

Abstract
We have used whole genome paired-end Illumina sequence data to identify tandem duplications in 20 isofemale lines of Drosophila yakuba and 20 isofemale lines of D. simulans and performed genome wide validation with PacBio long molecule sequencing. We identify 1,415 tandem duplications that are segregating in D. yakuba as well as 975 duplications in D. simulans, indicating greater variation in D. yakuba. Additionally, we observe high rates of secondary deletions at duplicated sites, with 8% of duplicated sites in D. simulans and 17% of sites in D. yakuba modified with deletions. These secondary deletions are consistent with the action of the large loop mismatch repair system acting to remove polymorphic tandem duplication, resulting in rapid dynamics of gain and loss in duplicated alleles and a richer substrate of genetic novelty than has been previously reported. Most duplications are present in only single strains, suggesting that deleterious impacts are common. Drosophila simulans shows larger numbers of whole gene duplications in comparison to larger proportions of gene fragments in D. yakuba. Drosophila simulans displays an excess of high-frequency variants on the X chromosome, consistent with adaptive evolution through duplications on the D. simulans X or demographic forces driving duplicates to high frequency. We identify 78 chimeric genes in D. yakuba and 38 chimeric genes in D. simulans, as well as 143 cases of recruited noncoding sequence in D. yakuba and 96 in D. simulans, in agreement with rates of chimeric gene origination in D. melanogaster. Together, these results suggest that tandem duplications often result in complex variation beyond whole gene duplications that offers a rich substrate of standing variation that is likely to contribute both to detrimental phenotypes and disease, as well as to adaptive evolutionary change.
Keywords: tandem duplications, deletions, Drosophila yakuba, Drosophila simulans, evolutionary novelty
Introduction
Gene duplications are an essential source of genetic novelty that can be useful in adaptation and in the origins of developmental complexity across phyla (Conant and Wolfe 2008). Additionally, duplicate sequences are commonly found in mammalian stem cells (Liang et al. 2008), cancer cell lines (Inaki and Liu 2012), and are associated with autoimmune disease, HIV susceptibility, Crohn’s disease, asthma, allergies, and autism (Ionita-Laza et al. 2009). Distinguishing the propensity with which gene duplications serve as causative disease factors as opposed to a source of favorable variation depends heavily on accurate ascertainment of their occurance and frequencies in the population.
In the Drosophila, there is substantial variation in the number and types of duplicate genes that are present in the sequenced reference genomes (Hahn et al. 2007) though the extent to which selection might drive rapid fixation of duplicate genes or whether mutation rates differ across species remains uncertain. Furthermore, these surveys of single strains from each species may not be representative of the variation present in populations and offer only limited opportunities to study their role in adaptation. The advent of Illumina sequencing has made population genomics of complex mutations in nonmodel Drosophila readily tractable. Paired-end Illumina sequencing offers the opportunity to survey copy number variation using definitive sequence-based comparisons that are free from complications related to sole use of coverage or hybridization intensities. Through the identification of paired-end reads that map in abnormal orientations, we can identify a high-confidence data set describing tandem duplications in sample populations (Tuzun et al. 2005; Korbel et al. 2007; Cridland and Thornton 2010).
Drosophila yakuba and D. simulans offer the opportunity to compare the role of tandem duplications in species that have high levels of nucleotide diversity and large effective population sizes of 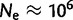 (Sawyer and Hartl 1992; Eyre-Walker et al. 2002; Bachtrog et al. 2006), allowing us to compare mutational and adaptive processes in independent systems where neutral forces of genetic drift should be minimal.
(Sawyer and Hartl 1992; Eyre-Walker et al. 2002; Bachtrog et al. 2006), allowing us to compare mutational and adaptive processes in independent systems where neutral forces of genetic drift should be minimal.
If different chromosomes produce tandem duplications at different rates, we may expect them to contribute differentially to adaptive changes. In D. melanogaster, the X chromosome contains greater repetitive content (Mackay et al. 2012), displays different gene density (Adams et al. 2000), has potentially smaller population sizes (Wright 1931; Andolfatto 2001), lower levels of background selection (Charlesworth 2012), and an excess of genes involved in female-specific expression (Ranz et al. 2003) in comparison to the autosomes. Furthermore, the X chromosome is hemizygous in males, exposing recessive mutations to the full effects of selection more often than comparable loci on the autosomes (Charlesworth et al. 1987). Hence, the incidence of duplications on the X and the types of genes affected may differ from the autosomes, and thereby produce different impacts on phenotypic evolution.
Many copy number variants are thought to be nonneutral (Hu and Worton 1992; Emerson et al. 2008; Cardoso-Moreira et al. 2011), especially when they capture partial gene sequences or create chimeric gene structures (Rogers and Hartl 2012) or result in recruitment of noncoding sequences (Lee and Reinhardt 2012). Such modifications are likely to change gene regulatory profiles (Rogers and Hartl 2012), increasing the likelihood of nonneutral phenotypes. Surveys in D. melanogaster have identified large numbers of such variants (Emerson et al. 2008; Rogers et al. 2009; Cardoso-Moreira et al. 2011, 2012; Lee and Reinhardt 2012). Establishing profiles of partial gene duplication, whole gene duplication, chimera formation, and recruitment of noncoding sequence are essential to a complete understanding of the roles tandem duplicates play in beneficial and detrimental phenotypic changes across species.
Here, we describe the number, types, and genomic locations of tandem duplications segregating in 20 strains of D. yakuba and in 20 strains of D. simulans and discuss differences across species and across chromosomes, as well as their potential to create novel gene constructs.
Results
We have sequenced the complete genomes of 20 isofemale lines of D. yakuba and 20 isofemale lines D. simulans each inbred in the lab for 9–12 generations to produce effectively haploid samples, as well as the reference genome stocks of each species (as a control for genome quality and false positives) (Drosophila Twelve Genomes Consortium 2007; Hu et al. 2013). Genomes are sequenced to high coverage of 50–150× for a total of 42 complete genomes (supplementary tables S1–S5, Supplementary Material online, see Materials and Methods). We have used mapping orientation of paired-end reads to identify recently derived, segregating duplications in these samples <25 kb in length that are supported by three or more divergently oriented read pairs (see Materials and Methods, supplementary text S1, tables S6 and S7, Supplementary Material online). We limit analysis to regions of the genome, which can be assayed with coverage depth of three or more reads across all strains, corresponding to the detection limit for tandem duplicates. We identify 1,415 segregating tandem duplications in D. yakuba and 975 segregating tandem duplications in D. simulans (fig. 1), including large numbers of gene duplications (table 1) with a low false positive rate (supplementary table S8, Supplementary Material online). We assess the numbers and types of gene duplications, differences in duplication rates and sizes across chromosomes, and describe evidence of secondary modification through deletions, which will influence the extent to which these variants can serve as a source of genetic novelty.
Fig. 1.

Tandem duplications in 20 sample strains of Drosophila yakuba. Regions spanned by divergently oriented reads are shown with sample strains plotted on different rows, whereas axes list genomic location in Mbp. Duplications are more common around the centromeres, especially on chromosome 2. Frequencies are shaded in grayscale according to frequency, with high-frequency variants shown in solid black. The D. simulans X chromosome appears to have an excess of high-frequency variants in comparison to the D. simulans autosomes and the D. yakuba X chromosome.
Table 1.
Duplicated Regions in Drosophila yakuba and D. simulans.
| D. yakuba | D. simulans | |
|---|---|---|
| Whole gene | 248 | 296 |
| Partial gene | 745 | 462 |
| Intergenic | 745 | 577 |
Genotyping and Quality Control
Divergently oriented paired-end reads are effective indicators of tandem duplications (Tuzun et al. 2005; Cridland and Thornton 2010; Mills et al. 2011; Zichner et al. 2013). We have used paired-end read orientation (fig. 2) combined with increased coverage in genomic sequencing (fig. 3) to identify tandem duplications in population samples of D. yakuba and D. simulans. Divergently oriented reads indicative of putative tandem duplications were clustered within a single strain, with three or more divergently oriented read pairs within the strain required to define each tandem duplication (see Materials and Methods). Duplications were then clustered across strains with coordinates defined as the maximum span of divergent reads across all strains. The distribution of supporting read pairs is highly skewed, with 3–4 supporting read pairs for many calls (supplementary fig. S1, Supplementary Material online). To account for duplications which may be undetected, we additionally included variants that showed 2-fold increases (fig. 3) in quantile normalized coverage (supplementary figs. S2 and S3, Supplementary Material online) and which are supported by one or more divergently oriented read pairs were also identified as having duplications if the duplicate was present in a second strain, thereby correcting sample frequency estimates for false negatives (see Materials and Methods, supplementary text S1, Supplementary Material online). We retained only those tandem duplicates which are not present in outgroup reference genomes of D. melanogaster, D. erecta, and D. yakuba or D. simulans as defined in a BLAST search (see Materials and Methods) suggesting recent origins.
Fig. 2.
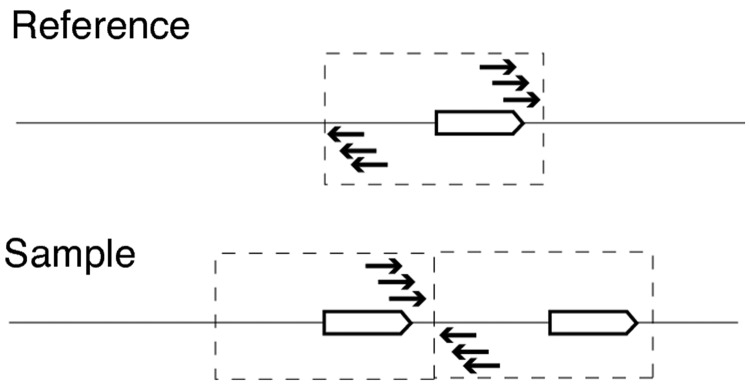
A tandem duplication in a sample that was then used to generate paired-end Illumina libraries. Duplications should be apparent through divergently oriented read pairs when mapping onto the reference genome. Tandem duplications require a minimum of three divergently oriented read pairs. Duplication span is recorded as the minimum and maximum coordinates spanned by divergent reads.
Fig. 3.

Coverage change for a duplication on chromosome 3 L in Line 9 of Drosophila yakuba. Regions spanned by divergently oriented reads are shaded. Sample coverage is shown in black, whereas reference genome coverage is shown in gray.
Using divergently oriented paired-end reads, we have identified 1,415 segregating tandem duplications across 20 sample strains of D. yakuba, in comparison to 975 segregating tandem duplications in 20 lines of D. simulans, with significantly more duplicates identified in D. yakuba than in D. simulans (one-sided t-test, 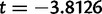 ,
, 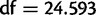 ,
, 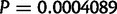 ). More variants are identified in D. yakuba in spite of higher coverage in D. simulans (supplementary table S4, Supplementary Material online), suggesting that the difference is likely to be biological rather than technical. In fact, the number of variants identified is only weakly correlated with coverage per strain (fig. 4) a product of sequencing to the saturation point of coverage (supplementary table S9, Supplementary Material online). Downsampling reads from D. yakuba CY17C, which was sequenced to 151×, we find that the portion of the genome covered with three or more reads (the detection limit of our assay) plateaus at roughly 45× (supplementary table S9, Supplementary Material online) though lower coverage data used in previous studies (Alkan et al. 2009; Sudmant et al. 2010; Mills et al. 2011; Zichner et al. 2013) will be far from this plateau. The tandem duplications identified across these sample strains cover 2.574% of the assayable genome of the X and four major autosomes in D. yakuba and 1.837% of the assayable genome of the X and four major autosomes in D. simulans. We are able to identify tandem duplications as small as 66 bp in D. yakuba and 78 bp in D. simulans.
). More variants are identified in D. yakuba in spite of higher coverage in D. simulans (supplementary table S4, Supplementary Material online), suggesting that the difference is likely to be biological rather than technical. In fact, the number of variants identified is only weakly correlated with coverage per strain (fig. 4) a product of sequencing to the saturation point of coverage (supplementary table S9, Supplementary Material online). Downsampling reads from D. yakuba CY17C, which was sequenced to 151×, we find that the portion of the genome covered with three or more reads (the detection limit of our assay) plateaus at roughly 45× (supplementary table S9, Supplementary Material online) though lower coverage data used in previous studies (Alkan et al. 2009; Sudmant et al. 2010; Mills et al. 2011; Zichner et al. 2013) will be far from this plateau. The tandem duplications identified across these sample strains cover 2.574% of the assayable genome of the X and four major autosomes in D. yakuba and 1.837% of the assayable genome of the X and four major autosomes in D. simulans. We are able to identify tandem duplications as small as 66 bp in D. yakuba and 78 bp in D. simulans.
Fig. 4.

Number of variants versus coverage by line in Drosophila yakuba (A) and D. simulans (B). Regression line (blue) and 95% confidence interval (gray) are shown. Correlation between coverage and number of duplications is low (D. yakuba adjusted 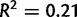 , D. simulans adjusted
, D. simulans adjusted 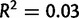 ).
).
We have successfully polymerase chain reaction (PCR)-amplified 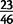 randomly chosen variants in D. yakuba and
randomly chosen variants in D. yakuba and 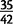 variants in D. simulans. The rate of PCR confirmation in D. simulans is not significantly different from previous studies of copy number variants, but we observe significant differences between D. yakuba and all other confirmation rates (supplementary table S10, Supplementary Material online). In view of this disparity, combined with difficulties of PCR primer design for variants whose precise structures are unknown, we generated PacBio long molecule sequencing data for four strains of D. yakuba in order to more reliably estimate the false positive rate (supplementary table S11, Supplementary Material online). PacBio long molecule sequencing has recently been used to validate targeted duplications in human genome data (Huddleston et al. 2014). We extend this approach to genome wide identification and validation of tandem duplications, and have generated PacBio reads for four different sample strains of D. yakuba. Across these four strains, we observe confirmation of 661 out of 688 mutations, for a maximum false positive rate of 3.9% (supplementary table S8, Supplementary Material online), though some variants may be unconfirmed due to low clone coverage in a region. Hence, the duplicates identified with paired-end reads in high coverage genomic sequence data are extremely accurate and comparable to or better than previous methods or attempts to identify and validate duplicates using lower coverage genomic sequences or microarrays (Alkan et al. 2009; Sudmant et al. 2010; Cardoso-Moreira et al. 2011; Mills et al. 2011; Zichner et al. 2013). Split read mapping with short Illumina reads performed poorly in comparison and failed to confirm 88.3% of variants, and breakpoint assembly was possible for <60% of variants in spite of high rates of confirmation with PacBio (see supplementary text S1, Supplementary Material online). Thus, requiring these criteria would exclude the majority of variant calls and would likely be biased against duplicates with formation facilitated by repetitive sequences. Where duplicate breakpoints contain repetitive or low complexity sequences, or where subsequent modification of alleles through deletion has altered surrounding sequence, PCRs are likely to fail, and we would suggest that confirmation using long molecule sequencing is far more reliable in the face of complex structures. Further description of genomic sequences, tandem duplications, and discussion of paired-end read performance in high-coverage genomic sequencing data in comparison to other methods is available in supplementary text S1, Supplementary Material online.
variants in D. simulans. The rate of PCR confirmation in D. simulans is not significantly different from previous studies of copy number variants, but we observe significant differences between D. yakuba and all other confirmation rates (supplementary table S10, Supplementary Material online). In view of this disparity, combined with difficulties of PCR primer design for variants whose precise structures are unknown, we generated PacBio long molecule sequencing data for four strains of D. yakuba in order to more reliably estimate the false positive rate (supplementary table S11, Supplementary Material online). PacBio long molecule sequencing has recently been used to validate targeted duplications in human genome data (Huddleston et al. 2014). We extend this approach to genome wide identification and validation of tandem duplications, and have generated PacBio reads for four different sample strains of D. yakuba. Across these four strains, we observe confirmation of 661 out of 688 mutations, for a maximum false positive rate of 3.9% (supplementary table S8, Supplementary Material online), though some variants may be unconfirmed due to low clone coverage in a region. Hence, the duplicates identified with paired-end reads in high coverage genomic sequence data are extremely accurate and comparable to or better than previous methods or attempts to identify and validate duplicates using lower coverage genomic sequences or microarrays (Alkan et al. 2009; Sudmant et al. 2010; Cardoso-Moreira et al. 2011; Mills et al. 2011; Zichner et al. 2013). Split read mapping with short Illumina reads performed poorly in comparison and failed to confirm 88.3% of variants, and breakpoint assembly was possible for <60% of variants in spite of high rates of confirmation with PacBio (see supplementary text S1, Supplementary Material online). Thus, requiring these criteria would exclude the majority of variant calls and would likely be biased against duplicates with formation facilitated by repetitive sequences. Where duplicate breakpoints contain repetitive or low complexity sequences, or where subsequent modification of alleles through deletion has altered surrounding sequence, PCRs are likely to fail, and we would suggest that confirmation using long molecule sequencing is far more reliable in the face of complex structures. Further description of genomic sequences, tandem duplications, and discussion of paired-end read performance in high-coverage genomic sequencing data in comparison to other methods is available in supplementary text S1, Supplementary Material online.
Complex Variation
We identified deletions that have occurred in duplicated alleles using long-spanning read pairs 600 bp or longer, corresponding approximately to the 99.9th percentile of fragment lengths in the reference genomes (supplementary table S5, Supplementary Material online). Out of 880 duplications 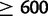 bp in length in D. yakuba, which could be surveyed for deletions within duplications using long-spanning reads, 151 (17%) contain long-spanning read pairs covering 50% or more of the duplicate sequence in one or more strain, indicative of subsequent deletion, multiple independent short-range dispersed duplications, or incomplete duplication (fig. 5). In D. simulans,
bp in length in D. yakuba, which could be surveyed for deletions within duplications using long-spanning reads, 151 (17%) contain long-spanning read pairs covering 50% or more of the duplicate sequence in one or more strain, indicative of subsequent deletion, multiple independent short-range dispersed duplications, or incomplete duplication (fig. 5). In D. simulans, 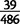 (8%) duplications
(8%) duplications 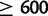 bp contain long-spanning reads covering 50% or more of duplicated sequence in one or more strains. Among 69 such modified variants in D. yakuba that are present in multiple strains, 66 have at least one strain that lacks these long-spanning reads, whereas 12 out of 14 variants in D. simulans lack long-spanning reads in one or more strains. Given large numbers of unaltered duplicates, the most parsimonious explanation is that deletions are most often secondary modification and that the majority of these constructs forms through full length duplication and subsequent deletion rather than independent dispersed duplications.
bp contain long-spanning reads covering 50% or more of duplicated sequence in one or more strains. Among 69 such modified variants in D. yakuba that are present in multiple strains, 66 have at least one strain that lacks these long-spanning reads, whereas 12 out of 14 variants in D. simulans lack long-spanning reads in one or more strains. Given large numbers of unaltered duplicates, the most parsimonious explanation is that deletions are most often secondary modification and that the majority of these constructs forms through full length duplication and subsequent deletion rather than independent dispersed duplications.
Fig. 5.

Complex breakpoints and subsequent modification of tandem duplications. Short-range dispersed duplications, duplication with insertion of novel sequence, and duplication with subsequent deletion will all display the same signals of divergently oriented reads. Although all of these indicate that duplication has occurred, signals solely from short sequence read pairs are unlikely to capture the full complexity of duplication events.
In one well-characterized example, we have identified a duplication which spans the chimeric retrogene jingwei (jgw) (Long and Langley 1993), which houses a deletion upstream from jgw (fig. 6). The duplication is defined by ten divergent read pairs and confirmed by split read mapping in PacBio long molecule sequencing, whereas the deletion is supported by 20 long-spanning read pairs in line NY66-2 and gapped alignment in PacBio reads (fig. 6). The same duplication and deletion are independently confirmed with PacBio sequences in line CY21B3. The duplication spanning jgw is found at a frequency of 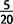 strains, whereas the deletion shown is observed only in CY21B3 and NY66-2, suggesting that the deletions is a secondary modification. A second independent duplication spans jgw in
strains, whereas the deletion shown is observed only in CY21B3 and NY66-2, suggesting that the deletions is a secondary modification. A second independent duplication spans jgw in 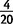 strains and is confirmed in PacBio data, indicating that the region has been modified multiple times in different strains.
strains and is confirmed in PacBio data, indicating that the region has been modified multiple times in different strains.
Fig. 6.

Read mapping patterns indicative of a modified duplication surrounding jingwei in Drosophila yakuba line NY66-2. Duplications are indicated with divergently oriented paired-end reads (blue) as well as with split read mapping of long molecule sequencing (purple). Deletions in one copy are suggested by gapped read mapping of long molecule reads (red) as well as multiple long-spanning read pairs at the tail of mapping distances in paired-end read sequencing (green) just upstream from jgw. Up to 20% of duplicates observed have long-spanning read pairs indicative of putative deletions in one or more alleles in the population.
Deletions are exceptionally common in Drosophila (Petrov et al. 1996), and several genetic mechanisms might offer means of excision in a short time frame after duplication. The large loop mismatch repair system can facilitate deletions of duplicated sequence to modify duplicated sequence as long as variants are polymorphic. The presence of unpaired duplicated DNA during meiosis or mitosis would commonly invoke the action of the large loop mismatch repair system, which if resolved imprecisely, could result in the construct observed (fig. 7). Deletions lying within a duplication have a median size of 3.6 kb in D. yakuba and 1.8 kb in D. simulans. Such large deletions are well outside the norm for genome wide large deletions in mutation accumulation lines of D. melanogaster, which show an average 409 bp and maximum of 2.6 kb (Schrider et al. 2013). Deletions of this size however are consistent with the size of excised fragments in large loop mismatch repair of several kilobases (Kearney et al. 2001). Deletion during nonhomologous end joining or homology-mediated replication slippage might produce deletions as well though it is unclear whether mutation rates are naturally high enough to operate in short time frames. Thus, we would expect modification of duplicated alleles to be extremely common, especially in deletion-biased Drosophila.
Fig. 7.

Secondary deletion via large loop mismatch repair. A tandem duplication forms via ectopic recombination or replication slippage. At some point prior to fixation in the population the duplication pairs with an unduplicated chromatid in meiosis or mitosis, invoking the action of the large loop mismatch repair system. Imprecise excision results in a modified duplicate with partially deleted sequence. Large loop mismatch repair requires that duplications are polymorphic, and would therefore produce secondary modification over short timescales, resulting in rapid modification of tandem duplicates.
Differences in Gene Duplications across Species
Duplicated coding sequences can diverge to produce novel peptides, novel regulatory profiles, or specialized subfunctions (Conant and Wolfe 2008). In order to determine the extent to which genes are likely to be duplicated and whether particular categories of gene duplications are more likely to be favored, we identified coding sequences captured by tandem duplications. We find large numbers of segregating gene duplications in both D. yakuba and D. simulans including hundreds of whole gene duplications (table 1). We used the maximum span of divergently oriented reads across all strains to identify tandem duplications that capture gene sequences in D. yakuba or D. simulans and to determine their propensity to capture whole and partial gene sequences.
We find that 47.3% of tandem duplications in D. yakuba and 40.8% in D. simulans capture coding sequences. The average duplicated gene in D. yakuba covers 45.9% of the gene sequence and 60.5% of the gene sequence in D. simulans. There are 670 duplications that capture gene sequences, spanning 845 different genes in D. yakuba, whereas 398 duplications span 478 genes in D. simulans. Some 103 genes in D. yakuba and 65 genes in D. simulans are captured in multiple independent duplications, with some genes falling in as many as six independent putative duplications as defined by divergently oriented reads in D. yakuba and 32 independent putative duplications in D. simulans. Such high rate of independent duplications in D. simulans is consistent with previous studies using microarrays (Cardoso-Moreira et al. 2012). In total 993 gene fragments in D. yakuba and 758 gene fragments in D. simulans exist as segregating copy number variants in the population, and 56 genes are duplicated in both species.
Assuming that unmodified duplications without deletions represent the original mutated state, in D. yakuba, 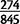 (27.6%) of duplicated gene fragments span more than 80% of gene sequence and capture the translation start site whereas 65 (7.7%) capture 20% or less and the translation start site. In D. simulans
(27.6%) of duplicated gene fragments span more than 80% of gene sequence and capture the translation start site whereas 65 (7.7%) capture 20% or less and the translation start site. In D. simulans
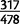 (66.3%) duplicated gene sequences capture 80% or more of the gene sequence and the translation start, and 34 (7.1%) capture 20% or less and include the translation start. Based on a resampling of gene duplications in D. yakuba, D. simulans houses an overabundance of whole or nearly whole gene duplications (
(66.3%) duplicated gene sequences capture 80% or more of the gene sequence and the translation start, and 34 (7.1%) capture 20% or less and include the translation start. Based on a resampling of gene duplications in D. yakuba, D. simulans houses an overabundance of whole or nearly whole gene duplications (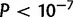 ) and an underrepresentation of small fragments (
) and an underrepresentation of small fragments (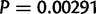 ), suggesting differences in the occurrence of whole gene duplications across species due either to mutational pressures or selection.
), suggesting differences in the occurrence of whole gene duplications across species due either to mutational pressures or selection.
Duplicate Genes and Rapidly Evolving Phenotypes
Biases in the rates at which duplications form in different genomic regions or a greater propensity for selection to favor duplications in specific functional classes can result in a bias in gene ontology (GO) categories among duplicated genes. We used DAVID GO analysis software to identify overrepresented functions among duplicate genes in D. yakuba and D. simulans. In D. yakuba, we observe 678 duplicated genes with orthologs in D. melanogaster. Overrepresented functional categories include immunoglobulins, extracellular matrix, chitins and aminoglycans, immune response and wound healing, drug and hormone metabolism, chorion development, chemosensory response and development, and morphogenesis (supplementary table S13, Supplementary Material online). In D. simulans, we observe 478 duplicated genes with orthologs in D. melanogaster. Overrepresented GO categories include cytochromes and oxidoreductases plus toxin metabolism, immune response to microbes, phosopholipid metabolism, chemosensory processing, carboxylesterases, glutathion transferase and drug metabolism, and sarcomeres (supplementary table S13, Supplementary Material online). In D. simulans, 65 genes were involved in multiple independent duplications that have distinct breakpoints. Overrepresented GO categories include immune response to bacteria, chorion development and oogenesis, chemosensory perception, and organic cation membrane transport (supplementary table S14, Supplementary Material online). In D. yakuba among 72 genes duplicated independently, chorion development and oogenesis, cell signaling, immune response, sensory processing, and development are overrepresented (supplementary table S14, Supplementary Material online).
There are 25 high-frequency variants found at a sample frequency of 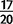 or greater in D. simulans, including lipases and endopeptidases expressed in male accessory glands and several genes involved in immune response to microbes (supplementary table S15, Supplementary Material online). One gene arose independently and has reached high frequency twice in D. simulans. In D. yakuba, we observe 13 high-frequency variants, including endopeptidases and adenosine monophosphate dependent ligases (supplementary table S15, Supplementary Material online). Both male reproductive proteins (Wong and Wolfner 2012) and immune response to pathogens (Lazarro and Clark 2012) are known for their rapid evolution, and therefore these genes are strong candidates to search for evidence of ongoing selective sweeps. Though mutational biases can produce similarities in GO categories, the overabundance of toxin metabolism genes and immune response peptides in both species as well as the overrepresentation of chemoreceptors, chitin cuticle genes, and oogenesis factors suggests that duplications are likely key players in rapidly evolving systems.
or greater in D. simulans, including lipases and endopeptidases expressed in male accessory glands and several genes involved in immune response to microbes (supplementary table S15, Supplementary Material online). One gene arose independently and has reached high frequency twice in D. simulans. In D. yakuba, we observe 13 high-frequency variants, including endopeptidases and adenosine monophosphate dependent ligases (supplementary table S15, Supplementary Material online). Both male reproductive proteins (Wong and Wolfner 2012) and immune response to pathogens (Lazarro and Clark 2012) are known for their rapid evolution, and therefore these genes are strong candidates to search for evidence of ongoing selective sweeps. Though mutational biases can produce similarities in GO categories, the overabundance of toxin metabolism genes and immune response peptides in both species as well as the overrepresentation of chemoreceptors, chitin cuticle genes, and oogenesis factors suggests that duplications are likely key players in rapidly evolving systems.
Chimeric Genes and Altered Coding Sequences
If only one boundary of a tandem duplication falls within a coding sequence and thereby copies the 5 end of a gene, the resulting construct will recruit formerly noncoding sequence to form the 3
end of a gene, the resulting construct will recruit formerly noncoding sequence to form the 3 end of the coding sequence (fig. 8A). We observe 143 cases of recruitment of noncoding sequence in D. yakuba (supplementary tables S16 and S17, Supplementary Material online) and 96 cases in D. simulans (supplementary tables S18 and S19, Supplementary Material online). Several of these are found at moderate frequencies >50%. Overrepresented GO categories among genes in D. simulans include immune defense and sarcomeres, whereas genes with recruited sequence in D. yakuba show an overrepresentation of genes involved in locomotory behavior. We observe one high-frequency variant in D. yakuba at a sample frequency of
end of the coding sequence (fig. 8A). We observe 143 cases of recruitment of noncoding sequence in D. yakuba (supplementary tables S16 and S17, Supplementary Material online) and 96 cases in D. simulans (supplementary tables S18 and S19, Supplementary Material online). Several of these are found at moderate frequencies >50%. Overrepresented GO categories among genes in D. simulans include immune defense and sarcomeres, whereas genes with recruited sequence in D. yakuba show an overrepresentation of genes involved in locomotory behavior. We observe one high-frequency variant in D. yakuba at a sample frequency of 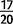 , an Adenylate cyclase involved in locomotor rhythm as well as two high-frequency variants in D. simulans LysB, an antimicrobial humoral response gene, and a gene of unknown function. These high-frequency chimeras are strong candidates for selective sweeps.
, an Adenylate cyclase involved in locomotor rhythm as well as two high-frequency variants in D. simulans LysB, an antimicrobial humoral response gene, and a gene of unknown function. These high-frequency chimeras are strong candidates for selective sweeps.
Fig. 8.

Abnormal gene structures. Duplicated sequence is highlighted with bold colors and is framed by the dashed box. (A) The partial duplication of a coding sequence (blue) results in the recruitment of previously upstream noncoding sequence (dashed lines) to create a novel open reading frame (blue and turquoise). (B) Tandem duplication where both boundaries fall within coding sequences results in a chimeric gene.
If both boundaries fall within different coding sequences, tandem duplications can create chimeric genes (fig. 8B) (Rogers et al. 2009). We find 130 tandem duplications in D. yakuba and 76 in D. simulans where both breakpoints fall within nonoverlapping coding sequences. Some 11 of the 130 duplications in D. yakuba and 30 of 76 in D. simulans have both breakpoints in gene sequences face one another and as such are not expected to create new open reading frames, as the constructs will lack promoters. Another 40 of the 130 duplications in D. yakuba and 8 of 76 in D. simulans have both breakpoints in gene sequences, and will have promoters that can potentially transcribe sequences from both strands of DNA (fig. 9, supplementary tables S20 and S21, Supplementary Material online). Only 78 chimeric coding sequences in D. yakuba (supplementary tables S22 and S23, Supplementary Material online) and 38 chimeric genes in D. simulans (supplementary table S24, Supplementary Material online) have parental genes in parallel orientation. Among the parental genes of these chimeras, cytochromes and genes involved in drug metabolism are overrepresented in D. yakuba. Other functional categories which are present but not overrepresented include endopeptidases, signaling glycopeptides, and sensory signal transduction peptides. Among parental genes in D. simulans, cytochromes and insecticide metabolism genes, sensory perception genes, and endopeptidase genes are overrepresented. Additional categories present include signal peptides, endocytosis genes, and oogenesis genes. Several such constructs are found at moderate frequencies above 10/20, suggesting that they are at least not detrimental. However, two chimeras in D. yakuba are found at high frequency. One formed from a combination of GE12441 and GE12442 is at a frequency of 16/20, and one formed from GE12353 and GE12354 is at a frequency of 19/20. In D. simulans one chimera, formed from CG11598 and CG11608, is at a frequency of 20/20. All of these genes are lipases or endopeptidases. These high-frequency variants are strong candidates for selective sweeps.
Fig. 9.
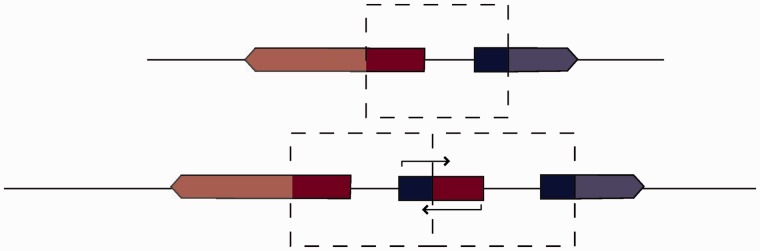
Dual promoter genes. Duplicated sequence is highlighted with bold colors and is framed by the dashed box. Tandem duplication where both boundaries fall within coding sequences results in a chimeric gene which contains two promoters, one which facilitates transcription in one direction, the other facilitating transcription from the opposite strand. The chimera is capable of making partial antisense transcripts.
Compared with the number of tandem duplications that capture coding sequences, the number of duplications which form chimeric genes indicates that chimeric constructs derived from parental genes in parallel orientation form as a result of 10.4% of tandem duplications that capture genes in D. yakuba and 9.5% of tandem duplications that capture coding sequences in D. simulans. These numbers are in general agreement with rates of chimeric genes formation estimated from a within-genome study of D. melanogaster of 16.0% compared with the rate of formation of duplicate genes (Rogers et al. 2009).
Association with Transposable Elements and Direct Repeats
Repetitive sequences are known to facilitate ectopic recombination events that commonly yield tandem duplications (Lim and Simmons 1994). In D. yakuba, 179 (12.7%) tandem duplications fall within 1 kb of a transposable element (TE) in at least one sample strain that has a duplication and 52 (3.7%) fall within 100 bp of a TE (supplementary table S25, Supplementary Material online). In D. simulans, 122 (12.5%) lie within 1 kb of a TE and 53 (5.4%) fall within 100 bp of a TE (supplementary table S25, Supplementary Material online). Additionally, 125 (8.8%) of duplications in D. yakuba have 100 bp or more of direct repeated sequence in the 500 bp up and downstream of duplication boundaries and 237 (16.7%) have 30 bp or more in the reference sequence as identified in a BLASTn comparison of regions flanking divergently oriented read spans at an E value 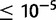 (supplementary table S25, Supplementary Material online). In D. simulans, 56 (5.7%) have 100 bp or more of direct repeated sequence in the 500 bp up and downstream of duplication boundaries in the reference and 150 (14.4%) have 30 bp or more of repeated sequence (supplementary table S25, Supplementary Material online). In total 371 duplications in D. yakuba and 243 duplications in D. simulans either lie within 1 kb of a TE in at least one strain or are flanked by 30 bp or more of direct repeated sequence. Hence, a maximum of 26.2% of duplications identified in D. yakuba and 24.9% of duplications identified in D. simulans may have been facilitated by ectopic recombination between large repeats, consistent with previous estimates from single genome studies of 30% in D. melanogaster but somewhat higher than those in D. yakuba of 12% (Zhou et al. 2008).
(supplementary table S25, Supplementary Material online). In D. simulans, 56 (5.7%) have 100 bp or more of direct repeated sequence in the 500 bp up and downstream of duplication boundaries in the reference and 150 (14.4%) have 30 bp or more of repeated sequence (supplementary table S25, Supplementary Material online). In total 371 duplications in D. yakuba and 243 duplications in D. simulans either lie within 1 kb of a TE in at least one strain or are flanked by 30 bp or more of direct repeated sequence. Hence, a maximum of 26.2% of duplications identified in D. yakuba and 24.9% of duplications identified in D. simulans may have been facilitated by ectopic recombination between large repeats, consistent with previous estimates from single genome studies of 30% in D. melanogaster but somewhat higher than those in D. yakuba of 12% (Zhou et al. 2008).
In D. yakuba, 14.4% of duplications with 100 bp or more of repetitive sequence and 21.1% of duplications with 30 bp or more are located on the X. In contrast, 46.4% of duplications in D. simulans with 100 bp or more of direct repeated sequence in the reference and 44.7% with more than 30 bp of repeated sequence in the D. simulans reference lie on the X chromosome. Based on a resampling of randomly chosen duplications, duplications on the X chromosome are overrepresented among duplications with direct repeats (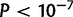 ) but the same is not true of duplicates with direct repeats in D. yakuba (
) but the same is not true of duplicates with direct repeats in D. yakuba (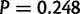 ). A genome wide BLASTn comparison shows that direct repeats are not overrepresented on the D. simulans X chromosome and cannot explain the observed association (supplementary table S26, Supplementary Material online). Hence, duplication via ectopic recombination may be exceptionally common on the X chromosome in D. simulans.
). A genome wide BLASTn comparison shows that direct repeats are not overrepresented on the D. simulans X chromosome and cannot explain the observed association (supplementary table S26, Supplementary Material online). Hence, duplication via ectopic recombination may be exceptionally common on the X chromosome in D. simulans.
Excess of Duplications on the D. simulans X
The distribution of duplication sizes was calculated for each major chromosomal arm in each species. Average duplicate size is 2,518 bp, in close agreement with that observed in mutation accumulation lines in D. melanogaster (Schrider et al. 2013) but somewhat larger than that observed using microarrays in D. simulans (Cardoso-Moreira et al. 2011). The X chromosome in D. yakuba displays an overabundance of small duplications in comparison to each of the autosomes in a Tukey’s Honestly Significant Difference (HSD) test after correction for multiple testing with 27% of duplicates 500 bp or less (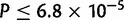 , figs. 1 and 10A, supplementary table S27, Supplementary Material online). Chromosome 2 R is also significantly different from the other three major autosomal arms (
, figs. 1 and 10A, supplementary table S27, Supplementary Material online). Chromosome 2 R is also significantly different from the other three major autosomal arms (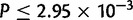 , fig. 10A, supplementary table S27, Supplementary Material online). However, in D. simulans there is no significant difference between the X and 2R, 2L, and 3R even though the X houses a greater density of duplications (fig. 10B). The D. simulans chromosome 3L is different from 2L (
, fig. 10A, supplementary table S27, Supplementary Material online). However, in D. simulans there is no significant difference between the X and 2R, 2L, and 3R even though the X houses a greater density of duplications (fig. 10B). The D. simulans chromosome 3L is different from 2L (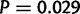 , fig. 10B, supplementary table S27, Supplementary Material online).
, fig. 10B, supplementary table S27, Supplementary Material online).
Fig. 10.

Cumulative distribution function for duplication sizes for the X and four major autosomal arms in (A) Drosophila yakuba and (B) D. simulans. The X chromosome in D. yakuba (red) is significantly different from all autosomes (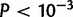 ) due to a large number of small duplications 500 bp or less. Chromosome 2 R (blue) is also different from Chr2L and 3 R (
) due to a large number of small duplications 500 bp or less. Chromosome 2 R (blue) is also different from Chr2L and 3 R (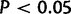 ). In D. simulans, chromosome 3 L (green) is significantly different 2 L, 3 R, and the X.
). In D. simulans, chromosome 3 L (green) is significantly different 2 L, 3 R, and the X.
We observe a significant effect in the number of duplications per mapped base pair by chromosome in both D. yakuba (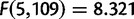 ,
, 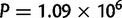 ) and D. simulans (
) and D. simulans (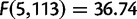 ,
, 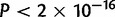 ). In a post hoc Tukey’s HSD test with correction for multiple testing, the D. simulans X chromosome contains more duplications per mapped base pair than any of the autosomes, with 316 duplications (
). In a post hoc Tukey’s HSD test with correction for multiple testing, the D. simulans X chromosome contains more duplications per mapped base pair than any of the autosomes, with 316 duplications (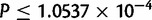 , supplementary table S28, Supplementary Material online, figs. 1 and 11). Chromosome 2 R contains an excess of duplicates in comparison to chromosome 3 R (
, supplementary table S28, Supplementary Material online, figs. 1 and 11). Chromosome 2 R contains an excess of duplicates in comparison to chromosome 3 R (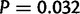 ), but all other pairwise comparisons of the four major autosomes are not significant. Chromosome 4 contains an excess of duplications per mapped base pair in comparison to all other chromosomes in both D. simulans and D. yakuba. In D. yakuba, the X is different from 3 L (
), but all other pairwise comparisons of the four major autosomes are not significant. Chromosome 4 contains an excess of duplications per mapped base pair in comparison to all other chromosomes in both D. simulans and D. yakuba. In D. yakuba, the X is different from 3 L (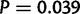 ) but not from any other autosome. Some 11 of the 25 duplicates in D. simulans are at a frequency of
) but not from any other autosome. Some 11 of the 25 duplicates in D. simulans are at a frequency of 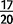 strains or greater (44%) on the X (fig. 1). In comparison, only 2 of the 13 high-frequency duplications in D. yakuba (15.4%) are located on the X, nor do we see a comparable overabundance of duplications on the D. yakuba X. These results point to a clear excess of duplications on the X chromosome in D. simulans in comparison to the autosomes, as well as an overabundance of duplications on the fourth chromosome in both species.
strains or greater (44%) on the X (fig. 1). In comparison, only 2 of the 13 high-frequency duplications in D. yakuba (15.4%) are located on the X, nor do we see a comparable overabundance of duplications on the D. yakuba X. These results point to a clear excess of duplications on the X chromosome in D. simulans in comparison to the autosomes, as well as an overabundance of duplications on the fourth chromosome in both species.
Fig. 11.

Number of Duplications per bp on the X and major autosomes in Drosophila yakuba and D. simulans. The X chromosome in D. simulans contains an excess of duplications in comparison with the autosomes. Chromosome 2 R also contains more duplications per bp than chromosome 3 R but no other autosomes are significantly different. Chromosome 4, the dot chromosome, has more duplications per mapped bp in both D. yakuba and D. simulans than any other chromosome.
Given the excess of duplications associated with repetitive content on the D. simulans X, repetitive elements may be an important factor in forming the observed overabundance of duplications on the D. simulans X. Although mutational and selective processes can lead to a bias in the number of duplications that form on different chromosomes, the excess of high-frequency variants on the D. simulans X at a frequency of 20 out of 20 would suggest that at least some of the overabundance on the D. simulans X is due to selective forces or demography resulting in duplicates spreading through the population.
Discussion
We have used paired-end reads to describe tandem duplications in sample strains of D. yakuba and D. simulans, their sample frequencies, and the genes that they affect. We use high coverage Illumina genomic sequencing data of 50× or greater to successfully identify tandem duplications among individually sequenced isofemale lines derived from natural populations. We have filtered tandem duplications to include recently derived segregating tandem duplicates that are not present in the reference genome of each species for genomic regions that have coverage of three or more read pairs across all sequenced strains. We show high rates of confirmation using long molecule PacBio sequences with 96.1% of variants showing evidence of confirmation.
We identify 1,415 tandem duplicates in D. yakuba and 975 in D. simulans, indicating that there is substantial standing variation segregating in populations that may contribute adaptive evolution and the instance of detrimental phenotypes. We identify hundreds of chimeric genes and cases where genes recruit formerly noncoding sequence. We have shown an excess of duplications on the D. simulans X chromosome as well as an overabundance of whole gene duplications in D. simulans, suggestive of selection acting on duplications.
Rapid Modification of Duplicated Alleles
Standing variation is expected to play a major role in adaptation and evolutionary change (Barrett and Schluter 2008). If the span of standing variation in populations is limited, the dynamics, genomic content, and variability of standing variation in populations are likely to play a defining role in evolutionary outcomes. The observed span of tandem duplications across strains is limited, with 2.574% of the assayable genome of the X and four major autosomes in D. yakuba and 1.837% of the assayable genome of the X and four major autosomes in D. simulans. Yet, the variation that is observed portrays a dynamic picture of gains and losses with evidence that duplications can induce subsequent deletions through large loop mismatch repair, suggesting that regions that are duplicated create genomic instability. The resulting expansion and contraction of genomic sequences will contribute to greater variability in these limited regions than has been suggested to date, offering wider variation upon which selection can act. Up to 17% of duplications in D. yakuba and 8% of duplications in D. simulans show long-spanning reads in one or more strains, indicative of complex changes such as subsequent deletion, insertion of foreign sequence, or incomplete or short-range dispersed duplication (fig. 5). These results are consistent with complex breakpoints previously observed in D. melanogaster (Cridland and Thornton 2010). Moreover, coverage changes for certain variants are consistent with duplication followed by subsequent deletion in one or both copies (supplementary fig. S4, Supplementary Material online). Hence, the current pool of genetic diversity will in fact be far greater than simple interpretation of divergently oriented reads or split read mapping might indicate. The majority of such changes has one or more strains with no signs of modification, suggesting that these variants are primarily duplications followed by deletions.
Secondary deletions of recently duplicated alleles may be exceptionally common, especially in deletion-biased genomes such as Drosophila. Deletion of excess unpaired DNA for polymorphic duplicates during large loop mismatch repair, excision of TEs, replication slippage, and deletion during nonhomologous end joining all offer common mechanisms that are likely to remove portions of duplicated alleles. Among these mechanisms, the large loop mismatch repair system specifically targets newly added DNA and is likely to be a driving force in the rapid modification of duplicated alleles. In the ideal case, precise excision would simply return the construct to singleton state resulting in a rapid cycle of mutations and reversions. However, when such removal is imprecise, these subsequent deletions are likely to modify duplicated sequences leaving incompletely duplicated segments. The average distance spanned by these putative deletions is over 2 kb, well above the mean for deletions observed in mutation accumulation lines of D. melanogaster (Schrider et al. 2013), but in agreement with the amount of DNA that can be efficiently removed by large loop mismatch repair (Kearney et al. 2001).
Duplications have the potential to induce secondary deletions quickly, whereas variants remain polymorphic, thereby offering mechanisms for rapid and potentially drastic genomic change that can potentially alter gene content, dosage, and regulation. Variation in populations, while limited in its genomic scope, may offer multiple variant forms at individual duplication sites. Thus, the substrate that is present for selection and adaptation will be far richer than a simple duplication or single copy, but rather can take on these complex forms of modified variants that remain largely unexplored in terms of their molecular and evolutionary impacts. Thus, although the observed amount of variation is limited to only a fraction of the genome, the level of variation at these duplicated sites portrays an exceptionally dynamic flux of duplications and deletions at these sites that will result in changes in the content and organization of the genome and therefore is expected to have a strong influence on evolutionary outcomes.
Drosophila yakuba displays 1.5 times as many duplications in comparison to D. simulans, as well as a 2-fold enrichment in the percentage of variants with signals of deletion in one or more copy and higher population level mutation rates. The rapid flux of duplication and deletion observed in D. yakuba has produced a wider array of standing variation, which is expected to have a significant effect on evolutionary trajectories. Drosophila yakuba will likely display not only a greater tendency toward pathogenic phenotypes associated with tandem duplicates (Hu and Worton 1992; Emerson et al. 2008) but also a greater source of standing variation that can be useful in adaptation and the development of novel traits (Conant and Wolfe 2008). Estimates of Ne in D. yakuba are higher than in D. simulans. We would thus expect greater instances of detrimental duplicates to be higher in D. simulans than in D. yakuba, but neutral mutations will collect more quickly across populations of D. yakuba due to high Ne. Hence, we suggest that the overabundance of duplicates in D. yakuba is not due to drift. Neither do we observe an excess of high-frequency variants in D. yakuba that might be suggestive of selection, especially with respect to polymorphic variants.
Based on birth–death models of gene families, D. simulans is suggested to have high rates of duplication, whereas D. yakuba showed only moderate rates of gene family evolution (Hahn et al. 2007). This may in fact be influenced by the overabundance of whole gene duplicates in D. simulans and not a reflection of genome wide mutation rates. The dichotomy between reference genomes and genome wide polymorphic variants might putatively be driven by selection for whole gene duplicates in D. simulans or mutational biases toward whole gene duplications.
Chimeric Genes
Chimeric genes are a known source of genetic novelty that are more likely to produce regulatory changes, alterations in cellular targeting and membrane bound domains, as well as selective sweeps in comparison to whole gene duplications (Rogers and Hartl 2012). Chimeric genes have been known to produce peptides with novel functions in Drosophila (Long and Langley 1993; Ranz et al. 2003; Zhang et al. 2004) and in humans (Zhang et al. 2009; Ohshima and Igarashi 2010) and many are associated with adaptive bursts of amino acid substitutions (Jones and Begun 2005; Jones et al. 2005). We observe large numbers of recently derived chimeric constructs within populations, with 222 chimeric genes or genes that recruit noncoding sequence in D. yakuba and 134 in D. simulans, even in a limited sample of 20 strains per species.
In spite of their known role in adaptation, the majority of copy number variants is thought to be detrimental (Hu and Worton 1992; Emerson et al. 2008; Cardoso-Moreira et al. 2011). Chimeric genes are associated with human cancers, and the molecular changes associated with chimera formation may contribute to their role as causative factors in human disease. The molecular changes that are facilitated by chimera formation (Rogers and Hartl 2012) likely contribute to their detrimental impacts on organisms and their role in disease as well as their potential for adaptation. We observe large numbers of chimeric genes that are identified as single variants in the population. Thus, chimeras may play the dual role of key players in adaptation to novel environments and as agents of detrimental phenotypic changes. The large amounts of standing variation observed may therefore contribute to disease alleles in populations, and proper identification is likely to be important for studies in human health.
Breakpoint Determination
Many variants have breakpoints that cannot be assembled de novo from Illumina sequences (supplementary table S30, Supplementary Material online). Yet, we observe a 96.1% confirmation rate using PacBio reads. These results imply that breakpoints are often repetitive, low complexity sequence or contain novel insertions and secondary events that are difficult to determine from paired-end read mapping alone or current naive de novo assembly methods. Hence, although paired-end Illumina read mapping is highly accurate, it cannot ascertain breakpoints to single base pair resolution in the majority of cases. Moreover, requiring breakpoint assembly to identify duplications will produce a strong ascertainment bias against up to 50% of all variants. This bias is more severe for small variants, even in Drosophila, which have compact genomes and few repeats in comparison to plants or vertebrates. Thus, short high-throughput Illumina reads orientation mapping offers an accurate but incomplete picture of variation present in the population, which can now be clarified with low coverage long read sequencing data.
Microarrays and coverage are subject to affects of misprobing, mismapping, and large amounts of noise relative to signal (supplementary fig. S4, Supplementary Material online). However, where accurate, arrays may reflect the span of duplicated segments more accurately than divergent reads alone as they would accurately reflect deletion after duplication. However, the presence of complex events such as subsequent deletions may, if not properly identified and accounted for, overestimate of the mutation rate of duplications and underestimate their frequency in the population by claiming a modified variant as an independent duplication. Here, the directional nature and spatial relationships of read pair mapping show advantages: Divergently oriented reads distinguish duplication, whereas long spanning properly oriented reads can indicate a deletion with greater clarity and properly identify subsequent modification. Identifying putative deletions in duplicated sequences requires a tight distribution of insert sizes during library preparation (supplementary table S5, Supplementary Material online) but offers a far more complete picture of variation that is segregating in populations and more accurate estimation of variant frequencies that is well worth the effort.
The X Chromosome
The D. simulans X chromosome appears to be unusual in that it contains an excess of duplications per mapped base pair in comparison to the autosomes, and an overabundance of duplications associated with long repeats. Within-genome surveys of nonsynonymous mutations in the D. simulans reference (Andolfatto et al. 2011) and large numbers of high-frequency derived variants among nonsynonymous sites and untranslated regions in D. simulans (Haddrill et al. 2008) indicate widespread selective sweeps acting on the X chromosome. Similarly, we identify an excess of variants identified at high frequency on the D. simulans X, consistent with previous work using microarrays (Cardoso-Moreira et al. 2011). In D. melanogaster, the X chromosome contains greater repetitive content (Mackay et al. 2012), displays different gene density (Adams et al. 2000), has potentially smaller population sizes (Wright 1931; Andolfatto 2001), lower levels of background selection (Charlesworth 2012), and an excess of genes involved in female-specific expression (Ranz et al. 2003) in comparison to the autosomes. Moreover, X chromosomes are subject to selfish genetic elements and often play a role in speciation (Presgraves 2008). Thus, the X chromosome may be exceptionally subject to widespread selection, and the role of tandem duplicates as responders to selective pressures deserves future exploration.
Similar patterns of high frequency variation have not been observed in D. yakuba, suggesting that evolution proceeds differently across the different species. The D. yakuba X chromosome has an excess of small duplications, which might potentially indicate selection acting against large duplications on the D. yakuba X. Tandem duplications are known to be detrimental, and given the hemizygous state of the X chromosome in males, we would expect purifying selection to act quickly on the X. The extent to which these patterns observed in D. yakuba may be driven by selection or demography remains to be seen and is an important open question deserving of future study.
Methodological Approach
Originally, high-throughput detection of copy number variation relied on microarrays or single nucleotide polymorphism chips available at the time (Dopman and Hartl 2007; Emerson et al. 2008; Ionita-Laza et al. 2009; Conrad et al. 2010) and therefore suffers from problems of misprobing, variable hybridization intensities, and dye effects, producing large amounts of noise relative to signal (Ionita-Laza et al. 2009). More recent studies have focused solely on changes in Illumina or 454 coverage analogous to changes in hybridization intensity (Sudmant et al. 2010). We have used coverage changes in combination with divergently oriented reads to identify variants using comparisons to a resequenced reference and quantile normalized coverage data to correct for stochastic coverage changes, repetitive content, GC bias, and low complexity sequence, resulting in robust variant calls. Furthermore, the common practice of retaining only variants that are present in multiple samples or at particular genotype frequencies (Conrad et al. 2010), detected by multiple independent methods (Alkan et al. 2009; Mills et al. 2011; Zichner et al. 2013), or that are larger than several kilobases (Alkan et al. 2009; Xie and Tammi 2009; Sudmant et al. 2010) can lead to severe ascertainment bias both with respect to the types of variants that are present, the estimation of their prevalence within populations and contribution to disease phenotypes, and the evolutionary impacts of duplicated sequences.
With more recent improvements in Illumina sequencing technology, we have been able to sequence strains individually to 50× or greater coverage. The use of paired-end reads in extremely high-coverage data provides clear advantages over previous work using roughly 3.6–25× with subsets sequenced to 30–42× (Sudmant et al. 2010; Mills et al. 2011; Zichner et al. 2013), or 16× (Alkan et al. 2009). The high-coverage data presented here cover the majority of the assayable genome and we exclude only a few percent of variant calls due to low coverage across strains (supplementary table S9, Supplementary Material online). Thus, we are able to identify hundreds of events that have three or more supporting divergent read pairs that might be missed at lower coverage (supplementary fig. S1, Supplementary Material online). We have additionally filtered sequences to exclude ancestral duplications, similarly to previous work in humans (Mills et al. 2011) allowing for identification of derived mutations. The result is a high-confidence data set for recently derived tandem duplications in data that effectively surveys the majority of the assayable genome. This high coverage is essential in ensuring valid results from the sole use of paired-end read orientation. However, the types of duplications that can be detected are highly dependent on sequencing library insert size. For breakpoints with large amounts of repetitive sequence, reads separated by 300 bp may not be sufficient to overcome difficulties of read mapping. Additionally, the minimum duplication size is also limited by insert size. Capturing these types of constructs using paired-end reads, especially in organisms with large amounts of repetitive content, including nested TEs, will require a more diverse range of library insert sizes, a factor that is likely to be important for surveys of larger, more repetitive genomes.
We are able to identify copy number variants as small as 66 bp using divergently oriented paired-end reads, even in cases where nucleotide divergence between paralogs or partially repetitive sequence might otherwise complicate their discovery through coverage changes or split read mapping using short reads. Divergently oriented reads are additionally reliable to detect duplications in regions where distributions of coverage are too irregular to allow for automated detection of duplicates. We observe a high 96.1% confirmation rate of variants among four sample strains of D. yakuba using PacBio reads up to 24 kb in length, suggesting that paired-end reads in high coverage genomic sequencing will grossly outperform previous methods which show high false positive and false negative rates. Moreover, the use of the newly annotated D. simulans genome (Hu et al. 2013) based on a single isofemale line will result in improved accuracy in comparison to previous studies in D. simulans (Cardoso-Moreira et al. 2011). The general principles of the paired-end approach should be broadly applicable and similar methods already have been used to identify chromosomal inversions in natural populations (Corbett-Detig et al. 2012). PacBio reads can confirm structural variants with high rates of success, even given low coverage and nucleotide high error rates (Huddleston et al. 2014) and we have extended this approach to our genome wide survey. However, the ambiguity of split read mapping in the face of repetitive elements can still complicate de novo duplicate discovery using split read mapping of long reads. Furthermore, for the present, generating high-coverage genomic sequencing equivalent to that of our paired-end Illumina data is not cost-effective. However, this technology and similar long read approaches are likely to offer advantages in confirming or discovering structural variants such as tandem duplications, as well as de novo assembly, that is, worth future exploration.
We are able to identify and confirm a large number of complex gene structures, such as chimeric genes, recruitment of adjacent noncoding sequence, potential coding sequence disruption, and potential selective silencing of expression. These complex mutations are often associated with cancer and other diseases (Ionita-Laza et al. 2009; Inaki and Liu 2012) and are most likely to cause pathogenic outcomes. Hence, the methods described here will be broadly applicable in genome-wide association studies and clinical studies as well as in evolutionary genetics of nonmodel systems where next generation sequencing has so recently made population genomics readily tractable.
Conclusions
Here, we have described the landscape of standing variation for tandem duplications in isofemale lines derived from natural populations of D. yakuba and D. simulans with high accuracy. The resulting portrait of hundreds to thousands of variants, including large numbers of complex breakpoints, modifying deletions, cases of recruited noncoding sequence, and dozens of chimeric genes per species reveals a rich substrate of segregating variation across populations. We show that although the span of duplications across the genome is quite limited duplicates can induce secondary mutations and result in dynamic changes, resulting in greater variation across mutated sites that offers more abundant variation for use in adaptation than has been previously portrayed. The ways in which this variation influences adaptive evolution and produces molecular changes will clarify the extent to which mutational profiles define evolutionary outcomes and the ways in which molecular changes associated with tandem duplications serve as causative factors in disease.
Materials and Methods
Population Samples
We surveyed variation in ten lines of D. yakuba from Nairobi, Kenya and ten from Nguti, Cameroon (collected by P. Andolfatto 2002) as well as ten lines of D. simulans from Madagascar (collected by B. Ballard in 2002) and ten strains from Nairobi, Kenya (collected by P. Andolfatto in 2006). Flies from these isofemale lines (i.e., descendants from a single wild-caught female) were inbred in the lab for 9–12 generations of sibling mating. These should provide effectively haploid samples of allelic variation representative of natural populations.
In addition to the 20 inbred lines derived from wild-caught flies, we also sequenced the reference strains for each species. For D. simulans, the reference strain is the w501 stock (UCSD stock center 14021-0251.011), whose sequence is described in Hu et al. (2013). For D. yakuba, the reference strain is UCSD stock center 14021-0261.01, and the genome sequence is previously described in Drosophila Twelve Genomes Consortium (2007). The majority of the wild-caught strains and the D. yakuba reference stock were sequenced with three lanes of paired-end sequencing at the UC Irvine Genomics High Throughput Facility (http://dmaf.biochem.uci.edu, last accessed April 2014). The sequencing of the D. simulans reference strain was described in Hu et al. (2013). The number of lanes and read lengths per lane are summarized in supplementary tables S1 and S2, Supplementary Material online.
Alignment to Reference
The sequencing reads were aligned to the appropriate reference genome (Drosophila Twelve Genomes Consortium 2007; Hu et al. 2013) using bwa version 0.5.9 (Li and Durbin 2009) with the following parameters, bwa aln −l 13 −m 50000000 −R 5000. The resulting paired-end mappings were resolved via the “sampe” module of bwa (bwa sampe −a 5000 −N 5000 −n 500), and the output was sorted and converted into a “bam” file using samtools version 0.1.18 (Li et al. 2009). In the alignment and resolution commands, −l is the hash sized used for seeding alignments, and the −R, −a, −N, and −n refer to how many alignments are recorded for reads mapping to multiple locations in the reference. After the paired-end mapping resolution, the bam files from each lane of sequencing were merged into a single bam file sorted according to position along the reference genome. A second bam file, sorted by read name, was then created for use as input into our clustering software.
Clustering Abnormal Mapping Events
Tandem duplications should be readily apparent among mapped reads as sequenced read pairs that map in divergent orientations (Tuzun et al. 2005; Cridland and Thornton 2010; Mills et al. 2011; Zichner et al. 2013), provided that tandem duplications with respect to a reference genome result in a single novel junction. Figure 2 shows a putative genomic sample that contains a tandem duplication of a gene that was used to generate paired-end sequencing reads. We allowed for up to two mismatches within mapped reads in order to capture divergent read calls in sample strains that have moderate numbers of nucleotide differences. Reads were required to map uniquely, and so if duplication breakpoints contain entirely repetitive sequences with no divergence across copies in the genome, they will not be found. These limitations will, however, have minimal effects (see supplementary text S1, Supplementary Material online).
Sets of read pairs in the same strain that are located within the 99.9th quantile of the mapping distance between properly mapped pairs from one another were clustered together into a single duplication. In practice, this threshold distance is roughly 1 kb. Tandem duplications were identified as regions where three or more divergently oriented read pairs cluster to the same location in a single strain. Allowing fewer divergent reads leads to a large number of false positive duplication calls due to cloning and sequencing errors. Further detail is offered in supplementary text S1, Supplementary Material online.
PacBio Alignment and Analysis
FASTQ files of PacBio reads were aligned to the D. yakuba reference (Drosophila Twelve Genomes Consortium 2007) using blasr (Chaisson and Tesler 2012), available from https://github.com/PacificBiosciences/blasr (last accessed October 2013), with default options and storing the resulting alignments in a bam file. Alignments from regions within 1 kb of putative tandem duplications called using short read data (divergent read orientation plus an increase in coverage) were extracted from the bam files using samtools 0.1.18 (Li et al. 2009). Reads falling within these regions were then pulled and realigned to the reference using a BLASTn (Altschul et al. 1990) with low complexity filters turned off (−F F) at an E value cutoff of 0.1 to allow for short alignments given the high error rate of PacBio sequencing. Alignment using BLASTn proved important because it revealed cases where the bam files resulting from blasr alignments failed to record secondary hits for a read, especially in cases where alignments are on the order of hundreds of base pairs. Here, confirmation benefits from long sequences which can anchor reads uniquely to a region, producing greater confidence in split read alignments. Variants were considered confirmed whether two segments of a single PacBio subread which do not overlap by more than 20% align to overlapping sections of the reference (supplementary fig. S5B, Supplementary Material online) or whether a single read aligns in split formation with the downstream end of the read aligning to an upstream region of the reference (supplementary fig. S5A and C–E, Supplementary Material online). An event was considered not confirmed whether there were no long reads showing any of the alignment patterns in supplementary figure S5, Supplementary Material online, within 1 kb of the variant. Variants were considered definite false positives if clearly contradicted by at least one read spanning the entire putatively duplicated region as defined by Illumina read mapping and adjacent reference. Some of these unconfirmed variants may be due to low clone coverage in the region or lack of sufficient read lengths rather than false positives.
HMM and Coverage Changes
To detect increases and decreases relative to reference resequencing, we quantile normalized coverage for each strain in R so that coverage displayed an equal median and variance across all strains (Bolstad et al. 2003). Such normalization renders tests of differing coverage robust in the face of differing sequence depth across samples or across sites and is essential for reliable confirmation of tandem duplicate calls (Bolstad et al. 2003).
We developed a hidden Markov model (HMM) to identify statistically significant increases in coverage at duplicated sites. In the HMM, hidden states are defined by copy number and they act to effect differential emission probabilities for the observed outcomes of coverage depth at duplicated or nonduplicated sites. We modeled differences in coverage for sample strains relative to the reference as the difference in two normal distributed random variables each with a mean and variance corresponding to the observed mean and variance in the reference in the given window. Detailed methods of the HMM and decoding are provided in supplementary text S1, Supplementary Material online.
Deletions and Complex Duplication Events
In order to determine the extent to which secondary deletions, incomplete duplications, or short-range dispersed duplicates result in complex structures that are not representative of classic duplications, we identified long-spanning read pairs that lie within tandem duplications. We identified read pairs with an estimated template length of 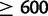 bp, corresponding to roughly the 99.9th percentile of fragment lengths in the reference genome (supplementary table S5, Supplementary Material online). Long-spanning reads whose end points lie within 200 bp of one another and which are fully contained within the duplication endpoints were clustered together. Clusters supported by five or more read pairs (corresponding to a putative P value of
bp, corresponding to roughly the 99.9th percentile of fragment lengths in the reference genome (supplementary table S5, Supplementary Material online). Long-spanning reads whose end points lie within 200 bp of one another and which are fully contained within the duplication endpoints were clustered together. Clusters supported by five or more read pairs (corresponding to a putative P value of  ) were recorded as signs of deletion or other complex rearrangement.
) were recorded as signs of deletion or other complex rearrangement.
Sample Frequency of Variants
Complex mutations that are present in two different strains and have divergent reads spanning regions on both the 5 and 3
and 3 ends that fall within 100 bp of one another in the two strains are considered to be equivalent duplications. Although divergently oriented reads reliably identify duplications, the true boundaries of the duplication may differ from the divergent read spans by several bases, especially in cases where repetitive or low complexity sequence lies at breakpoints, a complication which is not addressed in recent work (Mills et al. 2011; Zichner et al. 2013). For each duplication, the minimum of divergent read starts and the maximum of divergent read stops across all strains were recorded to indicate the span of the duplication. These breakpoints were assembled and confirmed in silico using phrap and lastz (supplementary text S1, Supplementary Material online).
ends that fall within 100 bp of one another in the two strains are considered to be equivalent duplications. Although divergently oriented reads reliably identify duplications, the true boundaries of the duplication may differ from the divergent read spans by several bases, especially in cases where repetitive or low complexity sequence lies at breakpoints, a complication which is not addressed in recent work (Mills et al. 2011; Zichner et al. 2013). For each duplication, the minimum of divergent read starts and the maximum of divergent read stops across all strains were recorded to indicate the span of the duplication. These breakpoints were assembled and confirmed in silico using phrap and lastz (supplementary text S1, Supplementary Material online).
In order to correct frequencies for the effects of false negatives, we used a combination of coverage and divergently oriented reads to identify tandem duplicates in additional strains. For each duplication present based on three or more pairs divergent reads in at least one strain, a different strain which had at least one divergently oriented read pair and displayed 2-fold or greater coverage increases as determined by the HMM across at least 75% of the duplication’s span was also recorded as having that particular duplication as well.
We retained tandem duplicates with divergent read pairs where the minimum start and the maximum stop after clustering were <25 kb apart. Although there may be some tandem duplications larger than 25 kb, divergent read pairs at a greater distance are substantially less likely to display 2-fold coverage changes across the entire span and are therefore more consistent with translocations within chromosomes (supplementary table S31, Supplementary Material online). Unannotated duplications in the reference are likely to be biased toward specific sizes, GO classes, or genomic locations and are likely to artificially influence statistical tests. We removed duplications that were also identified in the reference, and did not include these in downstream analyses of duplication sizes or numbers, GO, or site frequency spectra.
Polarizing Ancestral State
All tandem duplications identified are polymorphic in populations and are therefore expected to be extremely young. However, tandem duplications may be identified in sample strains relative to the references if they are new mutations in the sample or if they represent ancestral sequence that has returned to single copy in the reference through deletion. In order to identify duplications that represent the ancestral state, we pulled reference sequence corresponding to the maximum duplicate span and ran a BLASTn comparison of the sequence against the D. melanogaster, D. erecta, and D. yakuba or D. simulans reference genomes at an E value cutoff of  .
.
Ancestral tandem duplications were defined as any segment that has two hits on the same chromosome of the given reference that lie within 200 kb of one another, excluding unlocalized sequence and heterochromatin annotations where assembly and annotations are uncertain. Ancestral duplications that are shared across species should be separated by moderate numbers of nucleotide differences, and therefore are expected to be correctly assembled across outgroups. Hits must have at least 85% nucleotide identity and must span at least 80% of the contig spanned by divergently oriented reads in the sample. Based on these requirements, we removed 8.1% of duplications in D. simulans and 3.3% in D. yakuba, suggesting that the vast majority of duplicates identified are recently derived, as expected. Extended methods as well as detailed description of sequence data and confirmation are provided in supplementary text S1, Supplementary Material online. All data files are available at molpopogen.org/Data (last accessed April 11, 2014). Aligned bam files were deposited in the National Institutes of Health Short Read Archive under accession numbers SRP040290 and SRP029453. Sequenced stocks were deposited in the University of California, San Diego (UCSD) stock center with stock numbers 14021-0261.38–14021-0261.51 and 14021-0251.293–14021-0251.311.
TE Annotation and Repetitive Content
TEs were initially identified in the D. simulans and D. yakuba reference sequences by identifying all read pairs where one member of the pair aligned uniquely to the reference and the other member of the pair aligned multiply. This has been previously demonstrated to be an indication of a TE breakpoint (Mackay et al. 2012; Cridland et al. 2013). All locations to which these multiply aligning reads align were recorded and a fasta file of these putative TE locations was generated. Putative TE sequences were then aligned to the set of annotated TEs in version 5 of the D. melanogaster reference downloaded from flybase (www.flybase.org, last accessed December 2011) using tblastx (Altschul et al. 1990) with the following parameters (−f 999 −F ”” −e  −m 8). Regions of the reference sequence that aligned to a D. melanogaster TE with an E value of
−m 8). Regions of the reference sequence that aligned to a D. melanogaster TE with an E value of 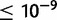 were kept and annotated as TEs. We then extracted 500 bp to either side of the annotated regions of the reference and aligned these regions to the set of D. melanogaster TEs, as earlier. This procedure was performed twice to capture the full length of TE sequence in the reference genomes.
were kept and annotated as TEs. We then extracted 500 bp to either side of the annotated regions of the reference and aligned these regions to the set of D. melanogaster TEs, as earlier. This procedure was performed twice to capture the full length of TE sequence in the reference genomes.
Once TEs were annotated in the reference genomes, TEs were detected in the 20 sample genomes (Mackay et al. 2012) which include both TE presence and TE absence calls. Briefly, initial TE detection was done by identifying all read pairs where one member of the pair aligned uniquely to the reference and the other member aligned to any known TE sequence were identified. Unique reads were clustered if they aligned to the same strand of the same chromosome, within a given threshold distance. This threshold was defined as the 99th quantile of mapping distances observed for uniquely mapping read pairs that are properly paired and lie in the expected orientation on opposite strands. At least three read pairs from each of the left and right estimates, in the correct orientation and correct strand, were required to indicate a TE. Phrap (version 1.090518) (Ewing and Green 1998) with the following parameters (−forcelevel 10 −minscore 10 −minmatch 10) was used to reassemble the local area around the TE insertion breakpoint. Contigs were classified according to TE family based on alignments in a BLASTn search (Altschul et al. 1990).
Following the initial TE detection phase we examined each position where a TE was identified, including TEs identified in the appropriate reference, in each other line in that species. At this stage, we were able to both identify TEs which had previously been missed by our pipeline as well as to make absence calls by reconstructing a contig that spans the TE insertion location. Repetitive content independent of TEs was defined using an all-by-all BLASTn comparison (Altschul et al. 1990) of sequence 500-bp upstream and downstream of duplication coordinates in the D. yakuba and D. simulans references at an E value 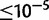 with no filter for low complexity or repetitive sequences. We used a resampling approach to identify overrepresentation of duplications associated with direct repeats. We performed 10,000,000 replicates choosing the same number of tandem duplications at random and determining whether an equal or greater number was identified on the X chromosome.
with no filter for low complexity or repetitive sequences. We used a resampling approach to identify overrepresentation of duplications associated with direct repeats. We performed 10,000,000 replicates choosing the same number of tandem duplications at random and determining whether an equal or greater number was identified on the X chromosome.
Tandem repeats which might putatively facilitate ectopic recombination may hinder identification of tandem duplicates if repeat sequences are identical. We performed an all-by-all BLASTn of all chromosomes for each of the reference genomes at an E value of 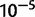 with low complexity filters turned off (−F F). Ignoring identical self hits, we identified all directly repeated sequences with >99.5% nucleotide identity which are >300 bp in length and which lie within 25 kb of one another, in accordance with the criteria for identifying duplicates.
with low complexity filters turned off (−F F). Ignoring identical self hits, we identified all directly repeated sequences with >99.5% nucleotide identity which are >300 bp in length and which lie within 25 kb of one another, in accordance with the criteria for identifying duplicates.
Identifying Duplicated Coding Sequence
Gene duplications were defined as any divergent read calls whose maximum span across all lines overlaps with the annotated CDS coordinates. Drosophila yakuba CDS annotations were based on flybase release D. yakuba r.1.3. Gene annotations for the recent reassembly of the D. simulans reference were produced by aligning all D. melanogaster CDS to the D. simulans reference in a tblastx. Percent coverage of the CDS was defined based on the portion of the corresponding genomic sequence from start to stop that was covered by the maximum span of divergent read calls across all strains.
Gene Ontology
We used DAVID GO analysis software (http://david.abcc.ncifcrf.gov/, last accessed March 2013) to determine whether any functional categories were overrepresented at duplicated genes. Functional data for D. yakuba and D. simulans are not readily available in many cases, and thus we identified functional classes in the D. melanogaster orthologs as classified in Flybase. GO clustering threshold was set to low and significance was defined using a cutoff of EASE 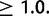 The DAVID clustering software uses Fuzzy Heuristic Partitioning to identify genes with related functional terms at all levels of GO from cellular processes to known phenotypes.
The DAVID clustering software uses Fuzzy Heuristic Partitioning to identify genes with related functional terms at all levels of GO from cellular processes to known phenotypes.
Differences among Chromosomes
We calculated the size of duplications that span <25 kb in each sample strain, excluding duplications identified in the reference, as incorrectly assembled duplicates are likely be biased toward repetitive and low complexity sequence. Significant differences in duplication sizes were identified using analysis of variance (ANOVA) and Tukey’s HSD test on the log normalized distribution of duplication sizes by chromosome.
We calculated the number of duplications that span <25 kb on each major chromosomal arm for each sample strain, excluding duplications identified in the reference. The number of duplications was then normalized by the number of mapped bases in the reference to adjust for different chromosome sizes and coverage. Differences in the number of duplications per base pair were identified using ANOVA and Tukey’s HSD test. There was no significant difference in the number of duplications present across lines.
Supplementary Material
Supplementary text S1, tables S1–S31, and figures S1–S5 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Elizabeth G. King, Anthony D. Long, and Alexis S. Harrison, and Trevor Bedford for helpful discussions, as well as Portola Coffee Labs for a supportive writing environment. J. J. Emerson gave valuable advice on the use of lastz and the University of California–San Cruz (UCSC) toolkit. B. Ballard shared D. simulans fly stocks collected from Madagascar. The authors also thank several anonymous reviewers whose comments substantially improved the manuscript. This work is supported by a National Institute of General Medical Sciences at the National Institute of Health Ruth Kirschtein National Research Service Award F32-GM099377 to R.L.R. Research funds were provided by National Institute of General Medical Sciences at the National Institute of Health grant R01-GM085183 to K.R.T. and R01-GM083228 to P.A. All sequencing and PacBio library preparations were performed at the UC Irvine High Throughput Genomics facility, which is supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA062203. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Andolfatto P. Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Mol Biol Evol. 2001;18:279–290. doi: 10.1093/oxfordjournals.molbev.a003804. [DOI] [PubMed] [Google Scholar]
- Andolfatto P, Wong KM, Bachtrog D. Effective population size and the efficacy of selection on the X chromosomes of two closely related Drosophila species. Genome Biol Evol. 2011;3:114–128. doi: 10.1093/gbe/evq086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D, Thornton K, Clark A, Andolfatto P. Extensive introgression of mitochondrial DNA relative to nuclear genes in the Drosophila yakuba species group. Evolution. 2006;60:292–302. [PubMed] [Google Scholar]
- Barrett RD, Schluter D. Adaptation from standing genetic variation. Trends Ecol Evol (Amst). 2008;23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Cardoso-Moreira M, Arguello JR, Clark AG. Mutation spectrum of Drosophila CNVs revealed by breakpoint sequencing. Genome Biol. 2012;13:R119. doi: 10.1186/gb-2012-13-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso-Moreira M, Emerson JJ, Clark AG, Long M. Drosophila duplication hotspots are associated with late-replicating regions of the genome. PLoS Genet. 2011;7:e1002340. doi: 10.1371/journal.pgen.1002340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics. 2012;13:238. doi: 10.1186/1471-2105-13-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The role of background selection in shaping patterns of molecular evolution and variation: evidence from variability on the Drosophila X chromosome. Genetics. 2012;191:233–246. doi: 10.1534/genetics.111.138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Coyne J, Barton N. The relative rates of evolution of sex chromosomes and autosomes. Am Nat. 1987;130:113–146. [Google Scholar]
- Conant GC, Wolfe KH. Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet. 2008;9:938–950. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett-Detig RB, Cardeno C, Langley CH. Sequence-based detection and breakpoint assembly of polymorphic inversions. Genetics. 2012;192:131–137. doi: 10.1534/genetics.112.141622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cridland JM, Macdonald SJ, Long AD, Thornton KR. Abundance and distribution of transposable elements in two Drosophila QTL mapping resources. Mol Biol Evol. 2013;30:2311–2327. doi: 10.1093/molbev/mst129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cridland JM, Thornton KR. Validation of rearrangement break points identified by paired-end sequencing in natural populations of Drosophila melanogaster. Genome Biol Evol. 2010;2:83–101. doi: 10.1093/gbe/evq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopman EB, Hartl DL. A portrait of copy-number polymorphism in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2007;104:19920–19925. doi: 10.1073/pnas.0709888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila Twelve Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Emerson JJ, Cardoso-Moreira M, Borevitz JO, Long M. Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science. 2008;320:1629–1631. doi: 10.1126/science.1158078. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD, Smith NG, Gaffney D. Quantifying the slightly deleterious mutation model of molecular evolution. Mol Biol Evol. 2002;19:2142–2149. doi: 10.1093/oxfordjournals.molbev.a004039. [DOI] [PubMed] [Google Scholar]
- Haddrill PR, Bachtrog D, Andolfatto P. Positive and negative selection on noncoding DNA in Drosophila simulans. Mol Biol Evol. 2008;25:1825–1834. doi: 10.1093/molbev/msn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MW, Han MV, Han SG. Gene family evolution across 12 Drosophila genomes. PLoS Genet. 2007;3:e197. doi: 10.1371/journal.pgen.0030197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu TT, Eisen MB, Thornton KR, Andolfatto P. A second generation assembly of the Drosophila simulans genome provides new insights into patterns of lineage-specific divergence. Genome Res. 2013;23:89–98. doi: 10.1101/gr.141689.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Worton RG. Partial gene duplication as a cause of human disease. Hum Mutat. 1992;1:3–12. doi: 10.1002/humu.1380010103. [DOI] [PubMed] [Google Scholar]
- Huddleston J, Ranade S, Malig M, Antonacci F, Chaisson M, Hon L, Sudmant PH, Graves TA, Alkan C, Dennis MY, et al. Reconstructing complex regions of genomes using long-read sequencing technology. Genome Res. 2014;24(4):688–696. doi: 10.1101/gr.168450.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaki K, Liu ET. Structural mutations in cancer: mechanistic and functional insights. Trends Genet. 2012;28:550–559. doi: 10.1016/j.tig.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Ionita-Laza I, Rogers AJ, Lange C, Raby BA, Lee C. Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis. Genomics. 2009;93:22–26. doi: 10.1016/j.ygeno.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CD, Begun DJ. Parallel evolution of chimeric fusion genes. Proc Natl Acad Sci U S A. 2005;102:11373–11378. doi: 10.1073/pnas.0503528102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CD, Custer AW, Begun DJ. Origin and evolution of a chimeric fusion gene in Drosophila subobscura, D. madeirensis and D. guanche. Genetics. 2005;170:207–219. doi: 10.1534/genetics.104.037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney HM, Kirkpatrick DT, Gerton JL, Petes TD. Meiotic recombination involving heterozygous large insertions in Saccharomyces cerevisiae: formation and repair of large, unpaired DNA loops. Genetics. 2001;158:1457–1476. doi: 10.1093/genetics/158.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarro B, Clark A. Rapid evolution of innate immune response genes. In: Singh RS, Kulathinal RJ, editors. Rapidly evolving genes and genetic systems. 2012. Oxford (United Kingdom): Oxford University Press. p. 203–208. [Google Scholar]
- Lee YC, Reinhardt JA. Widespread polymorphism in the positions of stop codons in Drosophila melanogaster. Genome Biol Evol. 2012;4:533–549. doi: 10.1093/gbe/evr113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Conte N, Skarnes WC, Bradley A. Extensive genomic copy number variation in embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105:17453–17456. doi: 10.1073/pnas.0805638105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JK, Simmons MJ. Gross chromosome rearrangements mediated by transposable elements in Drosophila melanogaster. Bioessays. 1994;16:269–275. doi: 10.1002/bies.950160410. [DOI] [PubMed] [Google Scholar]
- Long M, Langley CH. Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila. Science. 1993;260:91–95. doi: 10.1126/science.7682012. [DOI] [PubMed] [Google Scholar]
- Mackay TF, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, et al. The Drosophila melanogaster Genetic Reference Panel. Nature. 2012;482:173–178. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima K, Igarashi K. Inference for the initial stage of domain shuffling: tracing the evolutionary fate of the PIPSL retrogene in hominoids. Mol Biol Evol. 2010;27:2522–2533. doi: 10.1093/molbev/msq138. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Lozovskaya ER, Hartl DL. High intrinsic rate of DNA loss in Drosophila. Nature. 1996;384:346–349. doi: 10.1038/384346a0. [DOI] [PubMed] [Google Scholar]
- Presgraves DC. Sex chromosomes and speciation in Drosophila. Trends Genet. 2008;24:336–343. doi: 10.1016/j.tig.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science. 2003;300:1742–1745. doi: 10.1126/science.1085881. [DOI] [PubMed] [Google Scholar]
- Rogers RL, Bedford T, Hartl DL. Formation and longevity of chimeric and duplicate genes in Drosophila melanogaster. Genetics. 2009;181:313–322. doi: 10.1534/genetics.108.091538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, Hartl DL. Chimeric genes as a source of rapid evolution in Drosophila melanogaster. Mol Biol Evol. 2012;29:517–529. doi: 10.1093/molbev/msr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SA, Hartl DL. Population genetics of polymorphism and divergence. Genetics. 1992;132:1161–1176. doi: 10.1093/genetics/132.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Houle D, Lynch M, Hahn MW. Rates and genomic consequences of spontaneous mutational events in Drosophila melanogaster. Genetics. 2013;194:937–954. doi: 10.1534/genetics.113.151670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J. Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- Wong A, Wolfner M. Evolution of Drosophila seminal proteins and their networks. In: Singh RS, Kulathinal RJ, editors. Rapidly evolving genes and genetic systems. 2012. . Oxford (United Kingdom): Oxford University Press. p. 144–152. [Google Scholar]
- Wright S. Evolution in Mendelian Populations. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009;10:80. doi: 10.1186/1471-2105-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dean AM, Brunet F, Long M. Evolving protein functional diversity in new genes of Drosophila. Proc Natl Acad Sci U S A. 2004;101:16246–16250. doi: 10.1073/pnas.0407066101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lu S, Zhao S, Zheng X, Long M, Wei L. Positive selection for the male functionality of a co-retroposed gene in the hominoids. BMC Evol Biol. 2009;9:252. doi: 10.1186/1471-2148-9-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Zhang G, Zhang Y, Xu S, Zhao R, Zhan Z, Li X, Ding Y, Yang S, Wang W. On the origin of new genes in Drosophila. Genome Res. 2008;18:1446–1455. doi: 10.1101/gr.076588.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zichner T, Garfield DA, Rausch T, Stutz AM, Cannavo E, Braun M, Furlong EE, Korbel JO. Impact of genomic structural variation in Drosophila melanogaster based on population-scale sequencing. Genome Res. 2013;23:568–579. doi: 10.1101/gr.142646.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.