

Abstract
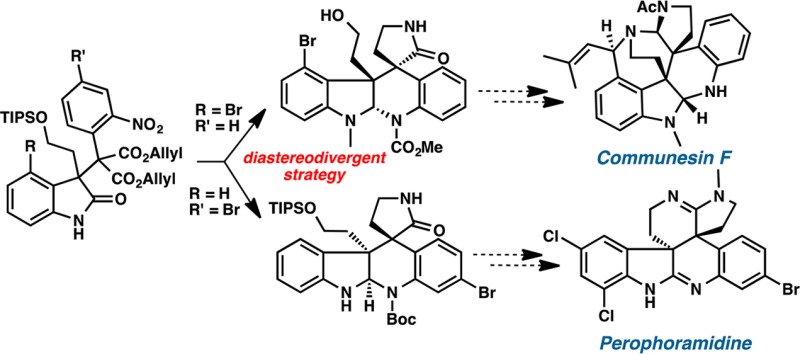
An efficient, unified, and stereodivergent approach toward communesin F and perophoramidine was examined. The C(3) all-carbon quaternary center of an oxindole was smoothly constructed by base-promoted indolone-malonate alkylation chemistry. The complementary relative stereochemistry of the crucial vicinal quaternary centers found in communesin F and perophoramidine was selectively installed by substrate-controlled decarboxylative allylic alkylations.
Communesins A (1a) and B (1b) were isolated in 1993 from a strain of Penicillium sp. found on a marine alga (Figure 1).1 Communesin B (1b) exhibits antiproliferative activity against P-388 lymphocytic leukemia cells (ED50 = 0.9 μM), LoVo (MIC = 3.9 μM), and KB cells (MIC = 8.8 μM).1,2 In the following years, communesins B–H (1b–1h) were isolated from related strains of Penicillium sp.3 Communesins A–F (1a–1f) show interesting biological activities such as insecticidal and antiproliferative activities against a variety of cancer cells.1−3 These complex, polycyclic, bioactive alkaloids possess several intriguing architectural features including vicinal quaternary carbon centers and bis-aminal functional groups.
Figure 1.

Communesins (1) and perophoramidine (2).
In 2002, structurally and biosynthetically related perophoramidine (2) was isolated from the ascidian Perophora namei by the Ireland group.4 Perophoramidine contains the equally unusual bis-amidine instead of bis-aminal functionality, possesses the alternate diastereomeric relationship between the vicinal quaternary centers, and lacks the azepine ring compared to communesins. Perophoramidine (2) exhibits cytotoxicity toward the HCT 116 human colon carcinoma cell line (IC50 = 60 μM) and induces apoptosis via PARP cleavage.5
These intriguing polycyclic alkaloids have attracted much attention from the synthetic community over the past decade.6 Herein, we report a unified, diastereodivergent approach toward the syntheses of communesin F (1f) and perophoramidine (2). As a first generation approach to communesin F (1f) and perophoramidine (2) we chose to pursue formal syntheses by intercepting key intermediates of previous routes. Our overarching plan for synthesis of these diastereodivergent series was to employ stereoselective enolate alkylations of substrates constructed using an oxindole coupling reaction developed in our laboratory for this purpose.7 Simultaneous to our work, Funk developed a similar oxindole based strategy, and more recently Lu has utilized this for the asymmetric synthesis spirocyclic oxindoles.8
Our initial strategic disconnections of pentacycle 3, an intermediate in Qin’s synthesis,6g involve late stage introduction of the cyclic aminal functionality by reductive cyclization and installation of γ-lactam by translactamization of lactone 4 (Scheme 1). The relative stereochemical relationship between C(7) and C(8) of lactone 4 was envisioned to be established by decarboxylative allylic alkylation. We anticipated that the congested vicinal quaternary centers on oxindole 5 could be constructed by the base-promoted alkylation of 3-bromooxindole 7 with aryl diallyl malonate 8 via in situ formation of o-azaxylylene intermediate 6.7,8
Scheme 1. Retrosynthesis of Communesin F (1f).
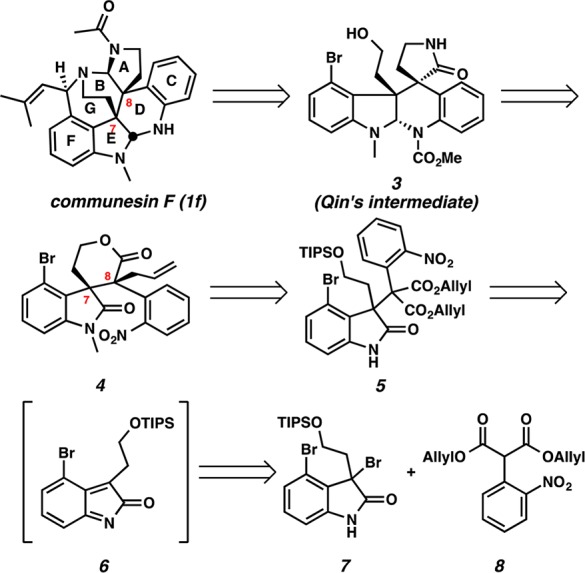
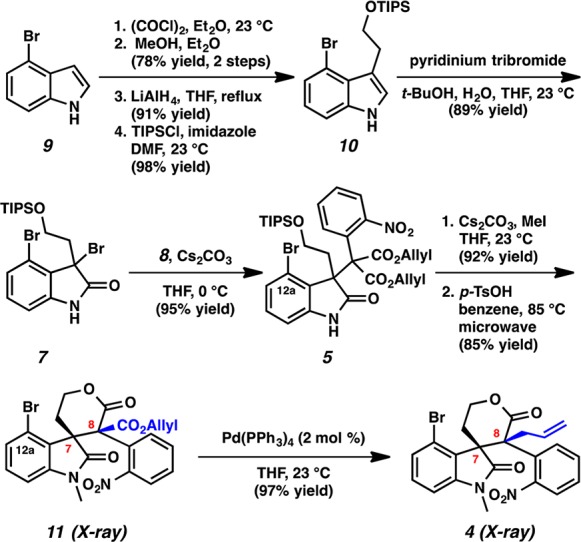
The diastereoselective synthesis of the key vicinal quaternary centers of lactone 4 is depicted in Scheme 2. 4-Bromoindole 9 was treated with oxalyl chloride and methanol to furnish the corresponding oxoacetate (78% yield, 2 steps), which was subjected to LiAlH4 reduction (91% yield)6g and subsequent silylation of the resultant alcohol with TIPSCl to afford silyl ether 10 (98% yield). Indole 10 was oxidized to dibromooxindole 7 with pyridinium tribromide in 89% yield.9 To our delight, alkylation of dibromooxindole 7 with diallyl malonate 8 smoothly installed the congested quaternary stereocenter on oxindole 5 in 95% yield despite the extra steric hindrance at C(12a) of the oxindole (communesin numbering) and the use of an unprecedented aryl substituted malonate derivative leading to vicinal quaternary centers. Methylation of the oxindole (92% yield) followed by microwave assisted lactonization afforded allyl ester 11 as a single diastereomer in 85% yield. Gratifyingly, decarboxylative allylic alkylation of 11 with catalytic Pd(PPh3)4 furnished lactone 4 as a single diastereomer. This remarkable reaction not only provides the vicinal quaternary centers needed for the communesin and perophoramidine effort at rt but also proceeds with complete diastereoselectivity. The relative stereochemistry of the vicinal quaternary centers of 11 and 4 was confirmed by single crystal X-ray analysis. Importantly, the diastereomer produced is in line with that needed for the communesins.
Scheme 2. Construction of the Vicinal Quaternary Centers.
Reduction of nitroarene 4 to the aniline by TiCl3 was followed by simultaneous lactone ring opening to furnish bis-oxindole 12 in 80% yield (Scheme 3). Silylation of primary alcohol 12 with TIPSCl (90% yield) and subsequent treatment with methyl chloroformate provided carbamate 13 in 98% yield. Ozonolysis of olefin 13 afforded aldehyde 14 in 94% yield. Next, reductive amination of aldehyde 14 and translactamization provided γ-lactam 16 in 97% yield.
Scheme 3. Synthesis of γ-Lactam 16.

We anticipated that the piperidine D ring of communesin F (1f) would be delivered under reductive cyclization conditions (Scheme 4). We attempted to activate oxindole 16 via an imidate by treatment with Tf2O. Surprisingly, these conditions delivered o-nitrobenzyl protected hexacyclic oxindole 17 in 75% yield. At this point, we envisaged that reductive cyclization of hexacycle 17 would produce the desired piperidine ring since the oxidation state at C(9) of 17 is identical to that of desired aminal 18. Gratifyingly, after extensive experimentation, we could successfully reduce the oxindole of 17 by treatment with a combination of DIBAL and Et2AlCl, and upon workup the propellane structure unravels to produce pentacyclic aminal 18. We were pleased to find that the o-nitrobenzyl group was cleaved by photolysis at 350 nm in 40% yield.10 Unexpectedly, we discovered that cleavage of the o-nitrobenzyl group was also achieved using 20% aq NaOH in methanol at 75 °C in 70% yield. To the best of our knowledge, this constitutes the first use of aqueous hydroxide for removal of an o-nitrobenzyl group.11 Aminal 3 proved identical to an intermediate previously advanced by the Qin group to communesin F,6g thus completing a formal synthesis of the natural product.
Scheme 4. Formal Synthesis of Communesin F (1f).

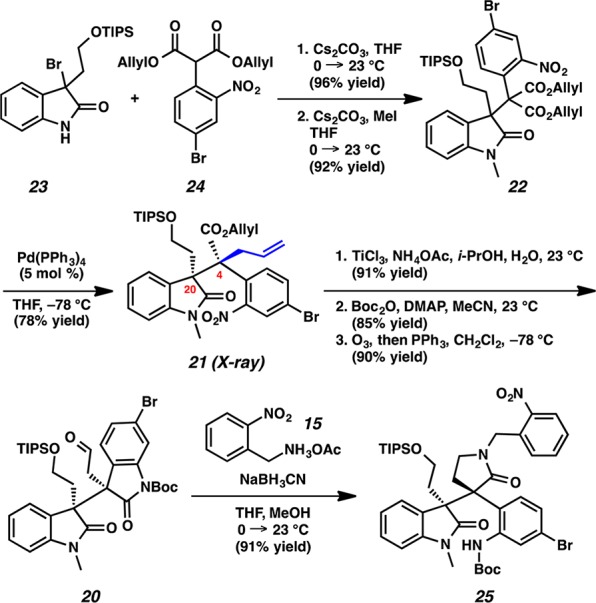
Having successfully completed a formal synthesis of communesin F (1f), our attention turned to perophoramidine (2) (Scheme 5). We envisaged that aminal 19 could be disconnected to afford aldehyde 20 (diastereomeric at the vicinal quaternary carbons compared to analogue 14) based on the expedient strategy that was used in our progress toward communesin F. Boc-protected oxindole 20 was excised to afford allyl ester 21. The vicinal quaternary centers of 21 was anticipated to be installed by decarboxylative allylic alkylation, although the relative stereochemistry was indeed an open question.
Scheme 5. Retrosynthesis of Perophoramidine (2).

In analogy to our communesin F synthesis, the quaternary centers of sterically congested diester 22 were constructed by alkylation of 3-bromooxindole 23 with aryl substituted malonate ester 24 (96% yield), followed by N-methylation of the resulting oxindole in 92% yield (Scheme 6). To our delight, direct decarboxylative allylic alkylation of diester 22 with catalytic Pd(PPh3)4 furnished allyl ester 21 in 78% yield as a single diastereomer. Interestingly, through X-ray analysis we discovered that the relative stereochemistry of the vicinal quaternary centers in this acyclic example was complementary to that of lactone 4 (Scheme 2) and, thus, suitable for elaboration to perophoramidine (2).12 Bis-oxindole 20 was obtained from allyl ester 21 by nitroarene reduction and simultaneous lactamization (91% yield),13 followed by Boc protection (85% yield) and subsequent ozonolysis (90% yield). Reductive amination of aldehyde 20 with o-nitroammonium acetate 15 provided lactam 25.
Scheme 6. Construction of the Contiguous Vicinal Quaternary Centers.
In contrast to the communesin system, we discovered that the desired aminal 26 was obtained directly by reduction with AlH3–Me2NEt in 42% yield (66% yield based on recovered starting material) (Scheme 7).14 The indoline methyl group was oxidized to a formyl functionality using PDC to produce 27 in 62% yield (93% yield based on recovered starting material).15 To our delight, the cleavage of both formyl and o-nitrobenzyl groups was achieved using 20% aq NaOH at 75 °C to deliver aminal 19 in 50% yield.16 Aminal 19 was advanced by Funk in his synthesis of perophoramidine,6o thus completing our formal synthesis of the natural product.
Scheme 7. Formal Synthesis of Perophoramidine (2).

In summary, we have completed stereocontrolled formal syntheses of communesin F (1f) and perophoramidine (2) using a stereodivergent alkylation approach. The highly congested all-carbon quaternary center of the oxindoles (5 and 22) was constructed by stabilized enolate alkylation of 3-bromooxindoles, a remarkably facile method discovered by our laboratory. The complementary relative stereochemistry of the contiguous vicinal quaternary centers found in communesin F and perophoramidine was introduced by substrate controlled diastereoselective decarboxylative allylic alkylation, again under exceedingly mild conditions. Several novel and intriguing intermediates such as the propellane hexacyclic oxindole were encountered toward the formal synthesis of communesin F. Finally, a previously unknown o-nitrobenzyl group cleavage protocol was discovered serendipitously and proved critical to the formal syntheses of both communesin F and perophoramidine.
Acknowledgments
The authors wish to thank NIH-NIGMS (R01GM080269), Amgen, the Gordon and Betty Moore Foundation, and Caltech for financial support. S.-J.H. thanks Fulbright (Foreign Student Program, No. 15111120) and the Ilju Foundation of Education & Culture (Predoctoral Research Fellowship) for financial support. F.V. thanks the German Academic Exchange Service (DAAD) (postdoctoral fellowship). S.K. thanks California TRDRP for financial support (postdoctoral fellowship 14FT-0002). J.A.M. is grateful for a fellowship by Bristol-Myers Squibb and Amgen. M.G. is grateful to the Swiss National Science Foundation for financial support (postdoctoral fellowship). Dr. David VanderVelde (Caltech) is gratefully acknowledged for assistance with the characterization of compounds by NMR spectroscopy. Lawrence Henling (Caltech) is acknowledged for X-ray crystallographic structural determination.
Supporting Information Available
The authors declare no competing financial interest. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Numata A.; Takahashi C.; Ito Y.; Takada T.; Kawai K.; Usami Y.; Matsumura E.; Imachi M.; Ito T.; Hasegawa T. Tetrahedron Lett. 1993, 34, 2355. [Google Scholar]
- a Ratnayake A. S.; Yoshida W. Y.; Mooberry S. L.; Hemscheidt T. K. J. Org. Chem. 2001, 66, 8717. [DOI] [PubMed] [Google Scholar]; b Retraction of this article was prompted by a revision of structure; see:Ratnayake A. S.; Yoshida W. Y.; Mooberry S. L.; Hemscheidt T. K. J. Org. Chem. 2003, 68, 1640. [DOI] [PubMed] [Google Scholar]; However, the biological activity reported in this manuscript was not specifically called into question and is distinct relative to previous reports.1
- a Jadulco R.; Edrada R. A.; Ebel R.; Berg A.; Schaumann K.; Wray V.; Steube K.; Proksch P. J. Nat. Prod. 2004, 67, 78. [DOI] [PubMed] [Google Scholar]; b Hayashi H.; Matsumoto H.; Akiyama K. Biosci. Biotechnol. Biochem. 2004, 68, 753. [DOI] [PubMed] [Google Scholar]; c Andersen B.; Smedsgaard J.; Frisvad J. C. J. Agric. Food Chem. 2004, 52, 2421. [DOI] [PubMed] [Google Scholar]; d Dalsgaard P. W.; Blunt J. W.; Munro M. H. G.; Frisvad J. C.; Christophersen C. J. Nat. Prod. 2005, 68, 258. [DOI] [PubMed] [Google Scholar]; e Blunt J. W.; Copp B. R.; Munro M. H. G.; Northcote P. T.; Prinsep M. R. Nat. Prod. Rep. 2006, 23, 26. [DOI] [PubMed] [Google Scholar]; f Wigley L. J.; Mantle P. G.; Perry D. A. Phytochemistry 2006, 67, 561. [DOI] [PubMed] [Google Scholar]
- Verbitski S. M.; Mayne C. L.; Davis R. A.; Concepcion G. P.; Ireland C. M. J. Org. Chem. 2002, 67, 7124. [DOI] [PubMed] [Google Scholar]
- Denault J.-B.; Salvesen G. S. Chem. Rev. 2002, 102, 4489. [DOI] [PubMed] [Google Scholar]
- a May J. A.; Zeidan R. K.; Stoltz B. M. Tetrahedron Lett. 2003, 44, 1203. [Google Scholar]; b Crawley S. L.; Funk R. L. Org. Lett. 2003, 5, 3169. [DOI] [PubMed] [Google Scholar]; c May J. A.; Stoltz B. Tetrahedron 2006, 62, 5262. [Google Scholar]; d Yang J.; Song H.; Xiao X.; Wang J.; Qin Y. Org. Lett. 2006, 8, 2187. [DOI] [PubMed] [Google Scholar]; e Crawley S. L.; Funk R. L. Org. Lett. 2006, 8, 3995. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Seo J. H.; Artman G. D. III; Weinreb S. M. J. Org. Chem. 2006, 71, 8891. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Yang J.; Wu H.; Shen L.; Qin Y. J. Am. Chem. Soc. 2007, 129, 13794. [DOI] [PubMed] [Google Scholar]; h George J. H.; Adlington R. M. Synlett 2008, 2093. [Google Scholar]; i Siengalewicz P.; Gaich T.; Mulzer J. Angew. Chem., Int. Ed. 2008, 47, 8170. [DOI] [PubMed] [Google Scholar]; j Liu P.; Seo J. H.; Weinreb S. M. Angew. Chem., Int. Ed. 2010, 49, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Zuo Z.; Xie W.; Ma D. J. Am. Chem. Soc. 2010, 132, 13226. [DOI] [PubMed] [Google Scholar]; l Zuo Z.; Ma D. Angew. Chem., Int. Ed. 2011, 50, 12008. [DOI] [PubMed] [Google Scholar]; m Belmar J.; Funk R. L. J. Am. Chem. Soc. 2012, 134, 16941. [DOI] [PubMed] [Google Scholar]; n Artman G. D. III; Weinreb S. M. Org. Lett. 2003, 5, 1523. [DOI] [PubMed] [Google Scholar]; o Fuchs J. R.; Funk R. L. J. Am. Chem. Soc. 2004, 126, 5068. [DOI] [PubMed] [Google Scholar]; p Sabahi A.; Novikov A.; Rainier J. D. Angew. Chem., Int. Ed. 2006, 45, 4317. [DOI] [PubMed] [Google Scholar]; q Evans M. A.; Sacher J. R.; Weinreb S. M. Tetrahedron 2009, 65, 6712. [Google Scholar]; r Wu H.; Xue F.; Xiao X.; Qin Y. J. Am. Chem. Soc. 2010, 132, 14052. [DOI] [PubMed] [Google Scholar]; s Schammel A. W.; Chiou G.; Garg N. K. Org. Lett. 2012, 14, 4556. [DOI] [PMC free article] [PubMed] [Google Scholar]; t Ishida T.; Takemoto Y. Tetrahedron 2013, 69, 4517. [Google Scholar]; u Ishida T.; Ikota H.; Kurahashi K.; Tsukano C.; Takemoto Y. Angew. Chem., Int. Ed. 2013, 52, 10204. [DOI] [PubMed] [Google Scholar]; v Zhang H.; Hong L.; Kang H.; Wang R. J. Am. Chem. Soc. 2013, 135, 14098. [DOI] [PubMed] [Google Scholar]
- a Krishnan S.; Stoltz B. M. Tetrahedron Lett. 2007, 48, 7571. [Google Scholar]; b Ma S.; Han X.; Krishnan S.; Virgil S. C.; Stoltz B. M. Angew. Chem., Int. Ed. 2009, 48, 8037. [DOI] [PubMed] [Google Scholar]
- a Fuchs J. R.; Funk R. L. Org. Lett. 2005, 7, 677. [DOI] [PubMed] [Google Scholar]; b Dou X.; Yao W.; Zhou B.; Lu Y. Chem. Commun. 2013, 49, 9224. [DOI] [PubMed] [Google Scholar]
- Marfat A.; Carta M. P. Tetrahedron Lett. 1987, 28, 4027. [Google Scholar]
- a Snider B. B.; Busuyek M. V. Tetrahedron 2001, 57, 3301. [Google Scholar]; b Gareau Y.; Zamboni R.; Wong A. W. J. Org. Chem. 1993, 58, 1582. [Google Scholar]; c Voelker T.; Ewell T.; Joo J.; Edstrom E. D. Tetrahedron Lett. 1998, 39, 359. [Google Scholar]
- Explorations in our laboratory regarding the generality of this deprotection method are ongoing.
- Despite the high levels of diastereocontrol observed in the allylic alkylations described in this manuscript (i.e., 11 → 4 and 22 → 21), a reasonable explanation for these selectivities has not been as forthcoming. Investigations into these fascinating reactions and the underlying principles guiding the observed stereoselectivities are ongoing.
- Fukuyama T.; Liu G. J. Am. Chem. Soc. 1996, 118, 7426. [Google Scholar]
- a Trost B. M.; Malhotra S.; Chan W. H. J. Am. Chem. Soc. 2011, 133, 7328. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li P.; Buchwald S. L. Angew. Chem., Int. Ed. 2011, 50, 6396. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Kong C.; Du Y.; Song H.; Zhang D.; Qin Y. Org. Biomol. Chem. 2012, 10, 2793. [DOI] [PubMed] [Google Scholar]
- Yamada Y.; Arima S.; Okada C.; Akiba A.; Kai T.; Harigaya Y. Chem. Pharm. Bull. 2006, 54, 788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.