

Abstract
Hypsizygus marmoreus is an industrialized edible mushroom. In the present paper, the genetic diversity among 20 strains collected from different places of China was evaluated by target region amplification polymorphism (TRAP) analysis; the common fragment of TRAPs was sequenced and analyzed. Six fixed primers were designed based on the analysis of H. marmoreus sequences from GenBank database. The genomic DNA extracted from H. marmoreus was amplified with 28 TRAP primer combinations, which generated 287 bands. The average of amplified bands per primer was 10.27 (mean polymorphism is 69.73%). The polymorphism information content (PIC) value for TRAPs ranged from 0.32 to 0.50 (mean PIC value per TRAP primer combination is 0.48), which indicated a medium level of polymorphism among the strains. A total of 36 sequences were obtained from TRAP amplification. Half of these sequences could encode the known or unknown proteins. According to the phylogenetic analysis based on TRAP result, the 20 strains of H. marmoreus were classified into two main groups.
1. Introduction
Hypsizygus marmoreus (Peck) H. E. Bigelow is an industrialized edible mushroom [1] and has become one of the most popular edible and medicinal mushrooms in East Asia because of its rich nutrition and medicinal value [2–7]. Quite a lot of studies on this edible fungus were focused on the nutrient substances, physiological characters, and healthcare functions for human, but genetic researches were not sufficient. The characteristics of strains are very important for the production output and quality of the cultivated mushrooms. Molecular markers can directly reflect the genetic polymorphisms in DNA levels and have been widely used to analyze the genetic diversities, construct the genetic linkage maps, identify strains, and assist in breeding [8, 9]. In the previous studies, the amplified fragment length polymorphism (AFLP) markers and sequence characterized amplified region (SCAR) markers were developed to discriminate the strains and analyse the genetic divergences among the strains of H. marmoreus [10, 11]. Although a few studies on molecular markers of H. marmoreus have been carried out, the tested strains and the applied analysis methods were limited [12–14].
Target region amplification polymorphism (TRAP) was developed on the basis of sequence-relation amplification polymorphism (SRAP) [15]. Compared with other marker techniques, TRAP has the advantages of easy operation, high stability and repeatability, strong targeting ability, and rich polymorphism information [16, 17]. It had been used to analyze the genetic diversity of Lentinula edodes [18], construct the genetic linkage map of Lentinula edodes [9], and analyze the relationship between phenotypic and genotypic traits of Auricularia auricula-judae [8]. However, it has not been applied in H. marmoreus.
In this study, the strains of H. marmoreus were extensively collected in China; the genetic diversities were evaluated with TRAP technique, and the genes of serine protease and fungal intracellular laccase were sequenced [19, 20]. The result will provide important genetic information for further studies on strain conservation, identification or discrimination of the strains, and new functional gene discovery.
2. Material and Methods
2.1. Fungus Materials
Twenty strains of H. marmoreus were collected from the Agricultural Culture Collection of China (Beijing, 6), provincial academies of agricultural science (Fujian, 2; Hunan, 2; Sichuan, 1), institutes of edible fungi and mushrooms (Gaoyou, 1; Mianyang, 1; Sanming, 1), Changbaishan (Jinlin, 1), and mushroom enterprises in different places (Shanghai, 2; Shandong, 1; Guangdong, 1; Hubei, 1) (Supplementary File 1 (Table S1) available online at http://dx.doi.org/10.1155/2014/619746), including 7 storage strains and 13 commercial strains (Supplementary File 1 (Table S1)). The mycelia of the strains were cultured on potato sucrose agar (PSA) medium at 25°C.
2.2. DNA Extraction
Genomic DNA was extracted from fresh mycelium using the Sangon Fungus Genomic DNA Extraction Kit (Sangon Biotech Co., Ltd., Shanghai). DNA concentration and purity were determined by spectrophotometry (BioSpec-nano, Shimadzu, Japan) and electrophoresis in 1.4% agarose gels with known DNA marker.
2.3. Primers Design and TRAP Analysis
Initially 4 sequences of the H. marmoreus were selected to design the fixed primers [19, 21, 22]. All fixed primers were designed by web-based software “Primers 3” [23, 24] (http://frodo.wi.mit.edu/). The arbitrary primers were designed according to [16, 25, 26]. All fixed primers and arbitrary primers were synthesized by Sangon (Sangon Biotech Co., Ltd., Shanghai, China). The information of all primers was showed in Supplementary File 1 (Table S2).
PCR was performed in a 25 μL reaction mixture containing 30 ng template DNA, 1.5 μL 2.5 mM dNTP, 1.5 μL 25 mM MgCl2, 1.5 μL 10× buffer (0.1 mM EDTA, 10.0 mM KCl, and 20 mM Tris-HCl in pH 8.0), 0.75 μL 10 mM primers, and 0.5 μL 2.5 U of Taq DNA polymerase (Tiangen Biotech, Beijing, China). A blank control and three negative controls were designed to improve the accuracy and reliability of TRAP results. The three negative controls contained DNA from Coprinus comatus, no primers, and blank control. To ensure the specificity of the experiment, only the bands specific to H. marmoreus were counted; that is, the bands amplified from both strains of H. marmoreus and not in strain of C. comatus were excluded from analyses. Controls without primers were used to detect and eliminate reagent contamination.
Amplification was performed in a Biometra TProfessional Standard (Biometra, GmbH, Germany). The TRAP-PCR reaction was modified based on Hu and Vick [15] as follows: 94°C for 10 min, 10 cycles of 94°C for 1 min, 35°C for 1 min, and 72°C for 2 min 30 s, then 30 cycles of 94°C for 1 min, 51.5°C for 1 min, and 72°C for 2 min 30 s, and a final extension step of 10 min at 72°C, and then stored at 4°C.
Amplified products were electrophoresed in 2% agarose gel with 0.5 Tris/Borate/EDTA buffer at 120 V for 45 min and stained with ethidium bromide (0.5 g/mL). Gels with amplification fragments were visualized and photographed under ultraviolet light using a GE Image Quant Digital Imaging System (GE Healthcare Bio-Sciences AB, Sweden). The DL2000 DNA ladder was used as a marker (Tiangen Biotech, Beijing, China).
2.4. Data Analysis
TRAP amplification products were compared with DNA ladder markers and scored using a binary code (present, 1; absent, 0) of each genotype using manual score; then the data of matrix were recorded in excel files for further analysis. Only well-separated bands with a high intensity of polymorphism were selected as markers. Pairwise comparisons were calculated using Jaccard's coefficient [27]. The similarity values were used to generate a consensus tree using the unweighted pair group method with arithmetic mean (UPGMA) algorithm [28]. Analyses were performed with NTSYS-pc version 2.1 [29]. The PIC, MI, qualitative nature of data, and effective MI were calculated as follows.
PIC values measure the information of a given DNA marker, PICi = 1 − ∑(P ij)2, where P ij is the frequency of the ith pattern revealed by the jth primer summed across all patterns revealed by the primers [30, 31]. The MI is the product of the total number of loci per primer (n). The MI is calculated for each ISSR primer as MI = PIC × ηβ, where PIC is the mean PIC value, η is the number of bands, and β is the proportion of polymorphism [32].
2.5. Sequencing Analyses
The common fragments only in all TRAP-PCR amplifications with H. marmoreus were extracted from agarose gels using spin column DNA extraction kit and cloned into pMD19-T vector (TaKaRa, Dalian, China). The clones were sequenced by Beijing Genomics Institute (BGI) after PCR check using universal primers (GV-M, 5′-GAGCGGATAACAATTTCACACAGG-3′ and M13–47, 5′-CGCCAGGGTTTTCCCAGTCACGAC-3′). Sequences were assembled using program of SeqMan (software of DNASTAR package) and submitted to GenBank.
The homology of sequences was predicted by web-based tool BLAST search [33–36]. The extrons or introns of sequence analyses were predicted by web-based program FENGE SH [37–39] (http://linux1.softberry.com/berry.phtml).
3. Results and Discussion
3.1. TRAP Analysis
After primary screening, 6 fixed primers and 20 arbitrary primers were selected to second screening. Twenty-eight primer combinations could produce clear bands with good polymorphisms and reproducibility after second screening (Table 1). These primer combinations generated a total of 287 scorable fragments, 202 of which were polymorphic. The average of fragments per primer combination was 10.27, and the total of fragments amplified by each TRAP primer combination ranged from 4 to 17. The percentages of polymorphic fragments ranged from 33.3% (JN00L1-ME11) to 100% (JN00R1-ME6) with a mean of 69.73% (Table 1). The size of the detected fragments ranged from 100 to 2100 bp (Table 1, Figure 1).
Table 1.
Primer combinations and some genetic information generated by TRAP markers.
| Primer combination | Total bands | Polymorphism bands | Percentage of polymorphism | Polymorphism information content | Marker index | Fragment range size |
|---|---|---|---|---|---|---|
| EF69R1-EM10 | 12 | 9 | 75.00 | 0.50 | 5.94 | 100–2000 |
| EU94L1-EM9 | 12 | 5 | 41.67 | 0.50 | 5.96 | 150–1900 |
| GQ76R1-EM1 | 8 | 4 | 50.00 | 0.46 | 3.70 | 300–1100 |
| GQ76R1-EM2 | 8 | 3 | 37.50 | 0.38 | 3.05 | 170–1200 |
| GQ76R1-EM4 | 6 | 4 | 66.67 | 0.50 | 3.00 | 150–2100 |
| GQ76R1-EM5 | 11 | 7 | 63.64 | 0.49 | 5.38 | 100–1200 |
| GQ76R1-EM6 | 9 | 6 | 66.67 | 0.50 | 4.46 | 100–750 |
| GQ76R1-EM17 | 17 | 11 | 64.71 | 0.50 | 8.50 | 120–2100 |
| GQ76R1-ME1 | 13 | 10 | 76.92 | 0.49 | 6.38 | 250–2000 |
| GQ76R1-ME2 | 12 | 7 | 58.33 | 0.50 | 5.99 | 150–1700 |
| GQ76R1-ME4 | 14 | 11 | 78.57 | 0.48 | 6.72 | 150–1900 |
| GQ76R1-ME7 | 10 | 4 | 40.00 | 0.48 | 4.84 | 120–1800 |
| GQ76R1-ME8 | 5 | 3 | 60.00 | 0.49 | 2.46 | 300–1200 |
| GQ76R1-ME9 | 7 | 6 | 85.71 | 0.39 | 2.75 | 150–1200 |
| GQ76R1-ME11 | 13 | 9 | 69.23 | 0.50 | 6.48 | 150–1800 |
| GQ76R1-ME13 | 11 | 7 | 63.64 | 0.48 | 5.28 | 120–1700 |
| JN00L1-EM5 | 16 | 13 | 81.25 | 0.50 | 7.99 | 100–1600 |
| JN00L1-EM10 | 14 | 10 | 71.43 | 0.50 | 6.99 | 200–900 |
| JN00L1-EM16 | 10 | 8 | 77.78 | 0.50 | 4.48 | 100–900 |
| JN00L1-EM26 | 10 | 8 | 80.00 | 0.50 | 5.00 | 120–1000 |
| JN00L1-ME6 | 4 | 2 | 75.00 | 0.50 | 2.00 | 150–500 |
| JN00L1-ME11 | 6 | 2 | 33.33 | 0.32 | 1.92 | 150–500 |
| JN00R1-EM2 | 13 | 11 | 84.62 | 0.49 | 6.41 | 120–1700 |
| JN00R1-EM6 | 10 | 9 | 90.00 | 0.45 | 4.52 | 200–700 |
| JN00R1-EM24 | 9 | 8 | 88.89 | 0.46 | 4.15 | 150–1000 |
| JN00R1-ME2 | 11 | 9 | 81.82 | 0.50 | 5.45 | 100–1200 |
| JN00R1-ME4 | 10 | 9 | 90.00 | 0.49 | 4.94 | 100–1100 |
| JN00R1-ME6 | 7 | 7 | 100.00 | 0.50 | 3.49 | 300–1500 |
| Total | 287 | 202 | ||||
| Minimum | 4 | 2 | 33.33 | 0.32 | 1.92 | |
| Maximum | 17 | 13 | 100.00 | 0.50 | 8.50 | |
| Mean | 10.27 | 7.21 | 69.73 | 0.48 | 4.94 |
Figure 1.

Fragment profiles of Hypsizygus marmoreus generated by primer combination of TRAP. ((a) EU94L1-EM9, (b) GQ76R1-ME1, BC, C1, and C2 correspond to blank control, no primers, and Coprinus comatus; the spots from 1 to 20 correspond to the strains of H. marmoreus (see Supplementary Table S1)).
The PIC values for TRAP markers ranged from 0.32 to 0.50, and the mean PIC value per TRAPs was 0.48. The MI for TRAPs ranged from 1.92 to 8.50, and the mean MI per ISSR was 4.94 (Table 1).
The PIC value provides an evaluation of the discriminating power of the TRAP marker to polymorphism. Generally, PIC value between 0.00 and 0.25 implied a very low genetic diversity among samples, PIC value between 0.25 and 0.50 shows a medium level of genetic diversity, and PIC value above 0.50 suggests a high level of genetic diversity [30, 31, 40, 41]. In this study, the PIC values for TRAP markers ranged from 0.32 to 0.5, with a mean value of 0.48 (Table 1), which indicated a medium level of genetic diversity among the H. marmoreus strains.
3.2. Phylogenetic Analysis and Principal Coordinates Analysis (PCoA) Based on TRAP Markers
A phylogenetic tree was constructed by UPGMA cluster analysis using 287 TRAP fragments from the 20 strains of H. marmoreus (Figure 2). All strains were classified into two clusters (at Jaccard's similarity coefficient of 0.718): cluster A consisted of 18 strains of H. marmoreus and JSGY-2 and HN-2 with white basidiocarps were members of cluster B. Cluster A formed three subgroups, A1, A2, and A3, at Jaccard's similarity coefficients of 0.575 and 0.435. Both subgroups A1 and A3 included two strains, ACCC5047 and SM-4, in subgroup A1 and ACCC51149 and FJNK-1 in subgroup A3; the remaining strains were clustered in subgroup A2.
Figure 2.
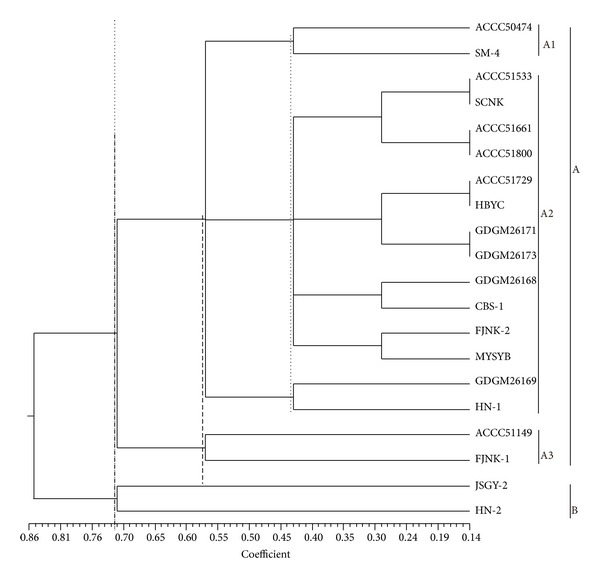
Phylogenetic tree based on TRAP data. The Hypsizygus marmoreus strains were classified into two subgroups at Jaccard's similarity coefficient of 0.718. Group B included strains of JSGY-2 and HN-2. Group A had the remaining 18 strains. Group A was classified into 3 subgroups at Jaccard's coefficients of 0.575 and 0.435.
PCoA analysis based on the fragments of TRAP amplification was shown in Figure 3. The data indicated that the similar clusters were those with UPGMA. The three most informative PCoA components accounted for 35.48% of the variations observed (Figure 3). Most of the commercial cultivars (storage strains) could not cluster together to one group like the previous studies by ISSR and SRAP [13, 14].
Figure 3.
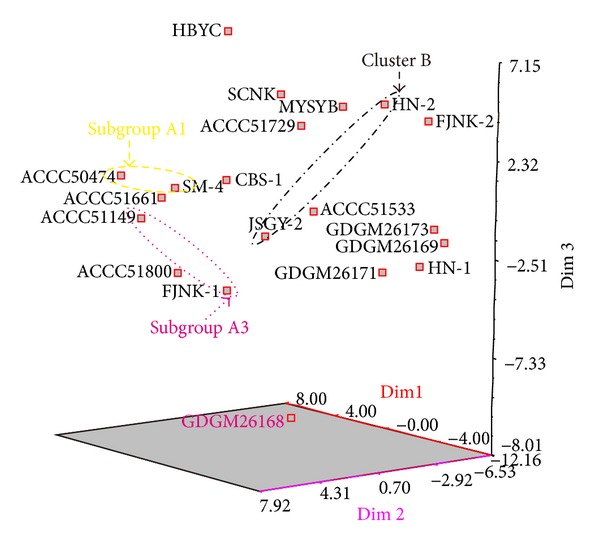
Three-dimensional map of principal coordinates analysis based TRAP data (black ellipse, cluster B; yellow ellipse, subgroup A1; pink ellipse, subgroup A3.e).
The strains of H. marmoreus might be from two ancestors according to the result of phylogenetic analysis based on TRAP. The genetic diversity in cluster A was more polymorphic than that in cluster B; however, the strains in cluster A2 were different from the others (Figure 2). In the TRAP phylogenetic analysis, the clusters were not consistent with strain origin and the color of fruit body, as well as the previous studies with ISSR and SRAP [13, 14], possibly because TRAP technology focuses on polymorphism of the function genes, while ISSR and SRAP, respectively, focus on microsatellite diversities and intron and extron diversities. Compared with the previous studies [13, 14], most strains with white basidiocarps were clustered in a subbranch in this study, possibly because of the TRAP technique associating with the phenotypes controlled by some particular genes.
3.3. Sequence Analyses
Among the fragments amplified with TRAP, 60 fragments were recycled and cloned into pMD19-T vector. Thirty-six sequences were obtained and then submitted to GenBank. The accession numbers of TRAP sequences were from KC906193 to KC906227 and KC906229. The lengths of the sequences ranged from 308 to 1338 bp. Eighteen sequences could encode the unknown function proteins based on BLAST prediction and open reading frame (ORF) analysis by web-based SoftBerry (Supplementary File 2).
Sequences of KC906206, KC906216, KC906218, KC906224, and KC906225 differed from each other and were partial sequences of heat shock protein 70 (HSP70) predicted by three models of BLAST search (BLAST N, BLAST X, and tBLAST X), which suggested a high diversity in genes of HSP70 of H. marmoreus. Based on BLAST X search, some other sequences of TRAP could be predicted to encode HSP protein, glycoside hydrolase, and unknown proteins, which coincided with the fragments selected to design fixed primers. Although the fixed primers were designed from the genes of H. marmoreus, only a small part of TRAP sequences could correspond with the chosen gene segments. There might be two reasons for that: one is the arbitrary primers amplifying fragments from genomic section matching with sequences of primers and the other may be that the low annealing temperature in PCR amplified many unspecific fragments [42].
The results of this study showed that TRAP was more effective than the other molecular markers to evaluate the genetic diversities of H. marmoreus strains.
4. Conclusion
The TRAP analysis was successfully applied to study the genetic diversities among the H. marmoreus strains. A total of 36 gene sequences of H. marmoreus were obtained. The results of this study indicated that the strains of H. marmoreus have medium level of genetic diversity.
Supplementary Material
Figure S1: Result of specific sequence by BLAST N
Figure S2: Result of specific sequence by BLAST X
Table S1: Information of the strains of Hypsizigus marmoreus
Table S2: Fixed and arbitrary primers used in the TRAP analysis of Hypsizigus marmoreus
Table S3: Information of the sequences used to designed specific primers for Hypsizigus marmoreus
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (nos. 31370071 and 31370072), the Science and Technology Planning Project of Guangdong Province (STPPGD) (no. 2012B091100072), and the Science Foundation for Young Scientists of Guangdong Academy of Sciences (no. qnjjsq201110).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Chengshu Qiu carried out the molecular analyses, participated in the design of the study, performed the statistical analysis, and drafted the paper. Wangqiu Deng, Wenjuan Yan, Bin Song, and Taihui Li conceived and helped to draft the paper. All authors read and approved the final paper.
References
- 1.Sánchez C. Cultivation of Pleurotus ostreatus and other edible mushrooms. Applied Microbiology and Biotechnology. 2010;85(5):1321–1337. doi: 10.1007/s00253-009-2343-7. [DOI] [PubMed] [Google Scholar]
- 2.Jung EB, Jo JH, Cho SM. Nutritional component and anticancer properties of various extracts from Haesongi mushroom (Hypsizigus marmoreus) Journal of the Korean Society of Food Science and Nutrition. 2008;37(11):1395–1400. [Google Scholar]
- 3.Lee Y-L, Jian S-Y, Mau J-L. Composition and non-volatile taste components of Hypsizigus marmoreus . LWT—Food Science and Technology. 2009;42(2):594–598. [Google Scholar]
- 4.Akihisa T, Franzblau SG, Tokuda H, et al. Antitubercular activity and inhibitory effect on Epstein-Barr virus activation of sterols and polyisoprenepolyols from an edible mushroom, Hypsizigus marmoreus . Biological & Pharmaceutical Bulletin. 2005;28(6):1117–1119. doi: 10.1248/bpb.28.1117. [DOI] [PubMed] [Google Scholar]
- 5.Krasnopolskaya LM, Leontieva MI, Avtonomova AV, et al. Antitumor properties of submerged cultivated biomass and extracts of medicinal mushrooms of genus Hypsizygus singer (Agaricomycetideae) International Journal of Medicinal Mushrooms. 2008;10(1):25–35. [Google Scholar]
- 6.Park JY, Oh W-J, Kwak D-M, et al. The anti-platelet activity of Hypsizygus marmoreus extract is involved in the suppression of intracellular calcium mobilization and integrin αIIbβ3 activation. Journal of Medicinal Plants Research. 2011;5(11):2369–2377. [Google Scholar]
- 7.Mori K, Kobayashi C, Tomita T, Inatomi S, Ikeda M. Antiatherosclerotic effect of the edible mushrooms Pleurotus eryngii (Eringi), Grifola frondosa (Maitake), and Hypsizygus marmoreus (Bunashimeji) in apolipoprotein E-deficient mice. Nutrition Research. 2008;28(5):335–342. doi: 10.1016/j.nutres.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Fan X-Z, Liu W, Xiao Y, Bian Y-B. Comparative analysis on the diversity of Auricularia auricula-judae by physiological characteristics, somatic incompatibility and TRAP fingerprinting. World Journal of Microbiology and Biotechnology. 2011;27(9):2081–2093. [Google Scholar]
- 9.Gong WB, Liu W, Lu YY, et al. Constructing a new integrated genetic linkage map and mapping quantitative trait loci for vegetative mycelium growth rate in Lentinula edodes . Fungal Biology. 2014;118(3):295–308. doi: 10.1016/j.funbio.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Lee CY, Park J-E, Lee J, Kim J-K, Ro H-S. Development of new strains and related SCAR markers for an edible mushroom, Hypsizygus marmoreus . FEMS Microbiology Letters. 2012;327(1):54–59. doi: 10.1111/j.1574-6968.2011.02458.x. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Hu XH, Feng ZY, Pan YJ. Development of AFLP markers and phylogenetic analysis in Hypsizygus marmoreus . The Journal of General and Applied Microbiology. 2009;55(1):9–17. doi: 10.2323/jgam.55.9. [DOI] [PubMed] [Google Scholar]
- 12.Lim YJ, Lee CY, Park JE, Kim SW, Lee HS, Ro HS. Molecular genetic classification of Hypsizigus marmoreus and development of strain-specific DNA Markers. The Korean Journal of Mycology. 2010;38(1):34–39. [Google Scholar]
- 13.Qiu CS, Yan WJ, Li P, Deng WQ, Song B, Li TH. Evaluation of growth characteristics and genetic diversity of commercial and stored lines of Hypsizygus marmoreus . International Journal of Agriculture & Biology. 2013;15(3):479–485. [Google Scholar]
- 14.Qiu CS, Yan WJ, Song B, Li P, Deng W, Li TH. Analysis of the genetic diversity and relationship within Hypsizygus marmoreus based on sequence-related amplification polymorphism. Journal of Pure and Applied Microbiology. 2013;7(1):1–9. [Google Scholar]
- 15.Hu JG, Vick BA. Target region amplification polymorphism: a novel marker technique for plant genotyping. Plant Molecular Biology Reporter. 2003;21(3):289–294. [Google Scholar]
- 16.Li G, Quiros CF. Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica . Theoretical and Applied Genetics. 2001;103(2-3):455–461. [Google Scholar]
- 17.Qiao LX, Liu HY, Sun JW, et al. Application of target region amplification polymorphism (TRAP) technique to Porphyra (Bangiales, Rhodophyta) fingerprinting. Phycologia. 2007;46(4):450–455. [Google Scholar]
- 18.Xiao Y, Liu W, Lu Y-Y, Gong W-B, Bian Y-B. Applying target region amplification polymorphism markers for analyzing genetic diversity of Lentinula edodes in China. Journal of Basic Microbiology. 2010;50(5):475–483. doi: 10.1002/jobm.201000018. [DOI] [PubMed] [Google Scholar]
- 19.Zhang XQ, Liu QH, Zhang GQ, Wang HX, Ng T. Purification and molecular cloning of a serine protease from the mushroom Hypsizigus marmoreus . Process Biochemistry. 2010;45(5):724–730. [Google Scholar]
- 20.Xu JZ, Zhang JL, Zhang WG, Hu KH. The novel role of fungal intracellular laccase: used to screen hybrids between Hypsizigus marmoreus and Clitocybe maxima by protoplasmic fusion. World Journal of Microbiology and Biotechnology. 2012;28(8):2625–2633. doi: 10.1007/s11274-012-1072-8. [DOI] [PubMed] [Google Scholar]
- 21.Li CC. Cloning and heterologous expression of heat shock HmHSP70 gene from Hypsizygus marmoreus [M.S. thesis] Qingdao, China: Qingdao Agriculture University; 2009. [Google Scholar]
- 22.Zhang J, Chen H, Zhiyong F, Chen M, Wang H. Cloning and sequence analysis of a glyceraldehyde 3-phosphate dehydrogenase gene from Hypsizygus marmoreus . Acta Edulis Fungi. 2011;18(2):5–9. [Google Scholar]
- 23.Primer3 (v.0.4.0) . Pick primers from a DNA sequence. http://frodo.wi.mit.edu/
- 24.Rozen S, Helen S. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Totowa, NJ, USA: Humana Press; 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 25.Budak H, Shearman RC, Parmaksiz I, Gaussoin RE, Riordan TP, Dweikat I. Molecular characterization of Buffalograss germplasm using sequence-related amplified polymorphism markers. Theoretical and Applied Genetics. 2004;108(2):328–334. doi: 10.1007/s00122-003-1428-4. [DOI] [PubMed] [Google Scholar]
- 26.Ren N, Liu J, Yang D, Chen J, Luan M, Hong J. Sequence-related amplified polymorphism (SRAP) marker as a new method for identification of endophytic fungi from Taxus . World Journal of Microbiology and Biotechnology. 2012;28(1):215–221. doi: 10.1007/s11274-011-0810-7. [DOI] [PubMed] [Google Scholar]
- 27.Jaccard P. Etude comparative de la distribution florale dans une portion des Alpes et des Jura. Bulletin de la Société Vaudoise des Sciences Naturelles. 1901;37(142):547–579. [Google Scholar]
- 28.Nei M. Analysis of gene diversity in subdivided populations. Proceedings of the National Academy of Sciences of the United States of America. 1973;70(12):3321–3323. doi: 10.1073/pnas.70.12.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rohlf FJ. Numerical Taxonomy and Multivariate Analysis System Version 2.1. New York, NY, USA: Applied Biostatistics; 2000. [Google Scholar]
- 30.Botstein D, White RL, Skolnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. The American Journal of Human Genetics. 1980;32(3):314–331. [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson JA, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME. Optimizing parental selection for genetic linkage maps. Genome. 1993;36(1):181–186. doi: 10.1139/g93-024. [DOI] [PubMed] [Google Scholar]
- 32.Powell W, Morgante M, Andre C, et al. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Molecular Breeding. 1996;2(3):225–238. [Google Scholar]
- 33.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 34.Zhang JH, Madden TL. PowerBLAST: a new network BLAST application for interactive or automated sequence analysis and annotation. Genome Research. 1997;7(6):649–656. doi: 10.1101/gr.7.6.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10, article 421 doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Research. 2000;10(4):516–522. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathé C, Sagot M-F, Schiex T, Rouzé P. Current methods of gene prediction, their strengths and weaknesses. Nucleic Acids Research. 2002;30(19):4103–4117. doi: 10.1093/nar/gkf543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solovyev VV, Shindyalov IN. Finding genes by computer: probabilistic and discriminative approaches. In: Jiang T, Xu Y, Zhang MQ, editors. Current Topics in Computational Biology. Cambridge, Mass, USA: The MIT Press; 2002. pp. 365–401. [Google Scholar]
- 40.Smith JSC, Chin ECL, Shu H, et al. An evaluation of the utility of SSR loci as molecular markers in maize (Zea mays L.): comparisons with data from RFLPS and pedigree. Theoretical and Applied Genetics. 1997;95(1-2):163–173. [Google Scholar]
- 41.Tams SH, Melchinger AE, Bauer E. Genetic similarity among European winter triticale elite germplasms assessed with AFLP and comparisons with SSR and pedigree data. Plant Breeding. 2005;124(2):154–160. [Google Scholar]
- 42.Liu ZH, Anderson JA, Hu J, Friesen TL, Rasmussen JB, Faris JD. A wheat intervarietal genetic linkage map based on microsatellite and target region amplified polymorphism markers and its utility for detecting quantitative trait loci. Theoretical and Applied Genetics. 2005;111(4):782–794. doi: 10.1007/s00122-005-2064-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Result of specific sequence by BLAST N
Figure S2: Result of specific sequence by BLAST X
Table S1: Information of the strains of Hypsizigus marmoreus
Table S2: Fixed and arbitrary primers used in the TRAP analysis of Hypsizigus marmoreus
Table S3: Information of the sequences used to designed specific primers for Hypsizigus marmoreus