

Abstract
PH domain leucine-rich repeat protein phosphatase (PHLPP) directly dephosphorylates and inactivates Akt and protein kinase C and is therefore a prime target for pharmacological intervention of two key signaling pathways, the phosphatidylinositol 3-kinase and diacylglycerol signaling pathways. Here we report on the first biochemical characterization of the phosphatase domain of a PHLPP family member. The human PHLPP1 and PHLPP2 phosphatase domains were expressed and purified from bacteria or insect cells and their activities compared to that of full-length proteins immunoprecipitated from mammalian cells. Biochemical analyses reveal that the PHLPP phosphatase domain effectively dephosphorylates synthetic and peptidic substrates, that its activity is modulated by metals and lipophilic compounds, and that it has relatively high thermal stability. Mutational analysis of PHLPP2 reveals an unusual active site architecture compared to the canonical architecture of PP2C phosphatases and identifies key acidic residues (Asp 806, Glu 989, and Asp 1024) and bulky aromatic residues (Phe 783 and Phe 808) whose mutation impairs activity. Consistent with a unique active site architecture, we identify inhibitors that discriminate between PHLPP2 and PP2Cα. These data establish PHLPP as a member of the PP2C family of phosphatases with a unique active site architecture.
Reversible phosphorylation of proteins provides a major mechanism by which cells control the transfer of information. Kinases and phosphatases act in dynamic opposition to switch signaling pathways on or off and to control the duration, magnitude, and localization of responses to signals. Members of the PHLPP family of phosphatases oppose oncogenic and proliferative pathways by directly dephosphorylating at least four kinases: the pro-survival kinases Akt, protein kinase C (PKC), S6 kinase, and pro-apoptotic kinase Mst1.1,2 In the case of Akt, PKC, and S6 kinase, PHLPP dephosphorylates a conserved regulatory site termed the hydrophobic motif, thus inactivating the kinases and inducing apoptosis.3−5 In the case of Mst1, PHLPP dephosphorylates an inhibitory site to activate Mst1 and promotes apoptosis.6
The PHLPP family consists of three isozymes encoded by two genes: PHLPP1α, PHLPP1β, and PHLPP2. All isozymes share the same domain structure of an N-terminal PH domain followed by a leucine-rich repeat segment, a phosphatase domain, and a C-terminal PDZ-binding motif.1,7 The phosphatase domains are 68% identical and group into a subfamily of the PP2C family of phosphatases.8,9 The PP2Cs are the principal members of the PPM (protein phosphatase manganese/magnesium-dependent) family of serine/threonine phosphatases, which also includes pyruvate dehydrogenase phosphatase.9 Similar to the members of the PPP family (phosphoprotein phosphatase family that includes PP1, PP2A, PP2B, etc.), PPM family members require a divalent cation for activity with a preference for Mn2+ or Mg2+. The main biochemical difference from other phosphatase families, however, is that the PPM proteins are not inhibited by okadaic acid or other natural products such as microcystin LR or cyclosporin A. Sequence alignments in the eukaryotic PP2C family reveal the conservation of 11 motifs and 4 aspartic acids that coordinate the metal ions.9 Phosphatases of the PP2C family have been shown to regulate stress in plants and bacteria.10,11 In mammals, PP2Cs regulate the stress response as well, by dephosphorylating key components of the stress-induced cascade.13 PP2C enzymes are attracting an increasing amount of attention as their role in cancer emerges.12 Notably, the phosphatase Wip1 (PP2Cδ/PPM1D)14 is normally induced by the p53 tumor suppressor after DNA damage following ionizing radiation or exposure to UV light. This protein is overexpressed, and the corresponding gene is amplified in different types of human cancers, including breast cancer and neuroblastoma. Furthermore, Ppm1d deletion delays or limits tumorigenesis in different mouse cancer models, defining WIP1 as an oncoprotein. Conversely, PHLPP has recently been shown to be a tumor suppressor in a mouse model.15 In humans, PHLPP is frequently lost or reduced, at the gene or protein level, in diverse cancers, including colon16−18 and breast19,20 cancers and glioblastoma.21,22 Dysregulation of PHLPP correlates not only with cancer but also with diseases such as obesity, where PHLPP1 is upregulated.23 It also plays a regulatory role in the heart via its negative regulation of Akt.24 Given the broad role of PHLPP in a wide spectrum of diseases, understanding its catalytic mechanisms is essential to its development as a therapeutic target.
This contribution provides a biochemical and structural characterization of the intrinsic catalytic activity of PHLPP. Using isolated PP2C domains purified from bacteria, insect cells, or mammalian cells, as well as full-length proteins from mammalian expression, we determine the metal-dependent, pH-dependent activation. We examine the effects of various factors on activity, such as reducing potential or lipophilic compounds. We also describe inactivating mutations in the catalytic domain.
Experimental Procedures
BL21(DE3)pLysS competent cells were purchased from EMD4Biosciences. EGF was purchased from Upstate. The monoclonal antibody against HA (Covance) and the polyclonal antibody against PHLPP2 (Bethyl) were used to assess expression. A monoclonal anti-HA antibody (Roche) and protein A/G Ultralink resin (Pierce) were used for immunoprecipitation. Palmitoleic and linoleic acids were obtained from Acros, and stearic acid was from AlfaAesar, oleic acid from Sigma, arachidonic acid from Biomol, and elaidic acid from MP. Ammonium molybdate (Fisher) and malachite green oxalate (Acros) were obtained at the highest grade possible. Protein phosphatase peptide substrate (RRAPTVA) and protein tyrosine phosphatase peptide substrate (ENDPYINASL) were obtained from Enzo, and HFPQFPSYSAS was ordered from Anaspec. COS7 cells are maintained in DMEM (Cellgro) supplemented with 5% FBS (Hydroclone) and 1% penicillin/streptomycin at 37 °C in 5% CO2.
Purification of the GST-Tagged Phosphatase Domain of PHLPP1 and PHLPP2
The GST-tagged fusion protein of the phosphatase domain of human PHLPP1 (Pro 627–Pro 990, PHLPP1α numbering) or PHLPP2 (Gly 745–Asn 1102) was expressed in competent BL21(DE3)pLysS cells and purified according to the method previously described.3 Purification yielded 15 ± 1 or 14 ± 1 mg/(L of culture), respectively. Purity was assessed on a sodium dodecyl sulfate (SDS) gel and was >90%. The protein was then either kept in a desalting buffer [20 mM Tris and 100 mM NaCl (pH 7.0)] or dialyzed against 1 L of 20 mM Tris-HCl, 2 mM MnCl2, or EDTA (pH 7.0). The enzymes were stored at 4 °C.
Purification of the His-Tagged Phosphatase Domain of PHLPP2
His-tagged PHLPP2 PP2C was expressed and purified from baculovirus-infected Sf21 cells. Sf21 cells were maintained in Hink’s TNM-FH medium (Cellgro), supplemented with 10% FBS and 1% penicillin/streptomycin, and incubated for 4 days with a baculovirus encoding His-PHLPP2 PP2C. Purification was conducted using the IMAC purification kit on Profinia (Bio-Rad), yielding 87 ± 5 μg of purified protein/L of culture medium. Purity was assessed by Coomassie staining of SDS gels and was >90%.
Purification of the HA-Tagged Phosphatase Domain and the Full-Length Protein of PHLPP
The isolated phosphatase domains of human PHLPP1 and full-length PHLPP1α, PHLPP1β, and PHLPP2 were overexpressed in COS7 cells as HA-tagged fusion proteins using FuGENE 6 transfection reagent (Promega). Cells were treated with 10 ng of EGF/mL of DMEM for 15 min prior to lysis and immunoprecipitated as previously described.3
Plasmid Constructs
Mutant constructs were generated by site-directed mutagenesis of pcDNA3-HA-PHLPP24 using the QuikChange protocol (Agilent Technologies).
Enzymatic Activity Assay
The enzymatic reaction was conducted at room temperature in a reaction mixture containing the appropriate pH buffer at 100 mM (see Table 1 of the Supporting Information), 0.02 mg/mL BSA, 4 mM DTT, and 100 mM NaCl, at the pH values noted in the legends. Divalent cations or inhibitors were added as noted. In the case of the isolated PP2C domain purified from bacteria or insect cells, an exact volume of a solution of a known concentration of protein was added to the reaction mixture. When enzymes purified from mammalian cells were used, the A/G beads supporting the proteins were added to the reaction mixture. The relative amount of protein in the assay was determined by Western blot. To determine the kinetic parameters kcat and KM, all data were fit to a Michaelis–Menten model, using nonlinear regression.
p-Nitrophenyl Phosphate (pNPP) Assay
This assay was previously described.25 Briefly, each well contained 125 μL of reaction mix (see above), 1 μM enzyme, and various concentrations of PNPP. Optical density was recorded over time at 405 nm. Absorbance was then plotted over time, and the slope was calculated.
Malachite Green Assay
The malachite green assay used in this paper was a slightly modified version of the protocol described previously.26 A fresh solution of reagent was prepared from 3 volumes of a 34 mM solution of ammonium molybdate in 4 M hydrochloric acid mixed with 1 volume of 518 μM malachite green in water. Tween 20 was added to a final concentration of 1% (v/v). The reaction was stopped by addition of 65 μL of the reagent to 65 μL of reaction mixture. The reagent was added to the reaction mixture at different time points, and the color was allowed to develop for 15 min. Absorbance was recorded at 650 nm. A calibration curve was obtained each time. The measured OD was converted to the amount of inorganic phosphate released, and data were plotted versus time. The initial slope (picomoles per second), measured between 0 and 25% conversion, was divided by the amount of protein (picomoles) to determine the velocity (inverse seconds).
Inhibitors
The following compounds from NCI Diversity Set I were tested: NSC 170008, 7-acetyl-6-ethyl-3,5,8-trihydroxy-9,10-dioxoanthracene-1,2-dicarboxylic acid; NSC 73101, 1,2,3,4,7,7-hexachloro-6-(9H-fluoren-2-ylcarbamoyl)bicyclo[2.2.1]hept-2-ene-5-carboxylic acid; NSC 11241, C.I. Basic Red 6; NSC 270127, N-(2-benzoyl-4-chlorophenyl)-2-chloroacetamide
Results
Kinetic Data
The isolated phosphatase domains of PHLPP1 and PHLPP2 were expressed in bacteria as N-terminally tagged GST fusion proteins, purified by GST affinity chromatography, and their kinetic parameters were examined. The GST tag had no effect on the enzymatic activity of either construct (data not shown) and thus was not removed for the analyses presented. Using pNPP as a substrate, we determined the KM and kcat for purified PHLPP1 and PHLPP2 phosphatase domains (Figure 1a). The kinetic profiles of both isozymes were nearly identical: maximal turnover rates, reflected by the kcat values, were (1.8 ± 0.3) × 10–3 s–1 for PHLPP1 and (1.8 ± 0.1) × 10–3 s–1 for PHLPP2. Substrate binding energetics for pNPP were also very similar as reflected by KM values of 2.1 ± 0.2 and 1.80 ± 0.08 mM for PHLPP1 and PHLPP2, respectively.
Figure 1.

PHLPP activity is highly dependent on the substrate. (a) PHLPP1 and PHLPP2 have similar phosphatase activities in vitro. The saturating kinetics of the phosphatase domains of PHLPP1 (empty diamonds) and PHLPP2 (gray squares) with increasing concentrations of the substrate pNPP is shown. The rates were determined by measuring the production of pNP at 405 nm in Tricine buffer (pH 7.5). The data are fit to the Michaelis–Menten equation. The graph shows mean values ± SEM for three separate experiments. (b) Activities of the phosphatase domain of PHLPP2 isolated from bacteria or insect cells are comparable on peptidic substrates. The saturating kinetics of the phosphatase domains of PHLPP2 isolated from Sf21 cells (black circles) or Escherichia coli BL21(DE3)pLysS (gray squares) with increasing concentrations of the substrate peptide is shown. The rates were determined by measuring the liberation of free phosphate by the malachite green assay in a Tricine buffer (pH 7.5). The data are fit to the Michaelis–Menten equation. The graph shows mean values ± SEM for three separate experiments.
We next compared the kinetic parameters of isolated phosphatase domains expressed in bacteria, baculovirus, and mammalian cells (Table 1). The GST-tagged PP2C domain of PHLPP2 was purified from Sf21 cells after infection by baculovirus; the HA-tagged PP2C domain of PHLPP1 was expressed in COS7 cells and immunoprecipitated using a HA antibody. The KM for pNPP for the PHLPP2 phosphatase domain purified from insect cells (1.3 ± 0.2 mM) or immunoprecipitated from mammalian cells (1.5 ± 0.5 mM) was comparable to that for bacterially expressed PHLPP2. In contrast, the kcat was 200-fold higher for the protein isolated from insect cells (0.37 ± 0.08 s–1) than for the protein purified from bacteria [(1.8 ± 0.1) × 10–3 s–1]. Because the concentration of phosphatase was not measured for mammalian phosphatase, kcat was not determined. These data reveal that the affinity of PHLPP for pNPP is similar regardless of the source of the protein; however, the catalytic activity is 2 orders of magnitude higher for phosphatase purified from insect cells (which better mimic the milieu of mammalian cells) than bacteria.
Table 1. Kinetic Parametersa.
| substrate | expression system | isoform | domain | kcat (s–1) | KM (M) | kcat/KM (s–1 M–1) |
|---|---|---|---|---|---|---|
| pNPP | bacteria | PHLPP1 | PP2C | (1.8 ± 0.3) × 10–3 | (2.1 ± 0.2) × 10–3 | 0.9 ± 0.2 |
| bacteria | PHLPP2 | PP2C | (1.8 ± 0.1) × 10–3 | (1.80 ± 0.08) × 10–3 | 1.0 ± 0.1 | |
| insect | PHLPP2 | PP2C | (370 ± 80) × 10–3 | (1.3 ± 0.2) × 10–3 | 285 ± 9 | |
| mammal | PHLPP1 | PP2C | not determined | (1.5 ± 0.5) × 10–3 | – | |
| mammal | PHLPP1 | full-length | not determined | (1.5 ± 0.5) × 10–3 | – | |
| RRAPTVA | bacteria | PHLPP2 | PP2C | 0.15 ± 0.04 | (64 ± 3) × 10–6 | (2.3 ± 0.7) × 103 |
| insect | PHLPP2 | PP2C | 1.1 ± 0.3 | (63 ± 3) × 10–6 | (18 ± 5) × 103 | |
| HFPQFPSYSAS | bacteria | PHLPP2 | PP2C | 1.1 ± 0.2 | (139 ± 1) × 10–6 | (8 ± 2) × 103 |
| insect | PHLPP2 | PP2C | 0.9 ± 0.2 | (40.5 ± 0.3) × 10–6 | (22 ± 5) × 103 |
PHLPP1 and PHLPP2 phosphatase domains and full-length proteins were obtained as fusion proteins using different expression systems: bacteria [BL21(DE3)pLysS cells], insect cells (Sf21), or mammalian cells (COS7). kcat and KM were determined using either pNPP or peptides as a substrate. Dephosphorylation of pNPP was measured by recording the OD at 405 nm, and dephosphorylation of peptides was assessed using the malachite green assay. Reactions occurred in Tricine buffer (pH 7.5). All data were fit to the Michaelis–Menten equation. Data are means ± SEM.
Phosphopeptides were also subjected to PHLPP dephosphorylation (Figure 1b). Two sequences were used: a threonine-phosphorylated peptide (RRAPTVA) that is a standard substrate for PP2C27 and a serine-phosphorylated peptide (HFPQFpSYSAS), corresponding to the sequence of the hydrophobic motif of Akt, a physiologically relevant substrate of PHLPP. Both peptides were significantly better substrates for PHLPP than the synthetic substrate pNPP. First, they were dephosphorylated more rapidly than pNPP as reflected by considerably higher kcat values, a difference most notable for the bacterially expressed enzyme where kcat was more than 600-fold higher for the hydrophobic motif peptide (1.1 ± 0.2 s–1) than for pNPP [(1.8 ± 0.1) × 10−3 s–1]. Second, the KM values were >1 order of magnitude lower. Taken together, the efficiency of the reaction, evaluated by the kcat/KM ratio, was enhanced by 2 to ∼4 orders of magnitude depending on the enzyme source [Table 1; kcat/KM ratio of (8 ± 2) × 103 for the hydrophobic motif peptide compared to a value of 1.0 ± 0.1 using bacterially expressed PHLPP2]. The most notable feature was that the catalytic rate of the bacterially expressed phosphatase domain was comparable to that of insect cell-expressed phosphatase when peptides were used as substrates. Thus, the phosphatase domain of PHLPP effectively dephosphorylates phosphopeptides.
pH Profiles
The pH dependence for the dephosphorylation of pNPP by the bacterially expressed phosphatase domains of PHLPP1 and PHLPP2 was examined in the absence of added Mn2+. Both enzymes had detectable catalytic activity between pH 5 and 9 (Figure 2a). kcat and kcat/KM were then determined for various pH values between 5.5 and 8.5. The profiles of activity of the two isozymes were very similar. The plot of kcat versus pH reveals two peaks of activity at pH 6.0 and 7.8 (Figure 2b). As a general trend, PHLPP1 activity decreased up to 5-fold under basic conditions (pH >8). The data could be fit to a model28 in which one residue is deprotonated. With this model, the ionization pH for PHLPP1 is 7.1 ± 0.2 and its pH-independent kcat value is (2.2 ± 0.5) × 10–3 s–1. With the same model, the kcat for PHLPP2 does not seem to depend on the pH, at least in this pH range.
Figure 2.

Effect of pH on the kinetic parameters. (a) Activity of PHLPP1 (empty diamonds) and PHLPP2 (gray squares) phosphatase domains was evaluated at different pH values. Reactions occurred at 10 mM pNPP and 100 mM pH-appropriate buffer (see Table 1 of the Supporting Information). Graphs show means ± SEM for at least three separate experiments. (b) kcat and (c) kcat/KM for PHLPP1 and PHLPP2 phosphatase domains evaluated at different pH values. Saturating kinetics were determined with increasing concentrations of the substrate pNPP (typically 1–15 mM) in 100 mM buffer, and data were fit to a Michaelis–Menten equation. pH data in panel c are fit to the equation v = C/(1+ H/Ka + H/Kb). Graphs show means ± SEM.
The kcat/KM versus pH plot (Figure 2c), however, confirms the existence of two important ionizations for catalysis set at pH 7.84 ± 0.05 and 8.28 ± 0.05 for PHLPP1 and pH 7.84 ± 0.07 and 8.2 ± 0.1 for PHLPP2. The pH-independent kcat/KM for PHLPP1 [(2.940 ± 0.001) × 10–2 M–1 s–1] is comparable to the value for PHLPP2 [(2.017 ± 0.008) × 10–2 M–1 s–1], stressing once again the strong similarity between the two isoforms.
Metal Requirement
Next we examined the effect of different metallic divalent cations on the enzymatic activity of PHLPP. EDTA, Mg2+, Ca2+, Cu2+, or Ni2+ was added at increasing concentrations to the reaction mixture, and the initial rate of dephosphorylation was determined at a saturating substrate concentration, using the phosphatase domain of PHLPP2 purified from bacteria (Figure 3a–c). In every case, the enzyme was active without addition of a metallic ion. This activity was insensitive to EDTA, even at a concentration of 100 mM. Mg2+ did not enhance activity; rather, modest inhibition was observed at concentrations of >10 mM. Ca2+, Cu2+, and Ni2+ inhibited the phosphatase, with half-maximal inhibition observed in the presence of approximately 40, 10, and 8 mM divalent cation, respectively. Zn2+ was the strongest inhibitor with complete inhibition observed in the presence of 1 mM ZnCl2 (data not shown).
Figure 3.

Influence of the cation on PHLPP activity. Relative activity of PHLPP2 phosphatase domains at 10 mM pNPP with increasing concentrations of (a) EDTA (red) or MgCl2 (yellow), (b) CaCl2, or (c) CuSO4 (blue) or NiSO4 (orange). The activity is given as a percentage of the activity of the protein in the absence of additive. (d) Relative activity of PHLPP2 phosphatase domains isolated from insect cells (black circles) or bacteria (gray squares) and full-length PHLPP1 immunoprecipitated from mammalian cells (white triangles) or the PHLPP1 phosphatase domain from bacteria (white diamonds) at 10 mM pNPP with increasing concentrations of MnCl2. k values for PHLPP1 (e) and PHLPP2 (f) phosphatase domains purified from BL21(DE3)pLysS cells were evaluated at 10 mM pNPP, at different pH values in the absence (empty symbols) or presence (black symbols) of 10 mM MnCl2. Graphs represent means ± SEM for three separate experiments.
The influence of Mn2+ on activity was more complex than expected (Figure 3d). According to the literature, Mn2+ is an activator of the PP2C family,29 to various degrees depending on the substrate.30,31 We indeed found that addition of MnCl2 to the reaction mixture at pH 7.5 increased PHLPP activity when the proteins were purified from insect or mammalian cells. A 7-fold increase was observed with the phosphatase domain of PHLPP2 purified from Sf21 cells, and a more modest 2-fold increase occurred in the case of PHLPP1 overexpressed in COS7 cells. However, when bacterial preparations were used as the enzyme, addition of MnCl2 to the reaction mixture at pH 7.5 resulted in a decrease in activity, by 70% in the case of PHLPP1 and 50% for PHLPP2. To understand this apparently conflicting result, we examined the dependence of this effect on the pH of the reaction mixture. MnCl2 was added to a final concentration of 1–20 mM to a reaction mixture buffered at various pH values, between pH 6 and 8.5 (Figure 3e,f). The data show that Mn2+ acts as an inhibitor in both cases, below pH 8.0. Addition of 10 mM MnCl2 to a reaction mixture resulted in an average 60% inhibition at these pH values. However, at pH 8.2 and 8.5, PHLPP1 showed a maximal 8-fold boost in activity (Figure 3e), and a 4-fold increase was observed for PHLPP2 (Figure 3f). Thus, Mn2+ is inhibitory below pH 8 and activating above pH 8.
Influence of Other Parameters
Varying the ionic strength of the reaction mixture (set by varying the concentration of sodium chloride in the reaction mixture from 1 to 500 mM) had no effect on the dephosphorylation rate of either PHLPP1 or PHLPP2 (data not shown). The chemical nature of the buffer also did not have any significant impact on the activity (Table 1 of the Supporting Information). However, the concentration of DTT in the solution plays a role in the enzymatic reaction (Figure 4a). Addition of 0.4–8 mM DTT to the reaction mixture significantly enhanced PHLPP1 activity with a maximal 3-fold increase at low DTT concentrations (<3 mM). On the other hand, DTT had a negligible effect on PHLPP2 activity at concentrations of <10 mM. More interestingly, DTT variation revealed the first small but significant difference between PHLPP2 and PHLPP1 activities, PHLPP2 being ∼2-fold more active than PHLPP1 in the absence of DTT.
Figure 4.

Activation of PHLPP by DTT and selected lipophilic compounds. (a) Effect of DTT on PHLPP1 and PHLPP2 activity. k values for PHLPP1 (white bars) and PHLPP2 (gray bars) were determined at 10 mM pNPP, in the absence of Mn2+ for different concentrations of DTT. (b) Effect of different lipophilic compounds on PHLPP2 phosphatase activity. Reactions were conducted in Tricine (pH 7.5) at 10 mM pNPP and 1 mM MnCl2 in the presence of 100 μM lipophilic compounds. (c) Relative activity of the PHLPP2 phosphatase domain with increasing concentrations of oleic (circles) and arachidonic (triangles) acid. Reactions were conducted in Tricine (pH 7.5) at 10 mM pNPP and 5 mM MnCl2. Graphs show means ± SEM for three separate experiments. *P < 0.01, and **P < 0.001 (Student’s t test).
It has been reported that PP2Cα and PP2Cβ are activated by fatty acids.31−33 Specifically, fatty acids enhance the affinity for Mn2+, such that they increase the activity recorded at subsaturating concentrations of Mn2+. We thus tested the effects of various fatty acids on the kinetic parameters of PHLPP, using the phosphatase domain of PHLPP2 purified from insect cells as an example. Fatty acids (100 μM) of various lengths (16–20 carbons), saturated state (zero to four double bonds), and configuration (cis vs trans double bond) were tested in the presence of 1 mM MnCl2 (Figure 4b). We found that only oleic acid (18:1 cis) and arachidonic acid (20:4) were able to increase PHLPP2 activity, by 30 and 50%, respectively. Palmitoleic acid was slightly inhibitory under these conditions. The effects of oleic and arachidonic acids were studied in more detail and revealed that activation of PHLPP started between 30 and 100 μM and increased with the concentration of the compound (Figure 4c). It should be noted that elaidic acid (18:1 trans), the isomer of oleic acid, did not change the PHLPP activity at any concentration tested (data not shown). These results are in good agreement with previous studies showing the importance of the configuration of the double bond.33 We also verified that the effects of fatty acids were linked to the presence of Mn2+. Indeed, addition of any fatty acid in solutions containing no supplementary Mn2+ had no effect on the activity of the protein (data not shown).
Thermal Stability of the Domain
The thermal stability of the isolated phosphatase domain, purified from Escherichia coli, was evaluated. First, we used circular dichroism (CD) to compare global structural states of PHLPP1 and PHLPP2 (Figure 5a). Little difference was observed in the CD spectra of the two proteins. The phosphatase domain of PHLPP2 was then submitted to increasing temperatures, and effects on protein structure were assessed using CD (Figure 5b). Changes in the slope of residue ellipticity versus temperature were interpreted as structural variations, in this case, unfolding of the protein. The PP2C domain of PHLPP2 had a temperature of denaturation of 50 °C in the reaction buffer. The same temperature of denaturation could be obtained using fluorescence polarization as a readout. PHLPP1 and PHLPP2 could then be compared, with PHLPP1 being unfolded at a slightly higher temperature (58 °C). We then measured the enzymatic activity of the PHLPP1 or PHLPP2 phosphatase domain using a fluorescent substrate, difluoromethyl umbeliferryl phosphate (DiFMUP). The dephosphorylation rate increased with an increase in temperature, up to a plateau, and then the enzyme slowly lost activity as heat denatured it (Figure 5c). The optimal temperature for activity was between 40 and 50 °C for PHLPP2, with PHLPP1 being more stable (Figure 5c). We then evaluated the importance of the metallic cations on thermal stability. To this end, we assessed the temperature of denaturation of the enzyme after dialysis against 5 mM EDTA. These conditions have been shown to efficiently remove the Mn2+ ions from the catalytic pocket of PP2Cα.34 We observed a transition in fluorescence at 37 °C, around 10 °C lower than that for the untreated protein. We then incubated the apoprotein in buffer containing 5 mM Mn2+, Mg2+, or Ca2+ for 48 h and determined the temperature of denaturation (Figure 5d). We first verified the Tm we found for Mn2+ was identical to that determined for proteins that had always been stored in the presence of Mn2+. All three metal ions increased thermal stability relative to that of the apoenzyme, in the following order: Mg2+ > Ca2+ > Mn2+. This increase was also observed when using the activity assay as a measure of thermal sensitivity (Figure 5e). These data indicate that both phosphatase domains are relatively stable proteins, with the phosphatase domain of PHLPP1 being slightly more stable than that of PHLPP2.
Figure 5.
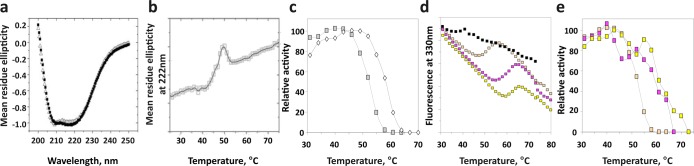
Structural stability of the isolated phosphatase domain of PHLPP. (a) Circular dichroism (CD) spectra for PHLPP1 (■) and PHLPP2 (△) phosphatase domains, isolated from bacteria. (b) The phosphatase domain of PHLPP2 was subjected to increasing temperatures, and variations of the mean residue ellipticity at 222 nm (by CD) were recorded. (c) Activity of the phosphatase domain of PHLPP1 (empty diamonds) or PHLPP2 (gray squares) was measured by fluorescence at different temperatures using DiFMUP as the substrate. (d) Phosphatase domain PHLPP2 treated with EDTA (black squares) and subsequently incubated with MnCl2 (orange squares), MgCl2 (yellow squares), or CaCl2 (pink squares) was subjected to increasing temperatures, and fluorescence emission at 330 nm was recorded. (e) Activity of PHLPP2 incubated with MnCl2 (orange squares), MgCl2 (yellow squares), or CaCl2 (pink squares) was measured by fluorescence at different temperatures using DiFMUP as the substrate.
Distinct Inhibitor Sensitivity of the PHLPP Phosphatase Domain
We next compared the inhibitor sensitivity of the PHLPP2 PP2C domain to that of PP2Cα using compounds we previously identified in a biochemical and virtual screen for PHLPP inhibitors.25 Compounds NSC 27157 and NSC 170008 were selective for the PHLPP2 PP2C domain, whereas compounds NSC 73101 and NSC 11241 were selective for PP2Cα (Figure 6). This selectivity underscores the unique architecture of the PHLPP PP2C domain compared to that of PP2Cα.
Figure 6.

Unique inhibitor sensitivity of the PHLPP PP2C domain. Relative activity of the PHLPP2 PP2C domain (gray squares) and PP2Cα (white circles) in the presence of different molecules (NCS 270156, NCS 170008, NCS 73101, and NCS 11241). Compounds NCS 270156 and NCS 170008 were selective for PHLPP2 compared to PP2Cα, whereas NCS 73101 inhibits PP2Cα preferentially. Reactions were conducted in Tricine (pH 7.5) at 10 mM pNPP at various inhibitor concentrations. Graphs show means ± SEM for three separate experiments.
Mutational Analysis of PHLPP2
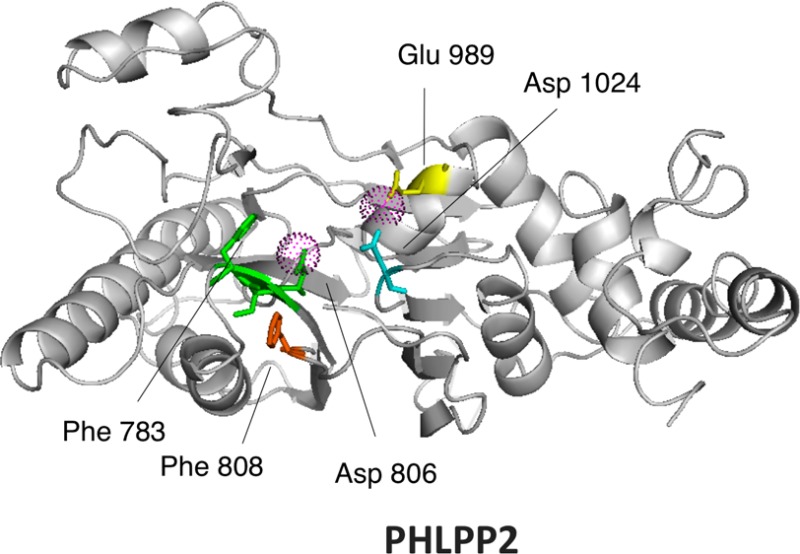
We have previously reported the construction of a homology model for PHLPP225 based on the crystal structure of PP2Cα34 (Figure 7b). We use this model to predict residues essential for catalysis. As noted above, PP2Cs share 11 conserved sequence motifs, with key conserved aspartic acids in motifs 1, 2, 8, and 11 (Figure 7c).9 We identified Asp 806 and Asp 1024 as corresponding to the catalytic Asp in the conserved 55DGxxG (PP2Cα numbering) of motif 2 (Figure 7c, green) and conserved Gxx282DN (PP2Cα numbering) of motif 11 (Figure 7c, cyan); these were mutated to Asn (D806N and D1024N). No Asp corresponding to that in the consensus Rxxx38D of motif 1 (Figure 7c, orange) was present; rather, the corresponding position was occupied by Trp 784. This and the preceding Phe 783 were mutated to Ala and Val, respectively, to test their role in maintaining the packing of the domain (F783V and W784A). The aspartic acid in the consensus 239DG in motif 8 (Figure 7c, yellow) was also not found in PHLPP2; rather, a lysine (Lys 985) aligned with this residue. This was mutated to leucine (K985L). In the homology model, we noticed that a glutamate (Glu 989) is positioned one helix turn from Lys 985, suggesting it could be the relevant acidic residue in motif 8; this was mutated to Gln (E989Q). Lastly, a previous sequence alignment identified Asp 820 as a conserved aspartic acid as the surrounding motif was conserved (motif 2, F/YDG conserved in most mammalian PP2Cs) . However, our homology model locates Asp 820 on the surface of the domain, away from the catalytic site. The D820N mutation was therefore introduced to assess if this residue indeed played a role in catalysis. We also mutated a third bulky aromatic, Phe 808 (F808V), as it could play a role in a π-stacking with Phe 782 and Tyr 784. The mutations were introduced into HA-tagged, full-length PHLPP2. The mutants were expressed in COS7 cells and partially purified by immunoprecipitation. Activity was assessed by both pNPP and malachite green assays (Figure 7a). Mutation of Asp 806 or Asp 1024 significantly impaired activity, confirming their equivalence to the conserved Asp in motifs 2 and 11. Interestingly, mutation of Glu 989 in motif 8 also impaired activity, suggesting its functional equivalence to the Asp in motif 8. Mutation of either Phe 783 or Phe 808, but not Trp 784, inhibited the enzyme, consistent with important structural roles for the two Phe residues. Mutation of Asp 820 did not alter activity, validating its positioning away from the catalytic pocket. Mutation of Lys 985 also had no effect on activity.
Figure 7.

Mutational analysis of PHLPP2. (a) Relative activity of point mutants of PHLPP2 using a peptide as the substrate. Point mutants of full-length PHLPP2 were overexpressed as HA-tagged fusion proteins in COS7 cells and immunoprecipitated. They were used to dephosphorylate the threonine-phosphorylated peptide RRAPTVA in Tricine (pH 7.5) in the presence of 5 mM MnCl2. Release of inorganic phosphate was monitored by the malachite green assay, and the speed of dephosphorylation was divided by the relative amount of protein, as determined by Western blotting. Activity is given relative to that of wild-type PHLPP2. The graph shows means ± SEM of three separate experiments. (b) Position of the mutated residues in the homology model of PHLPP2.25 (c) Sequence alignment of subdomains 1, 2, 8, and 11 of the PP2C family showing conserved RXXXD, DGXXG, DG, and GXXDN (colored orange, green, yellow, and cyan, respectively). Mn2+-coordinating acidic residues are colored red. Below is a schematic of PP2Cα (left) showing four asparates that coordinate the two bound Mn2+ ions (magenta spheres)34 and a homology model of PHLPP225 showing equivalent acidic residues and structurally important Phe residues.
Discussion
PHLPP Phosphatase Activity
PHLPP has the hallmarks of a PP2C. The phosphatase domain of PHLPP can be purified from various cells and demonstrates in every case the capacity to dephosphorylate synthetic and natural substrates. Consistent with a known property of PP2C enzymes, activity is insensitive to okadaic acid. Sequence alignment predicts that the phosphatase domains of PHLPP1 and PHLPP2 constitute a discrete subfamily of PP2C. Some distinctions indeed appear upon comparison to other members of the family. When the PP2C domains of PHLPP1 and PHLPP2 are expressed in bacteria, the enzyme is active and dephosphorylates pNPP with a kcat of 1.8 × 10–3 s–1 and a kcat/Km of ∼1 M–1 s–1. PP2Cα, which is a standard in the PP2C family, can also be purified from E. coli and hydrolyzes pNPP with a kcat of 1.02 s–1 and a kcat/Km of 859 M–1 s–1.28 PP2Cα is therefore almost 3 orders of magnitude more active and efficient at dephosphorylating this substrate. Purification from Sf21 cells increases the activity of the PHLPP phosphatase domain, but this enzyme is still 3-fold less active and less efficient than PP2Cα. However, when a phosphopeptide is used as the substrate, PHLPP as a phosphatase is almost as good as PP2Cα, even when purified from bacteria. PP2Cα dephosphorylates the PP2C preferred substrate peptide (RRAPTVA) with a kcat of 5.2 s–1 and a kcat/KM of 48 × 103 M–1 s–1 according to one study28 and 16.5 s–1 and 109 × 103 M–1 s–1 according to another.30 PHLPP from bacteria is therefore only 2–4-fold less active than PP2Cα when peptides are used as substrates. The proteins purified from insect cells are slightly more active than the bacterially expressed ones. The Km values are lower for all peptides than for pNPP, indicating a better recognition of peptides as substrates, as expected. The change in kcat, however, was more pronounced than anticipated. Indeed, in the case of PP2Cα, dephosphorylation occurred with an only 5–15-fold increase when using RRAPTVA as a substrate compared to pNPP, and kcat values were even decreased when other peptide sequences were used.36 The same reduction in kcat between pNPP and peptides was observed with PrpZ, a Salmonella enterica PP2C, for example.37 pNPP is supposed to mimic phosphorylated tyrosine residues, but the data obtained with PHLPP indicate it is a weak mimetic as a phosphotyrosine peptide was dephosphorylated as effectively as the Ser/Thr phosphopeptides (data not shown).
The major difference between PHLPP and other members of the PP2C family lies in their metal requirement. PP2C family members require either Mn2+ or Mg2+ for their activity. Mn2+ is most commonly preferred, but Mg2+ is often able to activate the enzymes and, in some instances, is the most efficient cofactor. We showed that PHLPP was active even in the absence of a cofactor and that we observed a residual activity in the presence of EDTA. In a majority of cases, PP2C family members had no in vitro activity in the absence of Mn2+ or Mg2+, on any substrate. A few exceptions exist. PP1MH is inactive toward pNPP and phosphopeptides without Mn2+ but was able to dephosphorylate casein in the absence of an additional metallic cation.30 Conversely, PP2Cβ has a small basal activity toward phosphopeptides but none toward casein.31 Nevertheless, this feature is an oddity in the family. In the case of PHLPP, the metal ions appear to play both a structural role and a catalytic role. On one hand, EDTA treatment causes a structural rearrangement, as observed by fluorescence (Figure 5d and SI), resulting in a conformation of the enzyme with a reduced thermal stability but unchanged catalytic activity. On the other hand, mutation of either key aspartic acid in the catalytic site (D806 or D1024) reduces PHLPP activity by 50%, suggesting that metal ions are involved in the catalytic process. These metals may be incorporated during protein expression (in a Mn2+-rich environment) and are likely to be tightly bound. Additional ions may be more loosely associated with the protein and play a structural role. Varying the nature of these ions affects the thermal stability of the recombinant enzyme (Figures 5d and 5e) but not its activity. PHLPP activity can be only enhanced by the addition of Mn2+. This is always the case for proteins expressed in insect cells or mammalian cells. In the case of bacterially expressed proteins, the influence of Mn2+ is more complex, being mainly inhibitory. However, this trend is overturned above pH 8.0 when these enzymes start to act like their counterparts purified in other systems. Activation by Mn2+ is enhanced by fatty acids as described previously,31−33 although the amplitude of the effect was attenuated.
Proteins expressed in E. coli showed some differences compared to proteins expressed in other systems. The most dramatic feature was their weak activity, as determined by the kcat, using pNPP as a substrate. However, this did not hold true for other substrates as shown in Table 1. We also found that those recombinant proteins were a good model for the metal requirement for PHLPP activity. Bacteria may lack the proper enzymes for post-translational modification or the proper chaperones for correct folding to ensure that the enzymes adopt conformations for maximal catalytic activity. We know that bacterially expressed protein displayed the odd characteristic of gaining activity upon being stored at 4 °C, consistent with slow structural rearrangements. Nonetheless, bacteria are still the preferred source of recombinant proteins as this system gives the highest yield of PHLPP. Care should be taken when analyzing results using bacterially expressed proteins, and comparison with proteins from mammalian cells should be undertaken to assess the physiological relevance of the results.
Mutational Analysis of PHLPP2
The archetypal mammalian PP2C is characterized by a central, buried β-sandwich composed by two antiparallel β-sheets, flanked by two antiparallel α-helices. Its catalytic core is composed of two Mn2+ ions coordinated by four invariant Asp residues and a nonconserved Glu.38 Mutational analysis of PP2Cα39 or PP2Cβ40 also identifies two important positively charged residues, an Arg and a His, the first one positioning the phosphate group correctly and the second one acting as a general acid for catalysis (Figure 7c). Using point mutations, we were able to identify three residues that are important for catalysis that are likely to coordinate metals: Asp 806 (corresponding to Asp 60 in PP2Cα), Asp 1024 (Asp 282 in PP2Cα), and Glu 989 (Asp 239 in PP2Cα). The exact alignment for residue 238 in PP2Cα is actually Lys 985. Mutation of Lys 985 to a neutral residue such as Leu has no effect, as shown, nor do mutations of this residue to an acidic residue such as Asp or Glu (data not shown). Glu 989 is predicted to be located one helix turn away compared to PP2Cα; therefore, we predict the distance between the two metallic centers to be significantly increased. We could not identify a homologue for Asp 38 (in motif 1) in PHLPP2, but this residue is not essential for catalysis.39 Mutation of Glu 37 or Asp 38 in PP2Cα does not result in a dramatic decrease in activity; indeed, the D38Q mutant is more active than wild-type PP2Cα. Asp 38 in PP2Cα is located at the beginning of the β3 sheet and coordinates the same Mn2+ as Asp 60, which is at the end of the β4 sheet34(Figure 7c). It is possible that this metallic complex adds stability to the structure. If so, this feature could be recapitulated in PHLPP2 by a putative π-stacking between Phe 783 (at the beginning of the β3 sheet) and Phe 808 (at the end of the β4 sheet) (Figure 7b,c). Supporting this hypothesis is the fact that mutation of either residue leads to almost complete inhibition of the PHLPP2 activity, even though the mutants are normally expressed.
The catalytic site we describe is missing elements compared to the canonical PP2C catalytic site9,34(Figure 7c). One possibility is that our homology model is inaccurate and the actual folding of the protein is providing additional coordination points for the metal that we missed. Still, this homology model performed well for determination of inhibitors and provided us with mechanistic details about inhibition.25 Another possibility is that not all residues are required for catalytic activity, contrary to previous reports.42 Different approaches can be developed by proteins to stabilize their structure or position or activate substrates. The low activity of the PP2C domain of PHLPP isolated from bacteria, when compared to other PP2Cs, could indicate that the active site of PHLPP is indeed different from the rest of the PPM family, making it less efficient or more specific toward substrates. Only resolution of the crystal structure of the phosphatase domain of PHLPP can definitively reveal the exact architecture of the active site.
In conclusion, we purified the catalytic domains of PHLPP1 and PHLPP2 from three different cell expression systems: bacteria, insect cells, and mammalian cells. These proteins dephosphorylate synthetic and peptidic substrates with kinetics that depend on both the substrate and the expression systems. We determined that PHLPP1 and PHLPP2 have almost similar in vitro activities and respond comparably to the presence of metallic ions. We also showed that metallic ions affect the structural stability of the domain. Finally, we probed for residues predicted to be important for catalysis in PHLPP2 by site-directed mutagenesis. We identified three residues that are likely involved in metal coordination and two that could be important for structural integrity. This work is only the beginning of the biochemical characterization of PHLPP, and many questions remain. One step is the identification of the residues involved in the active site of PHLPP1. Even though PHLPP1 and PHLPP2 are 58% identical in their PP2C domain amino acid sequences and present comparable kinetic profiles in vitro when using isolated domains purified from bacteria, differences are expected as, for example, the two critical Phe we identified in PHLPP2 have no equivalent in the PHLPP1 sequence. Another essential step is to understand the interactions between the domains and how this affects the activity of the phosphatase, the final goal being to ascertain how PHLPP phosphatase activity is regulated in the cell.
Acknowledgments
We thank Yann Gambin for help with the structural data collection and analyses, Bill Sinko and Corina Antal for help with sequence alignment and modeling, and David Brautigan and Jack Dixon for helpful advice on phosphatase assays.
Glossary
Abbreviations
- PHLPP
PH domain leucine-rich repeat protein phosphatase
- pNPP
p-nitrophenyl phosphate
- PKC
protein kinase C
- SEM
standard error of the mean.
Supporting Information Available
Buffers used at various pH values (Table 1) and fluorescence of the tryptophan for the phosphatase domain of PHLPP2 recorded as a function of time following treatment with 100 mM EDTA. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† E.S.: The University of Queensland, Institute for Molecular Bioscience, St. Lucia, Queensland 4072, Australia.
This work was supported by National Institutes of Health Grant GM067946 (A.C.N.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Warfel N. A.; Newton A. C. (2012) Pleckstrin Homology Domain Leucine-rich Repeat Protein Phosphatase (PHLPP): A New Player in Cell Signaling. J. Biol. Chem. 287, 3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton A. C.; Trotman L. C. (2014) Turning off AKT: PHLPP as a drug target. Annu. Rev. Pharmacol. Toxicol. 54, 537–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T.; Furnari F.; Newton A. C. (2005) PHLPP: A Phosphatase that Directly Dephosphorylates Akt, Promotes Apoptosis, and Suppresses Tumor Growth. Mol. Cell 18, 13–24. [DOI] [PubMed] [Google Scholar]
- Brognard J.; Sierecki E.; Gao T.; Newton A. C. (2007) PHLPP and a Second Isoform, PHLPP2, Differentially Attenuate the Amplitude of Akt Signaling by Regulating Distinct Akt Isoforms. Mol. Cell 25, 917–931. [DOI] [PubMed] [Google Scholar]
- Gao T.; Brognard J.; Newton A. C. (2008) The Phosphatase PHLPP Controls the Cellular Levels of Protein Kinase C. J. Biol. Chem. 283, 6300–6311. [DOI] [PubMed] [Google Scholar]
- Qiao M.; Wang Y.; Xu X.; Lu J.; Dong Y.; Tao W.; Stein J.; Stein G. S.; Iglehart J. D.; Shi Q.; Pardee A. B. (2010) Mst1 is an interacting protein that mediates PHLPPs’ induced apoptosis. Mol. Cell 38, 512–523. [DOI] [PubMed] [Google Scholar]
- O’Neill A. K.; Niederst M. J.; Newton A. C. (2013) Suppression of survival signalling pathways by the phosphatase PHLPP. FEBS J. 280, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brognard J.; Newton A. C. (2008) PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol. Metab. 19, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. (2009) Serine/threonine phosphatases: Mechanism through structure. Cell 139, 468–484. [DOI] [PubMed] [Google Scholar]
- Rodriguez P. L. (1998) Protein phosphatase 2C (PP2C) function in higher plants. Plant Mol. Biol. 38, 919–927. [DOI] [PubMed] [Google Scholar]
- Schweighofer A.; Hirt H.; Meskiene I. (2004) Plant PP2C phosphatases: Emerging functions in stress signaling. Trends Plant Sci. 9, 236–243. [DOI] [PubMed] [Google Scholar]
- Lammers T.; Lavi S. (2007) Role of type 2C protein phosphatases in growth regulation and in cellular stress signaling. Crit. Rev. Biochem. Mol. Biol. 42, 437–461. [DOI] [PubMed] [Google Scholar]
- Tamura S.; Toriumi S.; Saito J.; Awano K.; Kudo T. A.; Kobayashi T. (2006) PP2C family members play key roles in regulation of cell survival and apoptosis. Cancer Sci. 97, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guezennec X.; Bulavin D. V. (2010) WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem. Sci. 35, 109–114. [DOI] [PubMed] [Google Scholar]
- Chen M.; Pratt C. P.; Zeeman M. E.; Schultz N.; Taylor B. S.; O’Neill A.; Castillo-Martin M.; Nowak D. G.; Naguib A.; Grace D. M.; Murn J.; Navin N.; Atwal G. S.; Sander C.; Gerald W. L.; Cordon-Cardo C.; Newton A. C.; Carver B. S.; Trotman L. C. (2011) Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell 20, 173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Weiss H. L.; Rychahou P.; Jackson L. N.; Evers B. M.; Gao T. (2008) Loss of PHLPP expression in colon cancer: Role in proliferation and tumorigenesis. Oncogene 28, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser S.; Park Y.-K.; Franklin J.; Halberg R.; Yu M.; Jessen W.; Freudenberg J.; Chen X.; Haigis K.; Jegga A.; Kong S.; Sakthivel B.; Xu H.; Reichling T.; Azhar M.; Boivin G.; Roberts R.; Bissahoyo A.; Gonzales F.; Bloom G.; Eschrich S.; Carter S.; Aronow J.; Kleimeyer J.; Kleimeyer M.; Ramaswamy V.; Settle S.; Boone B.; Levy S.; Graff J. (2007) Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol. 8, R131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabates-Bellver J.; Van der Flier L. G.; de Palo M.; Cattaneo E.; Maake C.; Rehrauer H.; Laczko E.; Kurowski M. A.; Bujnicki J. M.; Menigatti M.; Luz J.; Ranalli T. V.; Gomes V.; Pastorelli A.; Faggiani R.; Anti M.; Jiricny J.; Clevers H.; Marra G. (2007) Transcriptome Profile of Human Colorectal Adenomas. Mol. Cancer Res. 5, 1263–1275. [DOI] [PubMed] [Google Scholar]
- Karnoub A. E.; Dash A. B.; Vo A. P.; Sullivan A.; Brooks M. W.; Bell G. W.; Richardson A. L.; Polyak K.; Tubo R.; Weinberg R. A. (2007) Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449, 557–563. [DOI] [PubMed] [Google Scholar]
- Richardson A. L.; Wang Z. C.; De Nicolo A.; Lu X.; Brown M.; Miron A.; Liao X.; Iglehart J. D.; Livingston D. M.; Ganesan S. (2006) X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 9, 121–132. [DOI] [PubMed] [Google Scholar]
- Bredel M.; Bredel C.; Juric D.; Harsh G. R.; Vogel H.; Recht L. D.; Sikic B. I. (2005) High-Resolution Genome-Wide Mapping of Genetic Alterations in Human Glial Brain Tumors. Cancer Res. 65, 4088–4096. [DOI] [PubMed] [Google Scholar]
- Parsons D. W.; Jones S.; Zhang X.; Lin J. C.-H.; Leary R. J.; Angenendt P.; Mankoo P.; Carter H.; Siu I. M.; Gallia G. L.; Olivi A.; McLendon R.; Rasheed B. A.; Keir S.; Nikolskaya T.; Nikolsky Y.; Busam D. A.; Tekleab H.; Diaz L. A. Jr.; Hartigan J.; Smith D. R.; Strausberg R. L.; Marie S. K. N.; Shinjo S. M. O.; Yan H.; Riggins G. J.; Bigner D. D.; Karchin R.; Papadopoulos N.; Parmigiani G.; Vogelstein B.; Velculescu V. E.; Kinzler K. W. (2008) An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 321, 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreozzi F.; Procopio C.; Greco A.; Mannino G. C.; Miele C.; Raciti G. A.; Iadicicco C.; Beguinot F.; Pontiroli A. E.; Hribal M. L.; Folli F.; Sesti G. (2011) Increased levels of the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP)-1 in obese participants are associated with insulin resistance. Diabetologia 54, 1879–1887. [DOI] [PubMed] [Google Scholar]
- Miyamoto S.; Purcell N. H.; Smith J. M.; Gao T.; Whittaker R.; Huang K.; Castillo R.; Glembotski C. C.; Sussman M. A.; Newton A. C.; Brown J. H. (2010) PHLPP-1 Negatively Regulates Akt Activity and Survival in the Heart. Circ. Res. 107, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierecki E.; Sinko W.; McCammon J. A.; Newton A. C. (2010) Discovery of Small Molecule Inhibitors of the PH Domain Leucine-Rich Repeat Protein Phosphatase (PHLPP) by Chemical and Virtual Screening. J. Med. Chem. 53, 6899–6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza R. C.; Junqueira J. C.; Rossoni R. D.; Pereira C. A.; Munin E.; Jorge A. O. (2010) Comparison of the photodynamic fungicidal efficacy of methylene blue, toluidine blue, malachite green and low-power laser irradiation alone against Candida albicans. Lasers in Medical Science 25, 385–389. [DOI] [PubMed] [Google Scholar]
- Donella Deana A.; Mac Gowan C. H.; Cohen P.; Marchiori F.; Meyer H. E.; Pinna L. A. (1990) An investigation of the substrate specificity of protein phosphatase 2C using synthetic peptide substrates; comparison with protein phosphatase 2A. Biochim. Biophys. Acta 2, 199–202. [DOI] [PubMed] [Google Scholar]
- Fjeld C. C.; Denu J. M. (1999) Kinetic Analysis of Human Serine/Threonine Protein Phosphatase 2Cα. J. Biol. Chem. 274, 20336–20343. [DOI] [PubMed] [Google Scholar]
- Barford D.; Das A. K.; Egloff M.-P. (1998) The Structure and Mechanism of Protein Phosphatases: Insights into Catalysis and Regulation. Annu. Rev. Biophys. Biomol. Struct. 27, 133–164. [DOI] [PubMed] [Google Scholar]
- Sugiura T.; Noguchi Y. (2009) Substrate-dependent metal preference of PPM1H, a cancer-associated protein phosphatase 2C: Comparison with other family members. BioMetals 22, 469–477. [DOI] [PubMed] [Google Scholar]
- Krieglstein J.; Selke D.; Maassen A.; Klumpp S. (2003) Activity of PP2Cb is increased by divalent cations and lipophilic compounds depending on the substrate. Methods Enzymol. 366, 282–289. [DOI] [PubMed] [Google Scholar]
- Krieglstein J.; Hufnagel B.; Dworak M.; Schwarz S.; Kewitz T.; Reinbold M.; Klumpp S. (2008) Influence of various fatty acids on the activity of protein phosphatase type 2C and apoptosis of endothelial cells and macrophages. Eur. J. Pharm. Sci. 35, 397–403. [DOI] [PubMed] [Google Scholar]
- Klumpp S.; Selke D.; Hermesmeier J. (1998) Protein phosphatase type 2C active at physiological Mg2+: Stimulation by unsaturated fatty acids. FEBS Lett. 437, 229–232. [DOI] [PubMed] [Google Scholar]
- Das A. K.; Helps N. R.; Cohen P. T.; Bradford D. (1996) Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 Å resolution. EMBO J. 15, 6798–6809. [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H.; Durell S. R.; Chatterjee D. K.; Anderson C. W.; Appella E. (2007) The Wip1 phosphatase PPM1D dephosphorylates SQ/TQ motifs in checkpoint substrates phosphorylated by PI3K-like kinases. Biochemistry 46, 12594–12603. [DOI] [PubMed] [Google Scholar]
- Lai S. M.; Le Moual H. (2005) PrpZ, a Salmonella enterica serovar Typhi serine/threonine protein phosphatase 2C with dual substrate specificity. Microbiology 151, 1159–1167. [DOI] [PubMed] [Google Scholar]
- Barford D. (1996) Molecular mechanisms of the protein serine/threonine phosphatases. Trends Biochem. Sci. 21, 407–412. [DOI] [PubMed] [Google Scholar]
- Jackson M. D.; Fjeld C. C.; Denu J. M. (2003) Probing the Function of Conserved Residues in the Serine/Threonine Phosphatase PP2Cα. Biochemistry 42, 8513–8521. [DOI] [PubMed] [Google Scholar]
- Kusuda K.; Kobayashi T.; Ikeda S.; Ohnishi M.; Chida N.; Yanagawa Y.; Shineha R.; Nishihira T.; Satomi S.; Hiraga A.; Tamura S. (1998) Mutational analysis of the domain structure of mouse protein phosphatase 2Cβ. Biochem. J. 332, 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner S. H.; Kular G.; Peggie M.; Sheperd S.; Schuttelkopf A. W.; Cohen P.; Van Aalten D. M. F. (2006) TAK1-binding protein 1 is a pseudophosphatase. Biochem. J. 399, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.