzene-degrading cultures (15).
In addition to Peptococcaceae and Azoarcus, other players in the nitrate-reducing benzene-degrading culture include Burkholderiales, Planctomycetes, and Chlorobi. Burkholderiales genes were not differentially transcribed on the two substrates (Fig. 4); therefore, these organisms are likely not directly involved in benzene and benzoate metabolism but, rather, are using electron donors external to these processes, such as lysed biomass or reduced sulfide provided in the medium. Based on the presence of Planctomycetes-related gene transcripts for the genes involved in anaerobic ammonium oxidation and transportation (see Table S2 in the supplemental material) (62), we confirm the role of this population within the nitrogen cycle (using ammonium and nitrite), as previously demonstrated for this culture (32). The low numbers of Chlorobi transcripts identified in this study and their sporadic detection in previous 16S rRNA clone libraries (29, 31) is indicative of an opportunistic role in the culture metabolism for this taxonomic group.
In summary, we have found that the Peptococcaceae organism in this culture grows specifically on benzene, likely in syntrophy with nitrate reducers, particularly an Azoarcus. Several genes encoding proteins homologous to putative aromatic carboxylases and decarboxylases were specifically transcribed on benzene, and these genes were most likely associated with Peptococcaceae. Hence, a Peptococcaceae organism must activate the benzene ring anaerobically via mechanisms that include carboxylation. We cannot rule out the possibility of a biotic or abiotic transformation of benzene to phenol having some role, though likely not a major one. This study also demonstrates that sequencing total RNA even without a reference metagenome can provide extremely valuable information on both the dominant taxa and major pathways in such enrichment cultures.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Center for Applied Genomics, Toronto, Canada for assistance with both pyrosequencing and Illumina sequencing.
Funding was provided by the Government of Canada through NSERC, Genome Canada, and the Ontario Genomics Institute (grant 2009-OGI-ABC-1405) and by the Government of Ontario through the ORF-GL2 program.
Footnotes
Published ahead of print 2 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00717-14.
REFERENCES
- 1.Botton S, Parsons JR. 2007. Degradation of BTX by dissimilatory iron-reducing cultures. Biodegradation 18:371–381. 10.1007/s10532-006-9071-9 [DOI] [PubMed] [Google Scholar]
- 2.Chiang CY, Salanitro JP, Chai EY, Colthart JD, Klein CL. 1989. Aerobic biodegradation of benzene, toluene, and xylene in a sandy aquifer—data analysis and computer modeling. Ground Water 27:823–834. 10.1111/j.1745-6584.1989.tb01046.x [DOI] [Google Scholar]
- 3.Burland SM, Edwards EA. 1999. Anaerobic benzene biodegradation linked to nitrate reduction. Appl. Environ. Microbiol. 65:529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coates JD, Chakraborty R, McInerney MJ. 2002. Anaerobic benzene biodegradation—a new era. Res. Microbiol. 153:621–628. 10.1016/S0923-2508(02)01378-5 [DOI] [PubMed] [Google Scholar]
- 5.Abu Laban N, Selesi D, Jobelius C, Meckenstock RU. 2009. Anaerobic benzene degradation by Gram-positive sulfate-reducing bacteria. FEMS Microbiol. Ecol. 68:300–311. 10.1111/j.1574-6941.2009.00672.x [DOI] [PubMed] [Google Scholar]
- 6.Edwards EA, Grbić-Galić D. 1992. Complete mineralization of benzene by aquifer microorganisms under strictly anaerobic conditions. Appl. Environ. Microbiol. 58:2663–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleinsteuber S, Schleinitz KM, Breitfeld J, Harms H, Richnow HH, Vogt C. 2008. Molecular characterization of bacterial communities mineralizing benzene under sulfate-reducing conditions. FEMS Microbiol. Ecol. 66:143–157. 10.1111/j.1574-6941.2008.00536.x [DOI] [PubMed] [Google Scholar]
- 8.Jahn MK, Haderlein SB, Meckenstock RU. 2005. Anaerobic degradation of benzene, toluene, ethylbenzene, and o-xylene in sediment-free iron-reducing enrichment cultures. Appl. Environ. Microbiol. 71:3355–3358. 10.1128/AEM.71.6.3355-3358.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovley D, Woodward J, Chapelle F. 1996. Rapid anaerobic benzene oxidation with a variety of chelated Fe(III) forms. Appl. Environ. Microbiol. 62:288–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nales M, Butler BJ, Edwards EA. 1998. Anaerobic benzene biodegradation: a microcosm survey. Bioremediat. J. 2:125–144. 10.1080/10889869891214268 [DOI] [Google Scholar]
- 11.Grbic-Galic D, Vogel TM. 1987. Transformation of toluene and benzene by mixed methanogenic cultures. Appl. Environ. Microbiol. 53:254–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakai N, Kurisu F, Yagi O, Nakajima F, Yamamoto K. 2009. Identification of putative benzene-degrading bacteria in methanogenic enrichment cultures. J. Biosci. Bioeng. 108:501–507. 10.1016/j.jbiosc.2009.06.005 [DOI] [PubMed] [Google Scholar]
- 13.Caldwell ME, Suflita JM. 2000. Detection of phenol and benzoate as intermediates of anaerobic benzene biodegradation under different terminal electron-accepting conditions. Environ. Sci. Technol. 34:1216–1220. 10.1021/es990849j [DOI] [Google Scholar]
- 14.Chakraborty R, Coates JD. 2005. Hydroxylation and carboxylation—two crucial steps of anaerobic benzene degradation by Dechloromonas strain RCB. Appl. Environ. Microbiol. 71:5427–5432. 10.1128/AEM.71.9.5427-5432.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ulrich AC, Beller HR, Edwards EA. 2005. Metabolites detected during biodegradation of 13C6-benzene in nitrate-reducing and methanogenic enrichment cultures. Environ. Sci. Technol. 39:6681–6691. 10.1021/es050294u [DOI] [PubMed] [Google Scholar]
- 16.Carmona M, Zamarro MT, Blazquez B, Durante-Rodriguez G, Juarez JF, Valderrama JA, Barragan MJL, Garcia JL, Diaz E. 2009. Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol. Mol. Biol. Rev. 73:71–133. 10.1128/MMBR.00021-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez Barragán MJ, Carmona M, Zamarro MT, Thiele B, Boll M, Fuchs G, García JL, Díaz E. 2004. The bzd gene cluster, coding for anaerobic benzoate catabolism, in Azoarcus sp. strain CIB. J. Bacteriol. 186:5762–5774. 10.1128/JB.186.17.5762-5774.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boll M, Fuchs G. 1995. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. Eur. J. Biochem. 234:921–933. 10.1111/j.1432-1033.1995.921_a.x [DOI] [PubMed] [Google Scholar]
- 19.Egland P, Pelletier D, Dispensa M, Gibson J, Harwood C. 1997. A cluster of bacterial genes for anaerobic benzene ring biodegradation. Proc. Natl. Acad. Sci. U. S. A. 94:6484–6489. 10.1073/pnas.94.12.6484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wischgoll S, Heintz D, Peters F, Erxleben A, Sarnighausen E, Reski R, Van Dorsselaer A, Boll M. 2005. Gene clusters involved in anaerobic benzoate degradation of Geobacter metallireducens. Mol. Microbiol. 58:1238–1252. 10.1111/j.1365-2958.2005.04909.x [DOI] [PubMed] [Google Scholar]
- 21.Kunapuli U, Griebler C, Beller HR, Meckenstock RU. 2008. Identification of intermediates formed during anaerobic benzene degradation by an iron-reducing enrichment culture. Environ. Microbiol. 10:1703–1712. 10.1111/j.1462-2920.2008.01588.x [DOI] [PubMed] [Google Scholar]
- 22.Abu Laban N, Selesi D, Rattei T, Tischler P, Meckenstock RU. 2010. Identification of enzymes involved in anaerobic benzene degradation by a strictly anaerobic iron-reducing enrichment culture. Environ. Microbiol. 12:2783–2796. 10.1111/j.1462-2920.2010.02248.x [DOI] [PubMed] [Google Scholar]
- 23.Holmes DE, Risso C, Smith JA, Lovley DR. 2011. Anaerobic oxidation of benzene by the hyperthermophilic archaeon Ferroglobus placidus. Appl. Environ. Microbiol. 77:5926–5933. 10.1128/AEM.05452-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogel TM, Grbìc-Galìc D. 1986. Incorporation of oxygen from water into toluene and benzene during anaerobic fermentative transformation. Appl. Environ. Microbiol. 52:200–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang T, Tremblay P-L, Chaurasia AK, Smith JA, Bain TS, Lovley DR. 2013. Anaerobic benzene oxidation via phenol in Geobacter metallireducens. Appl. Environ. Microbiol. 79:7800–7806. 10.1128/AEM.03134-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasai Y, Takahata Y, Manefield M, Watanabe K. 2006. RNA-based stable isotope probing and isolation of anaerobic benzene-degrading bacteria from gasoline-contaminated groundwater. Appl. Environ. Microbiol. 72:3586–3592. 10.1128/AEM.72.5.3586-3592.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang T, Bain TS, Nevin KP, Barlett MA, Lovley DR. 2012. Anaerobic benzene oxidation by Geobacter species. Appl. Environ. Microbiol. 78:8304–8310. 10.1128/AEM.02469-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kunapuli U, Lueders T, Meckenstock RU. 2007. The use of stable isotope probing to identify key iron-reducing microorganisms involved in anaerobic benzene degradation. ISME J. 1:643–653. 10.1038/ismej.2007.73 [DOI] [PubMed] [Google Scholar]
- 29.Ulrich AC, Edwards EA. 2003. Physiological and molecular characterization of anaerobic benzene-degrading mixed cultures. Environ. Microbiol. 5:92–102. 10.1046/j.1462-2920.2003.00390.x [DOI] [PubMed] [Google Scholar]
- 30.van der Zaan BM, Talarico Saia F, Stams AJM, Plugge CM, de Vos WM, Smidt H, Langenhoff AAM, Gerritse J. 2012. Anaerobic benzene degradation under denitrifying conditions: Peptococcaceae as dominant benzene degraders and evidence for a syntrophic process. Environ. Microbiol. 14:1171–1181. 10.1111/j.1462-2920.2012.02697.x [DOI] [PubMed] [Google Scholar]
- 31.Gitiafroz R. 2012. Microorganisms and metabolic pathways involved in anaerobic benzene biodegradation under nitrate-reducing conditions. Ph.D. thesis University of Toronto, Toronto, Canada [Google Scholar]
- 32.Nandi M. 2006. Biochemical and molecular characterization of anaerobic benzene-degrading cultures. M.A.Sc. thesis University of Toronto, Toronto, Canada [Google Scholar]
- 33.Edwards EA, Grbić-Galić D. 1994. Anaerobic degradation of toluene and o-xylene by a methanogenic consortium. Appl. Environ. Microbiol. 60:313–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waller AS, Krajmalnik-Brown R, Löffler FE, Edwards EA. 2005. Multiple reductive-dehalogenase-homologous genes are simultaneously transcribed during dechlorination by Dehalococcoides-containing cultures. Appl. Environ. Microbiol. 71:8257–8264. 10.1128/AEM.71.12.8257-8264.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blankenberg D, Kuster GV, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. 2010. Galaxy: a Web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. Chapter 19:Unit 19.10. 10.1002/0471142727.mb1910s89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A. 2005. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15:1451–1455. 10.1101/gr.4086505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goecks J, Nekrutenko A, Taylor J, Galaxy Team 2010. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11:R86. 10.1186/gb-2010-11-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urich T, Lanzen A, Qi J, Huson DH, Schleper C, Schuster SC. 2008. Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS One 3:e2527. 10.1371/journal.pone.0002527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 40.Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res. 17:377–386. 10.1101/gr.5969107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. 2011. Integrative analysis of environmental sequences using MEGAN 4. Genome Res. 21:1552–1560. 10.1101/gr.120618.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196. 10.1093/nar/gkm864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langmead B, Trapnell C, Pop M, Salzberg S. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chevreux BP, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, Suhai S. 2004. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14:1147–1159. 10.1101/gr.1917404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 46.Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A. 2011. Differential expression in RNA-seq: a matter of depth. Genome Res. 21:2213–2223. 10.1101/gr.124321.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neidhardt FC, Curtiss R, III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. (ed). 1996. Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed, vol 1 ASM Press, Washington, DC [Google Scholar]
- 48.Shinoda Y, Akagi J, Uchihashi Y, Hiraishi A, Yukawa H, Yurimoto H, Sakai Y, Kato N. 2005. Anaerobic degradation of aromatic compounds by Magnetospirillum strains: isolation and degradation genes. Biosci. Biotechnol. Biochem. 69:1483–1491. 10.1271/bbb.69.1483 [DOI] [PubMed] [Google Scholar]
- 49.Rabus R, Kube M, Heider J, Beck A, Heitmann K, Widdel F, Reinhardt R. 2005. The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch. Microbiol. 183:27–36. 10.1007/s00203-004-0742-9 [DOI] [PubMed] [Google Scholar]
- 50.Ettwig KF, Butler MK, Le Paslier D, Pelletier E, Mangenot S, Kuypers MMM, Schreiber F, Dutilh BE, Zedelius J, de Beer D, Gloerich J, Wessels HJCT, van Alen T, Luesken F, Wu ML, van de Pas-Schoonen KT, Op den Camp HJM, Janssen-Megens EM, Francoijs K-J, Stunnenberg H, Weissenbach J, Jetten MSM, Strous M. 2010. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464:543–548. 10.1038/nature08883 [DOI] [PubMed] [Google Scholar]
- 51.Gibson DT, Koch JR, Kallio RE. 1968. Oxidative degradation of aromatic hydrocarbons by microorganisms. I. Enzymic formation of catechol from benzene. Biochemistry 7:2653–2662 [DOI] [PubMed] [Google Scholar]
- 52.Tao Y, Fishman A, Bentley W, Wood T. 2004. Oxidation of benzene to phenol, catechol, and 1,2,3-trihydroxybenzene by toluene 4-monooxygenase of Pseudomonas mendocina KR1 and toluene 3-monooxygenase of Ralstonia pickettii PKO1. Appl. Environ. Microbiol. 70:3814–3820. 10.1128/AEM.70.7.3814-3820.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valderrama JA, Durante-Rodríguez G, Blázquez B, García JL, Carmona M, Díaz E. 2012. Bacterial degradation of benzoate: cross regulation between aerobic and anaerobic pathways. J. Biol. Chem. 287:10494–10508. 10.1074/jbc.M111.309005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Durante-Rodríguez G, Zamarro MT, García JL, Díaz E, Carmona M. 2008. New insights into the BzdR-mediated transcriptional regulation of the anaerobic catabolism of benzoate in Azoarcus sp. CIB. Microbiology 154(Pt 1):306–316. 10.1099/mic.0.2007/011361-0 [DOI] [PubMed] [Google Scholar]
- 55.Gescher J, Eisenreich W, Wörth J, Bacher A, Fuchs G. 2005. Aerobic benzoyl-CoA catabolic pathway in Azoarcus evansii: studies on the non-oxygenolytic ring cleavage enzyme. Mol. Microbiol. 56:1586–1600. 10.1111/j.1365-2958.2005.04637.x [DOI] [PubMed] [Google Scholar]
- 56.Fuerst JA, Sagulenko E. 2011. Beyond the bacterium: planctomycetes challenge our concepts of microbial structure and function. Nat. Rev. Microbiol. 9:403–413. 10.1038/nrmicro2578 [DOI] [PubMed] [Google Scholar]
- 57.Breinig S, Schiltz E, Fuchs G. 2000. Genes involved in anaerobic metabolism of phenol in the bacterium Thauera aromatica. J. Bacteriol. 182:5849–5863. 10.1128/JB.182.20.5849-5863.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schleinitz KM, Schmeling S, Jehmlich N, von Bergen M, Harms H, Kleinsteuber S, Vogt C, Fuchs G. 2009. Phenol degradation in the strictly anaerobic iron-reducing bacterium Geobacter metallireducens GS-15. Appl. Environ. Microbiol. 75:3912–3919. 10.1128/AEM.01525-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmeling S, Narmandakh A, Schmitt O, Gad'on N, Schuhle K, Fuchs G. 2004. Phenylphosphate synthase: a new phosphotransferase catalyzing the first step in anaerobic phenol metabolism in Thauera aromatica. J. Bacteriol. 186:8044–8057. 10.1128/JB.186.23.8044-8057.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meckenstock RU, Mouttaki H. 2011. Anaerobic degradation of non-substituted aromatic hydrocarbons. Curr. Opin. Biotechnol. 22:406–414. 10.1016/j.copbio.2011.02.009 [DOI] [PubMed] [Google Scholar]
- 61.Iwamoto M, Hirata J, Matsukami K, Kagawa S. 1983. Catalytic oxidation by oxide radical ions. 1. One-step hydroxylation of benzene to phenol over group 5 and 6 oxides supported on silica gel. J. Phys. Chem. 87:903–905 [Google Scholar]
- 62.Kuenen JG. 2008. Anammox bacteria: from discovery to application. Nat. Rev. Microbiol. 6:320–326. 10.1038/nrmicro1857 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.