

Abstract
Summary: Here we present the open-source R/Bioconductor software package BEAT (BS-Seq Epimutation Analysis Toolkit). It implements all bioinformatics steps required for the quantitative high-resolution analysis of DNA methylation patterns from bisulfite sequencing data, including the detection of regional epimutation events, i.e. loss or gain of DNA methylation at CG positions relative to a reference. Using a binomial mixture model, the BEAT package aggregates methylation counts per genomic position, thereby compensating for low coverage, incomplete conversion and sequencing errors.
Availability and implementation: BEAT is freely available as part of Bioconductor at www.bioconductor.org/packages/devel/bioc/html/BEAT.html. The package is distributed under the GNU Lesser General Public License 3.0.
Contact: akman@mpipz.mpg.de
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Bisulfite sequencing (BS-Seq) is a sequence-based method to accurately detect DNA methylation at specific loci, which involves treating DNA with sodium bisulfite (Frommer et al., 1992). The method is based on bisulfite conversion of unmethylated cytosines into uracil and has become a standard in DNA methylation profiling. Its advantage is accuracy, as the degree of methylation at each cytosine can be quantified with great precision (Fraga and Esteller, 2002). More recently, bisulfite sequencing has been applied in a genome-wide manner, which requires advanced computational analysis for determining DNA methylation patterns and changes therein. Thus far, such analysis has been limited to the comparison of individual CpG sites between samples. However, because of the bisulfite conversion of cytosines into uracils, and eventually thymines, sequence complexity is much reduced, with no account for incomplete conversion and/or sequencing errors. Bisulfite sequencing also often suffers from low coverage. Here, we present BS-Seq Epimutation Analysis Toolkit (BEAT), a novel tool for analyzing bisulfite-converted DNA sequences. To overcome the aforementioned limitations in the estimation of methylation rates, BEAT aggregates data from consecutive cytosines into regions by using a Bayesian binomial-beta mixture model. The model is derived, described in detail and evaluated in our Supplementary Material. For each region, it calculates a posterior methylation probability distribution that can be used for the comparison of DNA methylation between samples. Anticipating technological progress in the DNA methylation field, BEAT includes an error model adapted to single-cell BS-Seq data.
2 USAGE AND APPLICATION
The BEAT package can be used for estimating the true methylation levels of BS-Seq samples and for the calling of epimutations, which are differences in methylation states of a region in the genome. Pooling single CG counts into regions can be done with the function positions_to_regions, which reads a comma separated file and outputs a data.frame. The latter is the input to the BEAT model, which can be easily accessed via the core function generate_results.
We assume that all counts at a single CG position were obtained from pairwise different bisulfite-converted DNA templates, representing independent observations. Some of the most important parameters of our model are the false-positive and false-negative conversion rates. Let the false-positive rate 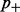 be the global rate of false methylation counts, which is identical to the non-conversion rate of non-methylated cytosines. Conversely, the false-negative rate
be the global rate of false methylation counts, which is identical to the non-conversion rate of non-methylated cytosines. Conversely, the false-negative rate 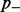 is the global rate of false non-methylation counts, which is identical to the inappropriate conversion rate of methylated cytosines. One can find an upper bound for
is the global rate of false non-methylation counts, which is identical to the inappropriate conversion rate of methylated cytosines. One can find an upper bound for 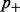 by considering all methylation counts at non-CG positions as false-positive results (resulting from non-conversion of presumably unmethylated cytosines). In the literature, false-negative rates were not described, therefore we recommend a conservative estimate of
by considering all methylation counts at non-CG positions as false-positive results (resulting from non-conversion of presumably unmethylated cytosines). In the literature, false-negative rates were not described, therefore we recommend a conservative estimate of 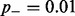 .
.
With the resulting estimates of methylation levels and methylation status from the model, which are returned as a data.frame, the function epimutation_calls can then determine epimutation differences between two samples and compute rates for demethylating and methylating epimutations. A sample use of BEAT follows. For a more detailed explanation of the required objects, we refer to the package vignette. Figure 1 graphically illustrates the output of BEAT. The R-objects required for this analysis are included in the BEAT package.
Fig. 1.
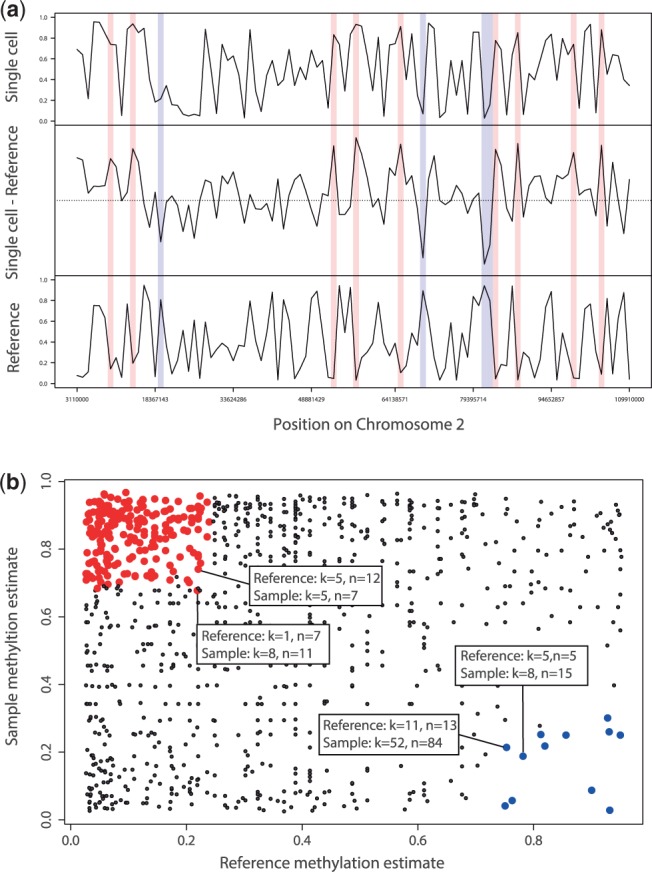
(a) Methylation estimates and epimutation calls on a DNA segment. For all regions with sufficient read coverage, the black curves show the methylation estimates for a single cell sample (top), a reference sample (bottom) and their difference (middle). Regions with methylating epimutations are marked in red, while regions with demethylating epimutations are marked in blue. Samples used for our analysis in this article were obtained from neuronal cells of young mice (data unpublished). (b) Scatterplot of methylation estimates of a multi-cell reference sample (x-axis) versus those of a sample (y-axis) for all common regions with sufficient coverage. Each dot represents a single region that is covered by both samples. Red dots indicate methylating epimutations in the sample, while blue dots indicate demethylating epimutations in the sample. Four dots representing exemplary regions with epimutations at the corresponding boundary value ranges for demethylating and methylating epimutations have been annotated with their values of methylated (k) and total (n) counts. Note that there exists no boundary line separating the red and the blue region because our Bayesian model assigns different methylation estimates to tuples (k1, n1), (k2, n2) with equal empirical methylation level k1/n1 = k2/n2
The most important step is the setting of the user-defined parameters.
# Load sample data
data(BEAT)
# Initialize working path
localpath <- system.file(’extdata’, package =’BEAT’)
# Set sample names and prefix of data files
> sampNames <- c(’reference’,’sample’)
# Set reference vs. non-ref status per sample
> is.reference <- c(TRUE, FALSE)
# Set BS-conversion rate per sample
> pplus <- c(0.2, 0.5)
> convrates <- 1 - pplus
# Create parameter object
> params <- makeParams(localpath, sampNames,
convrates, is.reference, pminus = 0.2,
regionSize = 10 000, minCounts = 5)
# Pool CG positions into genomic regions
> positions_to_regions(params)
# Model methylation levels and -status
> generate_results(params)
# Call epimutations
> epiCalls <- epimutation_calls(params)
3 CONCLUSION
The BEAT package delivers methods for the estimation of methylation levels, methylation status and for calling epimutation events in a two-sample comparison. To our knowledge, it is the first tool providing a rigid statistical model for handling BS-Seq samples. It has an in-built correction for conversion errors and is therefore tailored to the analysis of BS-Seq samples with possibly different BS-conversion rates.
Funding: NIH grants (AG17242, AG034421, AG032117 and SENS Foundation to J.V. and S.G.). Jeff Schell professorship for Computational Biology grant (SFB680) from the Deutsche Forschungsgemeinschaft (to A.T.). BMBF e:Bio grant (to K.A.).
Conflict of Interest: none declared
Supplementary Material
REFERENCES
- Fraga M, Esteller M. DNA methylation: a profile of methods and applications. Biotechniques. 2002;33:632, 634, 636–649. doi: 10.2144/02333rv01. [DOI] [PubMed] [Google Scholar]
- Frommer M, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, et al. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat. Methods. 2010;7:133–136. doi: 10.1038/nmeth.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.