

Abstract
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder with a prevalence of more than 1% after the age of 65 years. Mutations in the gene encoding leucine-rich repeat kinase 2 (LRRK2) have recently been linked to autosomal dominant, late-onset PD that is clinically indistinguishable from typical, idiopathic disease. LRRK2 is a multi-domain protein containing several protein interaction motifs as well as dual enzymatic domains of GTPase and protein kinase activities. Disease-associated mutations are found throughout the multi-domain structure of the protein. LRRK2, however, is unique among the PD-causing genes because a missense mutation, G2019S, is a frequent determinant of not only familial, but also of sporadic PD. Thus, LRRK2 has emerged as a promising therapeutic target for combating PD. This article reviews the current state of knowledge regarding the domain structure, amino acid substitutions, and potential functional roles of LRRK2.
Keywords: Parkinson’s disease, leucine-rich repeat kinase 2 (LRRK2), neurodegeneration, signal transduction
Introduction
Parkinson’s disease (PD)1 is a chronic and progressive neurodegenerative disorder of the central nervous system that affects control of voluntary movement. Pathologically, PD is characterized by the loss of dopaminergic neurons of the substantia nigra pars compacta (SNpc) and the presence of intraneuronal proteinacious cytoplasmic inclusions, termed Lewy bodies, in surviving neurons. Clinically, patients typically exhibit motor symptoms including tremor, rigidity, bradykinesia, and postural instability, and beneficially respond to dopamine replacement therapy. Common to multi-factorial diseases, the incidence of PD increases with age. The percentage of affected individuals within a population rises from ~1% at 65 years of age to ~5% by 85 years of age, with a mean age at onset of 55 years (Dauer and Przedborski 2003). Because of an aging population, improved diagnosis, and prolonged survival, especially in developing countries, the number of patients suffering from PD is predicted to double by the year 2030 (Dorsey et al. 2007).
While enormous steps have been taken over nearly the past 200 years in understanding the neurobiology of PD, the exact etiology of this devastating disease is still largely unknown. Currently, several mechanisms are implicated in the selective degeneration of dopaminergic neurons in PD (Cookson 2005). For example, failure or impairment in the ubiquitin-proteasome system, potentially leading to the aggregation of oligomeric proteins, may play a role in PD pathogenesis. Mitochondrial complex I toxins, such as 1-methyl-4-phenylpyridinium (MPP+) and paraquat, have shown that mitochondrial dysfunction and increasing oxidative stress also likely contribute to disease pathogenesis. Dopaminergic neurons may be selectively vulnerable to such cellular insults because of the endogenous elevated level of oxidative stress arising from the presence of dopamine in these cells (Schulz and Falkenburger 2004).
Although the majority of PD cases are sporadic, ~5–15% of cases are due to genetic alterations. A major breakthrough in recent years in understanding PD pathogenesis has been the mapping of ten chromosomal regions, named PARK1-11, and the subsequent cloning of seven genes involved in familial PD (Table 1). These include three autosomal dominant genes(α-synuclein, leucine-rich repeat kinase 2 [LRRK2], and Grb10-interacting GYF protein 2 [GIGYF2]), and three autosomal recessive genes (parkin, DJ-1, and phosphatase and tensin homolog (PTEN)-induced kinase 1 [PINK1]) (Bonifati et al. 2003; Chartier-Harlin et al. 2004; Kitada et al. 1998; Paisan-Ruiz C 2004; Polymeropoulos et al. 1997; Singleton et al. 2003; Valente et al. 2004; Zimprich A 2004). A mutation in the seventh gene, ubiquitin C-terminal hydrolase L1 (UCH-L1), has only been found in one family and the importance of this gene in familial PD is still uncertain (Leroy et al. 1998). Although mutations in these genes account for only a small percentage of all PD cases, studying the encoded proteins has enabled and supported many of the proposed mechanisms underlying the pathogenesis of PD. Identifying the causative genes associated with the other remaining PARK loci, for which linkage has been identified, will likely further contribute to elucidating the etiology of PD.
Table 1.
Parkinson’s Disease-Associated Genes
| Locus | MOI | Chromosomal Location | Protein Name | Mutations | Protein Function | Age at Onset |
|---|---|---|---|---|---|---|
| PARK1/4 | AD | 4q21 | α-synuclein | dominant A30P, E46K and A53T; genome duplication/triplication | DA transmission, SV dynamics | 30–60s |
| PARK2 | AR | 6q25 | parkin | numerous recessive homozygous/compound heterozygous missense, truncating, exon deletion/duplication/triplication | E3 ubiquitin ligase | 20–40s |
| PARK5 | AD | 4p14 | UCH-L1 | heterozygous I93M | ubiquitin hydrolase | 30–50s |
| PARK6 | AR | 1p35-37 | PINK1 | various recessive missense, exon deletion | mitochondrial kinase | 30–50s |
| PARK7 | AR | 1p38 | DJ-1 | various recessive homozygous/compound heterozygous missense, exon deletion | oxidative stress response | 20–40s |
| PARK8 | AD | 12q12 | LRRK2 | numerous dominant missense | protein kinase and GTPase | 40–60s |
| PARK11 | AD | 2q21.2 | GIGYF2 | several dominant missense | insulin signaling | 40–70s |
Abbreviations: MOI, mode of inheritance; AD, autosomal dominant; AR, autosomal recessive; UCH-L1, ubiquitin C-terminal hydrolase; PINK1, phosphatase and tension homolog (PTEN)-induced putative kinase 1; LRRK2, leucine-rich repeat kinase 2; GIGYF2, GRB-10-interacting GYF protein 2; DA, dopamine; SV, synaptic vesicle. Age of onset reported in years.
Although these key genetic discoveries have played an important role in understanding PD, mutations in most PD-associated genes have been linked to early-onset or pathologically atypical forms of the disease in only a few families. Distinctively, mutations in the most recently identified PD-associated gene, LRRK2, which is linked to the PARK8 locus, have been associated with late-onset, autosomal-dominantly inherited PD (Paisan-Ruiz C 2004; Zimprich A 2004). LRRK2 is also known as dardarin, after dardara, the Basque word for tremor. The LRRK2 gene spans 51 exons and encodes a rather large (2,527 amino acids) protein of 286KDa that contains multiple, independent domains. Because mutations in LRRK2 cause a familial form of PD that pathologically resembles idiopathic disease and are also found in up to 3% of sporadic PD cases, this protein has emerged as the most significant player in PD pathogenesis identified to date (Aasly et al. 2005; Berg D 2005; Taylor JP 2006).
Though much has been learned about LRRK2 in the last few years, many important questions remain unanswered. For example, what is the normal function of LRRK2? What functional effect do LRRK2 PD-associated mutations have on neurons? Why are dopaminergic neurons particularly sensitive to LRRK2 mutations when the protein has a widespread expression pattern? This article reviews the current state of knowledge regarding the domain structure, amino acid substitutions, and potential functional roles of LRRK2.
LRRK2 Multi-Domain Structure
LRRK2 is a member of the ROCO protein family. This class of multi-domain proteins is characterized by the presence of a 200–250 amino acid Roc (Ras of Complex protein) domain, immediately followed by a 300–400 amino acid domain termed COR (C-terminal of Roc). The Roc and COR domains are usually followed by a kinase domain, and various protein-protein interaction motifs may also exist within ROCO proteins. There are at least 40 members in the ROCO superfamily and these proteins are found in a variety of species including mammals, metazoa, dictyostelium, plants, and prokaryotes (Bosgraaf L 2003).
Sequence analysis predicts that LRRK2 comprises several independent domains including a leucine-rich repeat (LRR) domain, a Roc domain followed by its associated COR domain, a mitogen-activated protein kinase kinase kinase (MAPKKK) domain, and a C-terminal WD40 domain (Fig. 1) (Bosgraaf L 2003; Guo L 2006) Genetic variations are found throughout the functional domains of LRRK2 (Fig. 1), suggesting that this protein may serve as an upstream central integrator of multiple signaling pathways that are crucial for proper neuronal functioning. Also, the presence of both protein interaction domains (LRR and WD40) as well as enzymatic domains (Roc and MAPKKK) within LRRK2 suggests that this protein may serve as a scaffold for assembly of a multi-protein signaling complex. The predicted functions of these multiple domains are outlined in Figure 1.
Fig. 1. Schematic representation of the predicted LRRK2 domain structure.
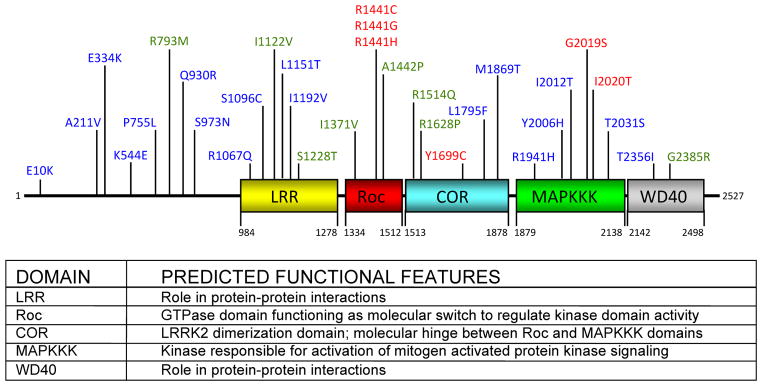
LRRK2 is composed of 13 LRRs, followed by the Roc, COR, and MAPKKK domains, as well as seven WD40 repeats at the C-terminus. Mutations that are considered definitively pathogenic are shown in red. Amino acid substitutions that can act as risk factors for disease and are putatively pathogenic are shown in green. Mutations that have been identified in PD patients but for which pathogenicity is uncertain are shown in blue. The amino acid residue numbers beneath each domain indicates the estimated domain boundaries. Predicted functions of each of the LRRK2 domains are listed. The GenBank accession number for human LRRK2 is AAV63975.
Leucine-rich repeat (LRR) domain
This motif is present in over 60 proteins and the primary function appears to be to provide a versatile structural framework for the formation of protein-protein interactions. LRR containing proteins participate in many biologically important processes, such as hormone-receptor interactions, enzyme inhibition, cell adhesion and cellular trafficking, early mammalian development, neural development, cell polarization, regulation of gene expression, apoptosis, and regulation of cytoskeletal dynamics (Kobe and Deisenhofer 1995). Based on homology modeling, many of the LRRK2 amino acid substitutions in the LRR domain are predicted to lie on the surface of this structural motif and, therefore, may be involved in forming protein-protein interactions (Mata et al. 2006b).
Ras of complex protein (Roc) and C-terminal of Roc (COR) domains
The Roc domain of ROCO family members stands out as a separate monophyletic group of the Ras-related superfamily of small GTPases (Bosgraaf L 2003). Three LRRK2 pathogenic amino acid substitutions in the Roc domain occur at the R1441 position (R1441C, R1441G, and R1441H). The COR domain is common to all ROCO proteins and is always found immediately C-terminal to the Roc domain. This Roc-COR module is conserved throughout evolution, suggesting a functional interdependence of these two motifs. Recent crystallographic structural determinations of Roc and Roc-COR suggest two different mechanisms of dimerization within these regions of LRRK2. Cookson and colleagues have reported that the Roc domain alone exists as a dimer with extensive domain swapping while Wittinghofer and colleagues have identified the COR domain as the dimerization motif (Deng et al. 2008; Gotthardt et al. 2008) Two PD-associated mutations, Y1699C and R1628P, are located in the COR domain.
Mitogen-activated protein kinase kinase kinase (MAPKKK) domain
Based on sequence similarity, the MAPKKK domain of LRRK2 belongs to the tyrosine kinase-like (TKL) subfamily of human protein kinases, whose members show sequence similarity to both serine/threonine and tyrosine kinases (Manning G 2002). The MAPKKK domain of LRRK2 most resembles receptor-interacting protein kinases (RIPKs), which are crucial sensors of cellular stress and can activate mitogen-activated protein kinase (MAPK) pathways (Meylan 2005). The PD-linked mutations G2019S and I2020T occur at the catalytic sites of the MAPKKK domain and are predicted to alter kinase activity (Guo L 2006; Marin 2006; Mata et al. 2006b).
WD40 domain
WD40 domains likely mediate protein-protein interactions. Interestingly, there are several examples in which WD40 domains are fused to domains that are predicted to have kinase activity, similar to the C-terminal domain arrangement of LRRK2 (Futey et al. 1995; Janda et al. 1996; Smith et al. 1999). The G2385R mutation lies within the WD40 domain (Mata et al. 2006b).
LRRK2 mRNA and Protein Expression
The expression pattern of LRRK2 mRNA in mouse and rat brains differs markedly from the cellular expression patterns of other genes linked to PD to date. For example, α-synuclein, parkin, UCHL-1, DJ-1, and PINK1 mRNA are all expressed in many, if not all, neurons of the brain, including dopamine neurons of the midbrain (Galter et al. 2007; Solano et al. 2000). Several recent reports, however, have found LRRK2 mRNA expression in dopamine-innervated areas such as the striatum, cortex, and olfactory tubercle, but expression was devoid in dopamine synthesizing neurons of the SNpc (Galter et al. 2006; Higashi et al. 2007b; Melrose et al. 2006; Taymans et al. 2006).
LRRK2 protein has been shown to be ubiquitously expressed and relatively abundant in most regions of the mouse and rat brain. LRRK2 protein was found to localize throughout the nigrostriatal dopaminergic pathway, with highest levels detected in the striatum but also at lower levels in the SNpc (Higashi et al. 2007b; Melrose et al. 2007). The apparent absence of LRRK2 mRNA in the SNpc despite the detection of low to moderate protein levels in this region may be due to several possibilities. For example, LRRK2 mRNA may have a short half-life or be transported to distal sites in nigral dopaminergic neurons. Alternatively, LRRK2 protein may have a particularly long half-life in these neurons so that only a small number of mRNA copies are required to maintain protein levels. LRRK2 protein expression was also significant in many non-dopaminergic areas such as the septal nuclei, hippocampus, and thalamus. LRRK2 staining was also present in the subventricular zone, a region containing stem cells that give rise to both neurons and glia, suggesting a potential role for LRRK2 in the regulation of neurogenesis (Melrose et al. 2007).
The expression of LRRK2 protein in mouse and rat brains has been further studied at a subcellular level. Despite the large size of LRRK2, in silico analysis failed to indicate the presence of hydrophobic transmembrane domains or obvious organelle targeting sequences (West et al. 2005). LRRK2 protein was found to localize throughout the cytoplasm of neuronal perikarya and dendritic processes where it associated with vesicular and membranous structures, the microtubule network, mitochondria, and other membrane-bound organelles (Biskup et al. 2006; Gandhi et al. 2008; Hatano et al. 2007). This is similar to the protein localization pattern found in LRRK2-overexpressing cell lines (Gloeckner et al. 2006; Greggio et al. 2006; Hatano et al. 2007; Smith et al. 2006; West et al. 2005). The R1441C and G2019S PD-associated mutations of LRRK2 did not alter this localization pattern in cultured cells (West et al. 2005). In mouse and rat brains, LRRK2 was specifically localized to Golgi transport vesicles, the plasma membrane, endosomes, lysosomes, mitochondria, synaptic vesicles, and microtubules (Biskup et al. 2006; Gandhi et al. 2008; Hatano et al. 2007). The membrane localization of LRRK2 was resistant to solubilization by non-ionic detergent, indicating that LRRK2 associates with lipid rafts, which are known to play important roles in signal transduction, membrane trafficking, and cytoskeletal organization (Brown and London 1998; Hatano et al. 2007). Taken together, these data suggest multiple possible roles for LRRK2: (1) a role in the biogenesis and/or regulation of vesicular structures throughout the neuron, (2) a role in vesicular transport, and (3) a role in membrane and protein turnover possibly through the lysosomal degradation pathway.
The expression of LRRK2 mRNA and protein has also been examined in neurologically normal human brains. Both mRNA and protein expression were found in brain regions of direct relevance to the pathogenesis of PD, including the cerebral cortex, caudate putamen, and SNpc (Biskup et al. 2006; Giasson et al. 2006; Greggio et al. 2006; Higashi et al. 2007a). LRRK2 protein localized to neuronal perikarya and dendritic and axonal processes in all brain regions studied consistent with reports from cultured cells, primary neurons, and rodent brains (Biskup et al. 2006; Giasson et al. 2006; Gloeckner et al. 2006; Greggio et al. 2006 Smith, 2006#516; Higashi et al. 2007a; West et al. 2005).
There is evidence suggesting that LRRK2 is localized to Lewy bodies in human PD brains. Several studies have demonstrated LRRK2 immunostaining of α-synuclein-positive brainstem and cortical Lewy bodies, the pathological hallmarks of PD and dementia with Lewy bodies, respectively (Alegre-Abarrategui et al. 2008; Greggio et al. 2006; Perry et al. 2008; Zhu et al. 2006a; Zhu et al. 2006b). However, there are some reports that fail to detect LRRK2 immunostaining in these pathological inclusions (Giasson et al. 2006; Higashi et al. 2007a; Melrose et al. 2007). This reported difference may be due to different epitopes recognized by LRRK2 antibodies used for immunohistochemistry, variations in preparation of brain tissues, and/or differences in brain pathologies of PD patients studied (Zhu et al. 2006a; Zhu et al. 2006b).
It remains to be seen if LRRK2 is essential to dopaminergic neurotransmission. Current evidence suggests that altered dopamine neurotransmission has no effect on LRRK2 protein expression. For example, no differences in LRRK2 mRNA signal were observed in the striatum of PD patients where clear dopamine D2 receptor depletion was evident (Galter et al. 2006). Also, vesicular amine transporter deficient mice have unaltered levels of LRRK2 mRNA expression despite extensive striatal dopamine depletion (Higashi et al. 2007b). The overall localization pattern of LRRK2 does, however, agree with a likely involvement in PD pathogenesis.
Putative Biologic Function of LRRK2 and Related Studies in Model Organisms
Dual enzymatic properties of LRRK2
Several groups have studied the kinase activity of the MAPKKK domain of LRRK2 and what effect PD-associated mutations may have on this enzymatic property. LRRK2 is a functional kinase, able to undergo both autophosphorylation and phosphorylation of the generic substrate myelin basic protein (MBP) (Gloeckner et al. 2006; West et al. 2005). One report, using bacterially expressed recombinant protein, demonstrated that autophosphorylation of LRRK2 is an inter-molecular reaction targeting two residues within the activation segment, T2031 and S2032, with the Ser residue of the G2019S mutation generating an additional autophosphorylation site (Luzon-Toro et al. 2007). In contrast, another study using mammalian expressed protein, demonstrated that dimeric LRRK2 undergoes intra-molecular autophosphorylation and that an intact C-terminus, but not N-terminus, is required for this activity (Greggio et al. 2008). There is also discrepancy in the literature regarding the effect PD-associated mutations have on LRRK2 kinase activity. For example, several studies demonstrated that the I1122V, R1441C, R1441G, R1514Q, Y1699C, G2019S, and I2020T familial PD-linked mutations of LRRK2 increased kinase activity as measured by autophosphorylation, suggesting a gain-of-function mechanism for LRRK2-linked disease (Gloeckner et al. 2006; Guo et al. 2007; Li et al. 2007; West et al. 2005; West et al. 2007). However, other reports found that only the G2019S mutation increased LRRK2 kinase activity, while all other mutations studied either did not influence or inhibited kinase activity (Jaleel et al. 2007; Lewis et al. 2007; Luzon-Toro et al. 2007). Interestingly, purified LRRK2 from transgenic mouse brain demonstrated kinase activity. Brain LRRK2 was associated with elevated kinase activity in comparison with that from transgenic lung or transfected cultured cells, which may help explain the brain-specific pathology in mutant LRRK2-induced PD (Li et al. 2007). As the physiological substrates of LRRK2 continue to be identified, the precise effect PD-associated mutations have on phosphorylation activity will likely be clarified.
Several groups have also studied the GTPase activity of the Roc domain of LRRK2 and what effect PD-associated mutations may have on this enzymatic property. LRRK2 is a GTP-binding protein, as measured by specific binding to GTP-agarose and radiolabeled GTP (Guo et al. 2007; Ito et al. 2007; Lewis et al. 2007; Smith et al. 2006). LRRK2 is also a functional GTPase, defined by the ability to bind and intrinsically hydrolyze GTP (Guo et al. 2007; Lewis et al. 2007; Li et al. 2007). Interestingly, purified LRRK2 from transgenic mouse brain also demonstrated GTPase activity (Li et al. 2007). According to one study, familial-linked mutations in LRRK2 within the Roc and COR domains (I1371V, R1441C, R1441G, and Y1699C) resulted in increased GTP-binding as measured by binding to GTP-agarose, while mutations outside these domains did not affect GTP-binding compared with wild-type LRRK2 (West et al. 2007). This is in contrast, however, to two reports that demonstrated that the Roc domain R1441C mutation did not increase GTP binding compared with wild-type LRRK2 (Guo et al. 2007; Lewis et al. 2007). Interestingly, though, the R1441C and R1441G mutations have a decreased rate of GTP hydrolysis compared with the wild-type protein, suggesting that these mutants spend more time in the activated GTP-bound state (Guo et al. 2007; Lewis et al. 2007; Li et al. 2007). This possibility is further corroborated by the structural studies of recombinant Roc and Roc-COR domains (Deng et al. 2008; Gotthardt et al. 2008).
A link exists between intrinsic GTPase activity and downstream kinase activity of LRRK2. Several studies have demonstrated that intact GTPase activity is required for LRRK2 kinase activity whereas GTPase activity functions independently of kinase activity (Guo et al. 2007; Ito et al. 2007; Smith et al. 2006; West et al. 2007). Also, GTP binding stimulates LRRK2 kinase activity, demonstrating intra-molecular regulation between the Roc and MAPKKK domains (Guo et al. 2007; Smith et al. 2006). Based on the newly appreciated dimeric structure of LRRK2, it is predicted that the dimeric Roc or Roc-COR domains act as binary switches to regulate kinase activation (Deng et al. 2008; Gotthardt et al. 2008). It is hypothesized that when in the GTP-bound conformation the dimerization of Roc or Roc-COR domains further induce self-association of the kinase domains, allowing for autophosphorylation and subsequent activation of downstream kinase activity (Deng et al. 2008; Gotthardt et al. 2008; Greggio et al. 2008).
PD-linked mutations of LRRK2 have been associated with neuronal cell toxicity. Several pathogenic mutations (I1122V, R1441C, Y1699C, G2019S, and I2020T) increased the tendency of LRRK2 to form inclusion bodies in multiple cell lines and rat primary neurons (Greggio et al. 2006). These pathogenic mutants of LRRK2 also caused neuronal degeneration, in both neuronal cell lines and primary neuronal cells, possibly through an apoptotic mechanism as measured by TUNEL staining (Greggio et al. 2006; Smith et al. 2006; Smith et al. 2005). The mitochondria-dependent apoptotic pathway, in which cytochrome c is released and caspase-3 is activated, is thought to mediate mutant LRRK2 toxicity in neuronal cells. This seems dependent on Apaf1, a scaffold protein participating in apoptosome formation (Iaccarino et al. 2007). Kinase-dead variants of LRRK2 blocked inclusion body formation and protected against neuronal cell death, suggesting that kinase activity is required for the dominant effects of mutant LRRK2 (Greggio et al. 2006; Smith et al. 2006; Smith et al. 2005). The G2019S pathogenic mutant of LRRK2 coupled with a GTP-binding defective alteration also reduced kinase activity and subsequent neuronal toxicity, further suggesting that GTPase activity regulates kinase activity, which in turn mediates the toxicity of mutant LRRK2 (Smith et al. 2006; West et al. 2007). Therefore, inhibiting activity of either the Roc or MAPKKK domains may serve as a potential therapeutic approach for combating LRRK2-mediated PD.
LRRK2 phosphorylation substrates
Kinase activity is clearly a critical component of LRRK2 function, however, little is known about the physiological substrates of LRRK2 and if aberrant phosphorylation of these substrates is involved in PD pathogenesis. Moesin, a member of the ERM (ezrin/radixin/moesin) family of proteins that functions to anchor the actin cytoskeleton to the plasma membrane, was recently identified as a potential substrate of LRRK2 (Bretscher et al. 2002; Jaleel et al. 2007). Moesin was efficiently phosphorylated by the G2019S variant of LRRK2 at Thr558, a previously characterized physiologically relevant phosphorylation site, which when phosphorylated enables moesin binding to F-actin (Huang et al. 1999; Jaleel et al. 2007). Moesin and radixin have been implicated in regulating neurite outgrowth (Paglini et al. 1998). Recent studies also demonstrated that overexpression of the G2019S LRRK2 mutant impedes neurite length and branching (MacLeod et al. 2006; Plowey et al. 2008)]. Taken together these data suggest that deregulation of moesin phosphorylation by mutant LRRK2 might contribute to the early loss of dopaminergic axon terminals in PD. The ERM-CD44 system has also been implicated as a GDI dissociation stimulator for the Rho family of Ras-related small GTPases. RhoGDI, the Rho subfamily specific GDI, directly interacts with ERM, initiating activation of the Rho GTPase by reducing RhoGDI activity (Takahashi et al. 1997). It is interesting to speculate that ERM may play a similar role for the Roc domain of LRRK2.
A second, weaker LRRK2 substrate was also identified: collapsing response mediator protein-2 (CRMP-2) (Jaleel et al. 2007). CRMP-2 is involved in regulation of growth cones and microtubule dynamics (Stenmark et al. 2007). CRMP-2 also interacts with Numb, an endocytosis-related protein that controls cell number in neurogenesis, and is found in active neurogenesis areas including the hippocampus and olfactory bulb (Johnson 2003; Stenmark et al. 2007). Localization of LRRK2 to these brain regions further suggests a role for LRRK2 in the regulation of neurogenesis (Melrose et al. 2007).
LRRK2 interacting proteins and related signaling cascades
Protein phosphorylation plays a pivotal role in MAPK-linked signal transduction pathways in both normal and pathologic states (Kyosseva 2004). The JNK and p38 MAPKs are two important branches of the MAPK signaling cascade that modulate stress and inflammatory responses and are activated during age-associated neurodegeneration (Ho et al. 2005; Silva et al. 2005). A recent study showed that activation of various MAPK signaling proteins (Src, Hsp27, and JNK) was reduced in patients with G2019S-associated PD, non-symptomatic G2019S carriers, and patients with sporadic PD compared with healthy controls (White et al. 2007). These results suggest that changes in MAPK-signaling may be common to PD pathophysiology, regardless of etiology. Another report demonstrated a reduction in LRRK2(G2019S)-induced neuritic autophagy and neurite shortening in SH-SY5Y cells, a dopaminergic neuroblastoma cell line, in response to an ERK inhibitor, U0126, further implicating involvement of MAPK-related signaling in LRRK2 function (Plowey et al. 2008). It still waits to be determined if various MAPK signaling proteins are direct substrates for LRRK2.
In addition to MAPK-related signaling, LRRK2 has been predicted to be involved in several other cellular pathways. For example, utilizing a combination of LRRK2-mediated protein precipitation and tandem mass spectrometry, several potential LRRK2 binding partners have been identified and subgrouped into various functional cellular pathways, including (1) chaperone pathway and cellular stress, (2) proteins associated with the cytoskeleton and trafficking, and (3) phosphorylation and kinase activity (Dachsel et al. 2007). Rab5b has been demonstrated to be a LRRK2-interacting protein and is suspected to play an important role in synaptic function by modulating the endocytosis of synaptic vesicles, further supporting a role for LRRK2 in trafficking (Shin et al. 2008). In addition, one study monitored changes in expression profiles in SH-SY5Y cells that were depleted of LRRK2 by RNAi and found that the differentially expressed genes could be grouped into signaling networks involved in axonal guidance, nervous system development, cell cycle, cell growth, cell differentiation, cell communication, MAPK cascades, and Ras signal transduction (Habig et al. 2008). Further characterization of these pathways is necessary to decipher the main functional roles of LRRK2.
Other studies have also identified an interaction of LRRK2 with specific chaperone proteins. For example, LRRK2 was found to interact, via the MAPKKK domain, with Hsp90 and its co-chaperone p50cdc37, which forms a chaperone system that has been shown to interact with several kinases, including the MAPKKKs Raf-1 and MLK-3 (Gloeckner et al. 2006; Grammatikakis et al. 1999; Zhang et al. 2004). In both instances, the Hsp90/p50cdc37 proteins do not serve as substrates but rather associate as chaperones assisting in both proper folding and activation of the kinases. It has been shown that inhibition of Hsp90 disrupts the association of this chaperone with LRRK2 and leads to proteosomal degradation of LRRK2, suggesting that Hsp90 inhibitors may be useful therapeutically to limit mutant LRRK2-mediated toxicity in neurons (Wang et al. 2008b).
Parkin, an E3 ubiquitin ligase, is mutated in recessive familial forms of PD and several of its substrates and interaction partners are also relevant in disease pathogenesis. Recently, wild-type and mutant forms of LRRK2 overexpressed in cultured cells were found to interact with parkin, but not with other PD-related proteins such as α-synuclein, DJ-1, or tau (Smith et al. 2005). The COR domain of LRRK2 interacts preferentially with the C-terminal R2 ring finger domain of parkin and while LRRK2 can be ubiquitinated, it is not a substrate for parkin. Co-overexpression of LRRK2 and parkin increased cytoplasmic protein aggregates containing LRRK2 and enhanced the ubiquitination of these aggregates (Smith et al. 2005). Parkin also associates with and ubiquitinates synphilin-1, a protein that interacts with α-synuclein, via the C-terminal R2 ring finger domain, the same domain necessary for interaction with LRRK2 (Chung et al. 2001). Similar to the results found with LRRK2, parkin also stimulates the formation of ubiquitinated aggregates when α-synuclein and synphilin-1 are co-overexpressed (Chung et al. 2001). Parkin is also highly enriched in Lewy bodies (Schlossmacher et al. 2002). To date, six heterozygous LRRK2 and parkin mutation carriers with parkinsonism have been identified (Dachsel et al. 2006; Lesage et al. 2006; Paisan-Ruiz et al. 2005). Compared with individuals carrying a single LRRK2 or parkin mutation, no significant variations regarding disease age at onset or severity of disease were observed. Although the clinical findings do not support a synergistic effect of LRRK2 and parkin, the modulating effects of other genes might conceal a potential genetic interplay.
LRRK2 orthologs in model organisms
Model organisms, both invertebrate and vertebrate, have been used to generate in vivo models of neurodegenerative diseases. An ideal in vivo model of LRRK2-linked pathogenesis should recapitulate key phenotypes of PD such as dopaminergic dysfunction and neurodegeneration, as well as cognitive and locomotive impairments. Thus far, only a few studies of LRRK2 and its orthologs in model organisms have been published.
LRRK is the Drosophila melanogaster ortholog of human LRRK2 and transgenic and loss-of-function mutants have been created in the fruit fly. One report revealed that a LRRK truncation mutant exhibited severely impaired locomotive activity and degeneration of dopaminergic neurons as seen by a reduction in tyrosine hydroxylase immunostaining and a shrunken morphology, demonstrating an endogenous role of LRRK2 in the maintenance of dopaminergic neurons (Lee et al. 2007). However, a subsequent study failed to confirm the neurodegenerative feature but instead showed increased sensitivity to oxidative stress in the same LRRK-deficient flies (Wang et al. 2008a). As this mutant line may still harbor a truncated protein with both LRR repeats and a Roc domain, further studies on additional LRRK-null mutants will be necessary to clarify the normal function of LRRK. A more recent study showed that overexpression of wild-type or LRRK2 (G2019S) in Drosophila resulted in selective loss of dopaminergic neurons, locomotor dysfunction, and early mortality. Interestingly, levodopa treatment improved locomotor impairment but did not prevent neuronal degeneration (Liu et al. 2008). This work provides an important in vivo gain-of-function model that recapitulates several features of human PD.
LRK-1 is the Caenorhabditis elegans ortholog of human LRRK2 and transgenic and deletion mutants have been created in the worm. It was reported that in LRK-1 deletion mutants, synaptic vesicle proteins mis-localized to both presynaptic and dendritic endings in neurons and this aberrant localization was dependent on the AP-1μ1 clathrin adaptor, UNC-101, which is involved in polarized dendritic transport. This study suggests that LRK-1 is involved in determining polarized sorting of synaptic vesicle proteins to axons by excluding these proteins from the dendritic specific transport machinery in the Golgi (Sakaguchi-Nakashima et al. 2007). Another study showed that overexpression of wild-type and LRRK2 (G2019S) in C. elegans was protective against rotenone toxicity while knockdown of endogenous LRK-1 by RNAi potentiated toxicity, suggesting a role for LRRK2 in mitochondrial regulation (Wolozin et al. 2008).
Prevalence and Pathological Features of LRRK2 Disease-Associated Mutations
To date, about 30 disease-associated variants are found throughout the LRRK2 gene (Fig. 1). The G2019S mutation, located in the MAPKKK domain, is by far the most common and was identified by several groups as a cause of both familial and sporadic PD. The frequency of this mutation in PD varies greatly across populations (Bonifati 2006). Studies from large series in the United States estimated a mutation frequency of ~3% in familial and ~1% in sporadic cases (Clark et al. 2006; Kay et al. 2006). The G2019S variant seems to be present at a lower frequency in patients from Northern Europe, than in those from Southern Europe such as Italy (~5% of familial and ~1% of sporadic cases), Spain, and Portugal (~6–18% of familial and ~3–6% of sporadic cases) (Aasly et al. 2005; Bialecka et al. 2005; Bras et al. 2005; Carmine Belin et al. 2006; Gaig et al. 2006; Goldwurm et al. 2005; Goldwurm et al. 2006; Infante et al. 2006; Mata et al. 2006a; Pchelina et al. 2006; Williams-Gray et al. 2006). However, an extremely high prevalence is found among Arab patients from North Africa (~37% of familial and ~41% of sporadic cases) and among Ashkenazi Jewish patients (~29% of familial and ~13% of sporadic cases) (Lesage et al. 2006; Ozelius et al. 2006). Therefore, a north to south gradient seems to exist for increasing frequency of the G2019S mutation. In contrast, the G2019S mutation has not been identified in three large series of Chinese patients and is rarely found in Indian, Japanese, and Greek patients (Fung et al. 2006a; Kalinderi et al. 2007; Lu et al. 2005; Punia et al. 2006; Spanaki et al. 2006; Tan et al. 2005; Tomiyama et al. 2006; Xiromerisiou et al. 2007; Zabetian et al. 2006b).
The G2019S mutation has been suggested to arise from at least three haplotypes. Most patients carrying this mutation and living in disparate countries in Europe and America share a common, ancient founder haplotype, which likely originated from North Africa or the Middle East ~2000 years ago or earlier (Goldwurm et al. 2005; Kachergus J 2005; Lesage et al. 2005; Zabetian et al. 2006b). A second haplotype has been detected in a few patients of European ancestry, while a third haplotype was found in Japanese patients (Zabetian et al. 2006a; Zabetian et al. 2006b). The occurrence of the G2019S mutation in at least three haplotypes suggests either an extremely old founder or a mutational hot spot.
The penetrance of the LRRK2 G2019S variant appears to be age-dependent, increasing from ~17% at age 50 to ~85% at age 70 (Kachergus J 2005). Recent estimates of the lifetime penetrance of this mutation in large series of PD patients from the United States and Europe yielded values of ~24–33% (Clark et al. 2006; Goldwurm et al. 2007; Ozelius et al. 2006). This incomplete penetrance may explain the high prevalence of the G2019S mutation among patients with sporadic PD and its rare occurrence in controls. It is important to note, however, that penetrance may vary across different ethnic populations.
The prevalence of various other LRRK2 amino acid substitutions is in general lower than that of the G2019S mutation but may also vary depending on ethnicity. For example, the R1441C and R1441G mutations, located in the Roc domain, were initially identified in several families (Paisan-Ruiz C 2004; Zimprich A 2004). The R1441G mutation has been associated with an apparently high incidence among Spanish and Basque PD patients, with a prevalence of up to 8% (Paisan-Ruiz C 2004). Another study on sixty European families with autosomal PD found a prevalence of 3.4% for the R1441C mutation (Di Fonzo A 2006). The R1628P and G2385R variants, located in the COR and WD40 domains respectively, have been identified as common risk factors for sporadic PD in Chinese and Asian populations, each with an attributable risk of ~2–4% (Di Fonzo et al. 2006; Farrer et al. 2007; Fung et al. 2006b; Mata IF 2005; Ross et al. 2008; Tan et al. 2008; Tan et al. 2007b; Tomiyama et al. 2006). Other putative mutations, located throughout the multi-domain structure of LRRK2 (R1067Q, I1122V, S1228T, R1441H, Y1699C, M1869T, and I2020T) have been reported with a frequency of < 1% (Punia et al. 2006). It is important to note that the majority of studies have focused on screening for mutations in exons 31 and 41, which encode the Roc and MAPKKK domains respectively, and, therefore, may prevent identification of novel variants in other regions of LRRK2. While some studies have done comprehensive mutation screening of all 51 exons of LRRK2, the continuation of such widespread studies will be crucial to the further identification and understanding of LRRK2 variants (Clarimon et al. 2008; Di Fonzo A 2006; Johnson et al. 2007; Nichols et al. 2007; Paisan-Ruiz et al. 2006; Paisan-Ruiz et al. 2008; Skipper et al. 2005).
There is a wide pathological spectrum associated with LRRK2 mutations. While age at disease onset ranges from ~40–80 years and clinical features are similar to those found in sporadic PD, some cases show various degrees of pathological heterogeneity. Presumably, mutations in different domains of LRRK2 could possibly result in different disease phenotypes.
Patients with the common G2019S mutation in the MAPKKK domain show no difference in the average age at disease onset and duration compared with those of sporadic cases (Goldwurm et al. 2005). Clinical and positron emission tomography (PET) studies have confirmed that the overall phenotype associated with the G2019S mutation is similar to that of idiopathic PD (Hernandez D 2005). Some patients with this mutation demonstrate classic PD with Lewy bodies, however, others are characterized by parkinsonism without Lewy bodies but demonstrate dystrophic neurites in the SNpc that stain for LRRK2 (Giasson et al. 2006).
Typical parkinsonism with nigral degeneration has also been reported in patients with other LRRK2 mutations, but in some cases the clinical and pathological features are more diverse. The I2020T mutation, located in the MAPKKK domain, is responsible for parkinsonism in a large Japanese family, the Sagamihara kindred, and these patients are afflicted with pure nigral degeneration without α-synuclein-positive Lewy body and Lewy neurite pathology (Funayama et al. 2005). Patients with the Y1699C mutation, located in the COR domain, are mainly from a large British kindred and displayed dementia and amyotrophy, in addition to parkinsonism (Khan NL 2005; Zimprich A 2004). Upon pathological examination, a patient with this mutation was found to display a few α-synuclein-positive Lewy body and Lewy neurite pathologies but had more extensive ubiquitin-positive cytoplasmic and nuclear inclusions (Zimprich A 2004). In patients with the Roc domain R1441C mutation, some individuals had classic α-synuclein-positive Lewy body and Lewy neurite pathology, while others had tau pathology reminiscent of progressive supranuclear palsy (PSP) and some patients did not have any Lewy bodies or Lewy neurites, but displayed asymmetric ubiquitin-positive inclusions (Haugarvoll et al. 2008; Zimprich A 2004). Moreover, it has been noted that in patients with the Y1699C and R1441C mutations, degeneration is not solely restricted to dopaminergic neurons of the SNpc, however, PET scan analysis indicated that the in vivo phenotype associated with these two mutations is indistinguishable from that of sporadic PD despite the pathological heterogeneity (Adams et al. 2005). Interestingly, mutations in LRRK2 have not been found in other primary neurodegenerative diseases such as Alzheimer’s disease (AD), PSP, or frontotemporal dementia (FTD) (Hernandez D 2005; Tan et al. 2007a; Toft et al. 2005). The diverse pathologies associated with LRRK2 mutations strongly suggest that this protein may be involved in multiple cellular processes in neurons.
Conclusions
PD is a devastating neurodegenerative disorder and despite nearly 200 years since its initial description, a definitive understanding of disease etiology and a preventive therapeutic approach are still missing. The discovery of LRRK2 mutations in PD has proven to be monumental to the field because, for the first time, gene mutations have been found to be a frequent determinant of both familial and sporadic PD. The pleomorphic pathology associated with LRRK2 mutations places this protein further upstream in disease-linked signaling, compared with previously identified PD-related genes. The identification of a vast number of mutations found throughout LRRK2 also suggests an upstream role in neuronal signaling. Because of the potential upstream role LRRK2 likely plays in neurons, therapeutic strategies aimed at this protein may be effective in combating a broad spectrum of PD. The unique multi-domain organization of LRRK2 may provide multiple avenues for therapeutic intervention, such as inhibiting GTP binding activity through the Roc domain or inhibiting kinase activity through the MAPKKK domain. It is still too early, but is certainly much desired, to reconcile the roles of LRRK2 and other disease-linked proteins (encoded by various PARK loci, Table 1) into overlapping neurodegenerative pathways. It is clear that further knowledge of LRRK2 is necessary to fully appreciate the role this multi-domain, dual enzyme plays in both normal neuronal function and the pathogenesis of PD.
Acknowledgments
This work was supported in part by the National Institutes of Health (grant -R21AG028797), National Parkinson Foundation (Mega Research Grants Program), National Science Foundation (Advance Institutional Transformation Grant SBE-0245054), and Johnson & Johnson Corporate Office of Science and Technology (Focused Funding Program).
Footnotes
Abbreviations
PD, Parkinson’s disease; SNpc, substantia nigra pars compacts; MPP+, 1-methyl-4-phenylpyridinium; LRRK2, leucine-rich repeat kinase 2; PINK1, phosphatase and tensin (PTEN)-induced kinase 1; UCH-L1, ubiquitin C-terminal hydrolase L1; Roc, Ras of Complex Protein; COR, C-terminal of Roc; LRR, leucine-rich repeat; MAPKKK, mitogen activated protein kinase kinase kinase; GDI, GDP dissociation inhibitor; TKL, tyrosine kinase-like; RIPK, receptor-interacting protein kinases; MAPK, mitogen activated protein kinase; ERK, extracellular-signal regulated kinase; JNK, c-Jun amino-terminal kinase; PET, positron emission tomography; PSP, progressive supranuclear palsy; AD, Alzheimer’s disease; FTD, frontotemperol demetia; MBP, myelin basic protein; TUNEL, Terminal deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling; ERM, ezrin/radixin/moesin; CRMP-2, collapsing response mediator protein-2.
References
- Aasly JO, Toft M, Fernandez-Mata I, Kachergus J, Hulihan M, White LR, Farrer M. Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann Neurol. 2005;57(5):762–765. doi: 10.1002/ana.20456. [DOI] [PubMed] [Google Scholar]
- Adams JR, van Netten H, Schulzer M, Mak E, McKenzie J, Strongosky A, Sossi V, Ruth TJ, Lee CS, Farrer M, Gasser T, Uitti RJ, Calne DB, Wszolek ZK, Stoessl AJ. PET in LRRK2 mutations: comparison to sporadic Parkinson’s disease and evidence for presymptomatic compensation. Brain. 2005;128(Pt 12):2777–2785. doi: 10.1093/brain/awh607. [DOI] [PubMed] [Google Scholar]
- Alegre-Abarrategui J, Ansorge O, Esiri M, Wade-Martins R. LRRK2 is a component of granular alpha-synuclein pathology in the brainstem of Parkinson’s disease. Neuropathol Appl Neurobiol. 2008;34(3):272–283. doi: 10.1111/j.1365-2990.2007.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg DSK, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brussel T, Schulte C, Maass S, Nagele T. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease*. Brain. 2005;128(Pt 12):3000–3011. doi: 10.1093/brain/awh666. [DOI] [PubMed] [Google Scholar]
- Bialecka M, Hui S, Klodowska-Duda G, Opala G, Tan EK, Drozdzik M. Analysis of LRRK 2 G 2019 S and I 2020 T mutations in Parkinson’s disease. Neurosci Lett. 2005;390(1):1–3. doi: 10.1016/j.neulet.2005.07.045. [DOI] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60(5):557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Bonifati V. Parkinson’s disease: the LRRK2-G2019S mutation: opening a novel era in Parkinson’s disease genetics. Eur J Hum Genet. 2006;14(10):1061–1062. doi: 10.1038/sj.ejhg.5201695. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Bosgraaf LVHP. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643(1–3):5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Bras JM, Guerreiro RJ, Ribeiro MH, Januario C, Morgadinho A, Oliveira CR, Cunha L, Hardy J, Singleton A. G2019S dardarin substitution is a common cause of Parkinson’s disease in a Portuguese cohort. Mov Disord. 2005;20(12):1653–1655. doi: 10.1002/mds.20682. [DOI] [PubMed] [Google Scholar]
- Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 2002;3(8):586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- Carmine Belin A, Westerlund M, Sydow O, Lundstromer K, Hakansson A, Nissbrandt H, Olson L, Galter D. Leucine-rich repeat kinase 2 (LRRK2) mutations in a Swedish Parkinson cohort and a healthy nonagenarian. Mov Disord. 2006;21(10):1731–1734. doi: 10.1002/mds.21016. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364(9440):1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7(10):1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- Clarimon J, Pagonabarraga J, Paisan-Ruiz C, Campolongo A, Pascual-Sedano B, Marti-Masso JF, Singleton AB, Kulisevsky J. Tremor dominant parkinsonism: Clinical description and LRRK2 mutation screening. Mov Disord. 2008;23(4):518–523. doi: 10.1002/mds.21771. [DOI] [PubMed] [Google Scholar]
- Clark LN, Wang Y, Karlins E, Saito L, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology. 2006;67(10):1786–1791. doi: 10.1212/01.wnl.0000244345.49809.36. [DOI] [PubMed] [Google Scholar]
- Cookson MR. The biochemistry of Parkinson’s disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Dachsel JC, Mata IF, Ross OA, Taylor JP, Lincoln SJ, Hinkle KM, Huerta C, Ribacoba R, Blazquez M, Alvarez V, Farrer MJ. Digenic parkinsonism: investigation of the synergistic effects of PRKN and LRRK2. Neurosci Lett. 2006;410(2):80–84. doi: 10.1016/j.neulet.2006.06.068. [DOI] [PubMed] [Google Scholar]
- Dachsel JC, Taylor JP, Mok SS, Ross OA, Hinkle KM, Bailey RM, Hines JH, Szutu J, Madden B, Petrucelli L, Farrer MJ. Identification of potential protein interactors of Lrrk2. Parkinsonism Relat Disord. 2007 doi: 10.1016/j.parkreldis.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Deng J, Lewis PA, Greggio E, Sluch E, Beilina A, Cookson MR. Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc Natl Acad Sci U S A. 2008;105(5):1499–1504. doi: 10.1073/pnas.0709098105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo ATC, De Mari M, Chien HF, Ferreira J, Rohe CF, Riboldazzi G, Antonini A, Albani G, Mauro A, Marconi R, Abbruzzese G, Lopiano L, Fincati E, Guidi M, Marini P, Stocchi F, Onofrj M, Toni V, Tinazzi M, Fabbrini G, Lamberti P, Vanacore N, Meco G, Leitner P, Uitti RJ, Wszolek ZK, Gasser T, Simons EJ, Breedveld GJ, Goldwurm S, Pezzoli G, Sampaio C, Barbosa E, Martignoni E, Oostra BA, Bonifati V Italian Parkinson’s Genetics Network. Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson’s disease. Eur J Hum Genet. 2006;14(3):322–331. doi: 10.1038/sj.ejhg.5201539. [DOI] [PubMed] [Google Scholar]
- Di Fonzo A, Wu-Chou YH, Lu CS, van Doeselaar M, Simons EJ, Rohe CF, Chang HC, Chen RS, Weng YH, Vanacore N, Breedveld GJ, Oostra BA, Bonifati V. A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson’s disease risk in Taiwan. Neurogenetics. 2006;7(3):133–138. doi: 10.1007/s10048-006-0041-5. [DOI] [PubMed] [Google Scholar]
- Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68(5):384–386. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- Farrer MJ, Stone JT, Lin CH, Dachsel JC, Hulihan MM, Haugarvoll K, Ross OA, Wu RM. Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord. 2007;13(2):89–92. doi: 10.1016/j.parkreldis.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, Tsuji S, Obata F. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol. 2005;57(6):918–921. doi: 10.1002/ana.20484. [DOI] [PubMed] [Google Scholar]
- Fung HC, Chen CM, Hardy J, Hernandez D, Singleton A, Wu YR. Lack of G2019S LRRK2 mutation in a cohort of Taiwanese with sporadic Parkinson’s disease. Mov Disord. 2006a;21(6):880–881. doi: 10.1002/mds.20814. [DOI] [PubMed] [Google Scholar]
- Fung HC, Chen CM, Hardy J, Singleton AB, Wu YR. A common genetic factor for Parkinson disease in ethnic Chinese population in Taiwan. BMC Neurol. 2006b;6:47. doi: 10.1186/1471-2377-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futey LM, Medley QG, Cote GP, Egelhoff TT. Structural analysis of myosin heavy chain kinase A from Dictyostelium. Evidence for a highly divergent protein kinase domain, an amino-terminal coiled-coil domain, and a domain homologous to the beta-subunit of heterotrimeric G proteins. J Biol Chem. 1995;270(2):523–529. doi: 10.1074/jbc.270.2.523. [DOI] [PubMed] [Google Scholar]
- Gaig C, Ezquerra M, Marti MJ, Munoz E, Valldeoriola F, Tolosa E. LRRK2 mutations in Spanish patients with Parkinson disease: frequency, clinical features, and incomplete penetrance. Arch Neurol. 2006;63(3):377–382. doi: 10.1001/archneur.63.3.377. [DOI] [PubMed] [Google Scholar]
- Galter D, Westerlund M, Belin AC, Olson L. DJ-1 and UCH-L1 gene activity patterns in the brains of controls, Parkinson and schizophrenia patients and in rodents. Physiol Behav. 2007;92(1–2):46–53. doi: 10.1016/j.physbeh.2007.05.046. [DOI] [PubMed] [Google Scholar]
- Galter D, Westerlund M, Carmine A, Lindqvist E, Sydow O, Olson L. LRRK2 expression linked to dopamine-innervated areas. Ann Neurol. 2006;59(4):714–719. doi: 10.1002/ana.20808. [DOI] [PubMed] [Google Scholar]
- Gandhi PN, Wang X, Zhu X, Chen SG, Wilson-Delfosse AL. The Roc domain of leucine-rich repeat kinase 2 is sufficient for interaction with microtubules. J Neurosci Res. 2008 doi: 10.1002/jnr.21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM. Biochemical and pathological characterization of Lrrk2. Ann Neurol. 2006;59(2):315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O’Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15(2):223–232. doi: 10.1093/hmg/ddi439. Epub 2005 Dec 2001. [DOI] [PubMed] [Google Scholar]
- Goldwurm S, Di Fonzo A, Simons EJ, Rohe CF, Zini M, Canesi M, Tesei S, Zecchinelli A, Antonini A, Mariani C, Meucci N, Sacilotto G, Sironi F, Salani G, Ferreira J, Chien HF, Fabrizio E, Vanacore N, Dalla Libera A, Stocchi F, Diroma C, Lamberti P, Sampaio C, Meco G, Barbosa E, Bertoli-Avella AM, Breedveld GJ, Oostra BA, Pezzoli G, Bonifati V. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. J Med Genet. 2005;42(11):e65. doi: 10.1136/jmg.2005.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldwurm S, Zini M, Di Fonzo A, De Gaspari D, Siri C, Simons EJ, van Doeselaar M, Tesei S, Antonini A, Canesi M, Zecchinelli A, Mariani C, Meucci N, Sacilotto G, Cilia R, Isaias IU, Bonetti A, Sironi F, Ricca S, Oostra BA, Bonifati V, Pezzoli G. LRRK2 G2019S mutation and Parkinson’s disease: a clinical, neuropsychological and neuropsychiatric study in a large Italian sample. Parkinsonism Relat Disord. 2006;12(7):410–419. doi: 10.1016/j.parkreldis.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Goldwurm S, Zini M, Mariani L, Tesei S, Miceli R, Sironi F, Clementi M, Bonifati V, Pezzoli G. Evaluation of LRRK2 G2019S penetrance: relevance for genetic counseling in Parkinson disease. Neurology. 2007;68(14):1141–1143. doi: 10.1212/01.wnl.0000254483.19854.ef. [DOI] [PubMed] [Google Scholar]
- Gotthardt K, Weyand M, Kortholt A, Van Haastert PJ, Wittinghofer A. Structure of the Roc-COR domain tandem of C. tepidum, a prokaryotic homologue of the human LRRK2 Parkinson kinase. EMBO J. 2008;27(17):2352. doi: 10.1038/emboj.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammatikakis N, Lin JH, Grammatikakis A, Tsichlis PN, Cochran BH. p50(cdc37) acting in concert with Hsp90 is required for Raf-1 function. Mol Cell Biol. 1999;19(3):1661–1672. doi: 10.1128/mcb.19.3.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23(2):329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Greggio E, Zambrano I, Kaganovich A, Beilina A, Taymans JM, Daniels V, Lewis P, Jain S, Ding J, Syed A, Thomas KJ, Baekelandt V, Cookson MR. The Parkinson’s disease associated Leucine rich repeat kinase 2 (LRRK2) is a dimer that undergoes intra-molecular autophosphorylation. J Biol Chem. 2008 doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG. The Parkinson’s disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007 doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo LWW, Chen SG. Leucine-rich repeat kinase 2: relevance to Parkinson’s disease. Int J Biochem Cell Biol. 2006;38(9):1469–1475. doi: 10.1016/j.biocel.2006.02.009. Epub 2006 Mar 1462. [DOI] [PubMed] [Google Scholar]
- Habig K, Walter M, Poths S, Riess O, Bonin M. RNA interference of LRRK2-microarray expression analysis of a Parkinson’s disease key player. Neurogenetics. 2008;9(2):83–94. doi: 10.1007/s10048-007-0114-0. [DOI] [PubMed] [Google Scholar]
- Hatano T, Kubo S, Imai S, Maeda M, Ishikawa K, Mizuno Y, Hattori N. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum Mol Genet. 2007;16(6):678–690. doi: 10.1093/hmg/ddm013. [DOI] [PubMed] [Google Scholar]
- Haugarvoll K, Rademakers R, Kachergus JM, Nuytemans K, Ross OA, Gibson JM, Tan EK, Gaig C, Tolosa E, Goldwurm S, Guidi M, Riboldazzi G, Brown L, Walter U, Benecke R, Berg D, Gasser T, Theuns J, Pals P, Cras P, De Deyn PP, Engelborghs S, Pickut B, Uitti RJ, Foroud T, Nichols WC, Hagenah J, Klein C, Samii A, Zabetian CP, Bonifati V, Van Broeckhoven C, Farrer MJ, Wszolek ZK. Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology. 2008;70(16 Pt 2):1456–1460. doi: 10.1212/01.wnl.0000304044.22253.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez DPRC, Crawley A, Malkani R, Werner J, Gwinn-Hardy K, Dickson D, Wavrant Devrieze F, Hardy J, Singleton A. The dardarin G 2019 S mutation is a common cause of Parkinson’s disease but not other neurodegenerative diseases. Neurosci Lett. 2005;389(3):137–139. doi: 10.1016/j.neulet.2005.07.044. [DOI] [PubMed] [Google Scholar]
- Higashi S, Biskup S, West AB, Trinkaus D, Dawson VL, Faull RL, Waldvogel HJ, Arai H, Dawson TM, Moore DJ, Emson PC. Localization of Parkinson’s disease-associated LRRK2 in normal and pathological human brain. Brain Res. 2007a;1155:208–219. doi: 10.1016/j.brainres.2007.04.034. [DOI] [PubMed] [Google Scholar]
- Higashi S, Moore DJ, Colebrooke RE, Biskup S, Dawson VL, Arai H, Dawson TM, Emson PC. Expression and localization of Parkinson’s disease-associated leucine-rich repeat kinase 2 in the mouse brain. J Neurochem. 2007b;100(2):368–381. doi: 10.1111/j.1471-4159.2006.04246.x. [DOI] [PubMed] [Google Scholar]
- Ho GJ, Drego R, Hakimian E, Masliah E. Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr Drug Targets Inflamm Allergy. 2005;4(2):247–256. doi: 10.2174/1568010053586237. [DOI] [PubMed] [Google Scholar]
- Huang L, Wong TY, Lin RC, Furthmayr H. Replacement of threonine 558, a critical site of phosphorylation of moesin in vivo, with aspartate activates F-actin binding of moesin. Regulation by conformational change. J Biol Chem. 1999;274(18):12803–12810. doi: 10.1074/jbc.274.18.12803. [DOI] [PubMed] [Google Scholar]
- Iaccarino C, Crosio C, Vitale C, Sanna G, Carri MT, Barone P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum Mol Genet. 2007;16(11):1319–1326. doi: 10.1093/hmg/ddm080. [DOI] [PubMed] [Google Scholar]
- Infante J, Rodriguez E, Combarros O, Mateo I, Fontalba A, Pascual J, Oterino A, Polo JM, Leno C, Berciano J. LRRK2 G2019S is a common mutation in Spanish patients with late-onset Parkinson’s disease. Neurosci Lett. 2006;395(3):224–226. doi: 10.1016/j.neulet.2005.10.083. [DOI] [PubMed] [Google Scholar]
- Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, Iwatsubo T. GTP Binding Is Essential to the Protein Kinase Activity of LRRK2, a Causative Gene Product for Familial Parkinson’s Disease. Biochemistry. 2007;46(5):1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem J. 2007;405(2):307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda L, Tichy P, Spizek J, Petricek M. A deduced Thermomonospora curvata protein containing serine/threonine protein kinase and WD-repeat domains. J Bacteriol. 1996;178(5):1487–1489. doi: 10.1128/jb.178.5.1487-1489.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J, Paisan-Ruiz C, Lopez G, Crews C, Britton A, Malkani R, Evans EW, McInerney-Leo A, Jain S, Nussbaum RL, Foote KD, Mandel RJ, Crawley A, Reimsnider S, Fernandez HH, Okun MS, Gwinn-Hardy K, Singleton AB. Comprehensive screening of a North American Parkinson’s disease cohort for LRRK2 mutation. Neurodegener Dis. 2007;4(5):386–391. doi: 10.1159/000105160. [DOI] [PubMed] [Google Scholar]
- Johnson JE. Numb and Numblike control cell number during vertebrate neurogenesis. Trends Neurosci. 2003;26(8):395–396. doi: 10.1016/S0166-2236(03)00166-8. [DOI] [PubMed] [Google Scholar]
- Kachergus JMI, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ, Toft M. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet. 2005;76(4):672–680. doi: 10.1086/429256. Epub 2005 Feb 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinderi K, Fidani L, Bostantjopoulou S, Katsarou Z, Kotsis A. The G2019S LRRK2 mutation is uncommon amongst Greek patients with sporadic Parkinson’s disease. Eur J Neurol. 2007;14(10):1088–1090. doi: 10.1111/j.1468-1331.2007.01867.x. [DOI] [PubMed] [Google Scholar]
- Kay DM, Zabetian CP, Factor SA, Nutt JG, Samii A, Griffith A, Bird TD, Kramer P, Higgins DS, Payami H. Parkinson’s disease and LRRK2: frequency of a common mutation in U.S. movement disorder clinics. Mov Disord. 2006;21(4):519–523. doi: 10.1002/mds.20751. [DOI] [PubMed] [Google Scholar]
- Khan NLJS, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, Gibbons V, Gandhi S, Vaughan J, Eunson LH, Katzenschlager R, Gayton J, Lennox G, Revesz T, Nicholl D, Bhatia KP, Quinn N, Brooks D, Lees AJ, Davis MB, Piccini P, Singleton AB, Wood NW. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128(Pt 12):2786–2796. doi: 10.1093/brain/awh667. Epub 2005 Nov 2784. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kobe B, Deisenhofer J. Proteins with leucine-rich repeats. Curr Opin Struct Biol. 1995;5(3):409–416. doi: 10.1016/0959-440x(95)80105-7. [DOI] [PubMed] [Google Scholar]
- Kyosseva SV. Mitogen-activated protein kinase signaling. Int Rev Neurobiol. 2004;59:201–220. doi: 10.1016/S0074-7742(04)59008-6. [DOI] [PubMed] [Google Scholar]
- Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun. 2007;358(2):534–539. doi: 10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395(6701):451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, Pollak P, Brice A. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med. 2006;354(4):422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- Lesage S, Leutenegger AL, Ibanez P, Janin S, Lohmann E, Durr A, Brice A. LRRK2 haplotype analyses in European and North African families with Parkinson disease: a common founder for the G2019S mutation dating from the 13th century. Am J Hum Genet. 2005;77(2):330–332. doi: 10.1086/432422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PA, Greggio E, Beilina A, Jain S, Baker A, Cookson MR. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun. 2007;357(3):668–671. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, Ross CA, Montell C, Smith WW. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A. 2008;105(7):2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CS, Simons EJ, Wu-Chou YH, Fonzo AD, Chang HC, Chen RS, Weng YH, Rohe CF, Breedveld GJ, Hattori N, Gasser T, Oostra BA, Bonifati V. The LRRK2 I2012T, G2019S, and I2020T mutations are rare in Taiwanese patients with sporadic Parkinson’s disease. Parkinsonism Relat Disord. 2005;11(8):521–522. doi: 10.1016/j.parkreldis.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Luzon-Toro B, de la Torre ER, Delgado A, Perez-Tur J, Hilfiker S. Mechanistic insight into the dominant mode of the Parkinson’s disease-associated G2019S LRRK2 mutation. Hum Mol Genet. 2007;16(17):2031–2039. doi: 10.1093/hmg/ddm151. [DOI] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52(4):587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Manning GWD, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Marin I. The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol Biol Evol. 2006;23(12):2423–2433. doi: 10.1093/molbev/msl114. [DOI] [PubMed] [Google Scholar]
- Mata IFKJ, Taylor JP, Lincoln S, Aasly J, Lynch T, Hulihan MM, Cobb SA, Wu RM, Lu CS, Lahoz C, Wszolek ZK, Farrer MJ. Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics. 2005;6(4):171–177. doi: 10.1007/s10048-005-0005-1. Epub 2005 Sep 2017. [DOI] [PubMed] [Google Scholar]
- Mata IF, Ross OA, Kachergus J, Huerta C, Ribacoba R, Moris G, Blazquez M, Guisasola LM, Salvador C, Martinez C, Farrer M, Alvarez V. LRRK2 mutations are a common cause of Parkinson’s disease in Spain. Eur J Neurol. 2006a;13(4):391–394. doi: 10.1111/j.1468-1331.2006.01256.x. [DOI] [PubMed] [Google Scholar]
- Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci. 2006b;29(5):286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Melrose H, Lincoln S, Tyndall G, Dickson D, Farrer M. Anatomical localization of leucine-rich repeat kinase 2 in mouse brain. Neuroscience. 2006;139(3):791–794. doi: 10.1016/j.neuroscience.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Melrose HL, Kent CB, Taylor JP, Dachsel JC, Hinkle KM, Lincoln SJ, Mok SS, Culvenor JG, Masters CL, Tyndall GM, Bass DI, Ahmed Z, Andorfer CA, Ross OA, Wszolek ZK, Delldonne A, Dickson DW, Farrer MJ. A comparative analysis of leucine-rich repeat kinase 2 (Lrrk2) expression in mouse brain and Lewy body disease. Neuroscience. 2007;147(4):1047–1058. doi: 10.1016/j.neuroscience.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Meylan EaTJ. The RIP kinases: crucial integrators of cellular stress. Trends Biochem Sci. 2005;30(3):151–159. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Nichols WC, Elsaesser VE, Pankratz N, Pauciulo MW, Marek DK, Halter CA, Rudolph A, Shults CW, Foroud T. LRRK2 mutation analysis in Parkinson disease families with evidence of linkage to PARK8. Neurology. 2007;69(18):1737–1744. doi: 10.1212/01.wnl.0000278115.50741.4e. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, Lipton RB, Soto-Valencia J, Risch N, Bressman SB. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2006;354(4):424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- Paglini G, Kunda P, Quiroga S, Kosik K, Caceres A. Suppression of radixin and moesin alters growth cone morphology, motility, and process formation in primary cultured neurons. J Cell Biol. 1998;143(2):443–455. doi: 10.1083/jcb.143.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Evans EW, Jain S, Xiromerisiou G, Gibbs JR, Eerola J, Gourbali V, Hellstrom O, Duckworth J, Papadimitriou A, Tienari PJ, Hadjigeorgiou GM, Singleton AB. Testing association between LRRK2 and Parkinson’s disease and investigating linkage disequilibrium. J Med Genet. 2006;43(2):e9. doi: 10.1136/jmg.2005.036889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz CJS, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Lang AE, Kawarai T, Sato C, Salehi-Rad S, Fisman GK, Al-Khairallah T, St George-Hyslop P, Singleton A, Rogaeva E. LRRK2 gene in Parkinson disease: mutation analysis and case control association study. Neurology. 2005;65(5):696–700. doi: 10.1212/01.wnl.0000167552.79769.b3. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Nath P, Washecka N, Gibbs JR, Singleton AB. Comprehensive analysis of LRRK2 in publicly available Parkinson’s disease cases and neurologically normal controls. Hum Mutat. 2008;29(4):485–490. doi: 10.1002/humu.20668. [DOI] [PubMed] [Google Scholar]
- Pchelina SN, Yakimovskii AF, Ivanova ON, Emelianov AK, Zakharchuk AH, Schwarzman AL. G2019S LRRK2 mutation in familial and sporadic Parkinson’s disease in Russia. Mov Disord. 2006;21(12):2234–2236. doi: 10.1002/mds.21134. [DOI] [PubMed] [Google Scholar]
- Perry G, Zhu X, Babar AK, Siedlak SL, Yang Q, Ito G, Iwatsubo T, Smith MA, Chen SG. Leucine-rich repeat kinase 2 colocalizes with alpha-synuclein in Parkinson’s disease, but not tau-containing deposits in tauopathies. Neurodegener Dis. 2008;5(3–4):222–224. doi: 10.1159/000113708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105(3):1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Punia S, Behari M, Govindappa ST, Swaminath PV, Jayaram S, Goyal V, Muthane UB, Juyal RC, Thelma BK. Absence/rarity of commonly reported LRRK2 mutations in Indian Parkinson’s disease patients. Neurosci Lett. 2006;409(2):83–88. doi: 10.1016/j.neulet.2006.04.052. [DOI] [PubMed] [Google Scholar]
- Ross OA, Wu YR, Lee MC, Funayama M, Chen ML, Soto AI, Mata IF, Lee-Chen GJ, Chen CM, Tang M, Zhao Y, Hattori N, Farrer MJ, Tan EK, Wu RM. Analysis of Lrrk2 R1628P as a risk factor for Parkinson’s disease. Ann Neurol. 2008;64(1):88–92. doi: 10.1002/ana.21405. [DOI] [PubMed] [Google Scholar]
- Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17(7):592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, Ochiishi T, Shimura H, Sharon R, Hattori N, Langston JW, Mizuno Y, Hyman BT, Selkoe DJ, Kosik KS. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol. 2002;160(5):1655–1667. doi: 10.1016/S0002-9440(10)61113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Falkenburger BH. Neuronal pathology in Parkinson’s disease. Cell Tissue Res. 2004;318(1):135–147. doi: 10.1007/s00441-004-0954-y. [DOI] [PubMed] [Google Scholar]
- Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J, Hatano T, Hattori N, Kim KS, Chang S, Seol W. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res. 2008 doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Silva RM, Kuan CY, Rakic P, Burke RE. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: a new therapeutic target in Parkinson’s disease. Mov Disord. 2005;20(6):653–664. doi: 10.1002/mds.20390. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Skipper L, Li Y, Bonnard C, Pavanni R, Yih Y, Chua E, Sung WK, Tan L, Wong MC, Tan EK, Liu J. Comprehensive evaluation of common genetic variation within LRRK2 reveals evidence for association with sporadic Parkinson’s disease. Hum Mol Genet. 2005;14(23):3549–3556. doi: 10.1093/hmg/ddi376. [DOI] [PubMed] [Google Scholar]
- Smith TF, Gaitatzes C, Saxena K, Neer EJ. The WD repeat: a common architecture for diverse functions. Trends Biochem Sci. 1999;24(5):181–185. doi: 10.1016/s0968-0004(99)01384-5. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9(10):1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A. 2005;102(51):18676–18681. doi: 10.1073/pnas.0508052102. Epub 12005 Dec 18613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solano SM, Miller DW, Augood SJ, Young AB, Penney JB., Jr Expression of alpha-synuclein, parkin, and ubiquitin carboxy-terminal hydrolase L1 mRNA in human brain: genes associated with familial Parkinson’s disease. Ann Neurol. 2000;47(2):201–210. [PubMed] [Google Scholar]
- Spanaki C, Latsoudis H, Plaitakis A. LRRK2 mutations on Crete: R1441H associated with PD evolving to PSP. Neurology. 2006;67(8):1518–1519. doi: 10.1212/01.wnl.0000239829.33936.73. [DOI] [PubMed] [Google Scholar]
- Stenmark P, Ogg D, Flodin S, Flores A, Kotenyova T, Nyman T, Nordlund P, Kursula P. The structure of human collapsin response mediator protein 2, a regulator of axonal growth. J Neurochem. 2007;101(4):906–917. doi: 10.1111/j.1471-4159.2006.04401.x. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Sasaki T, Mammoto A, Takaishi K, Kameyama T, Tsukita S, Takai Y. Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J Biol Chem. 1997;272(37):23371–23375. doi: 10.1074/jbc.272.37.23371. [DOI] [PubMed] [Google Scholar]
- Tan EK, Lee J, Chen CP, Wong MC, Zhao Y. Case control analysis of LRRK2 Gly2385Arg in Alzheimer’s disease. Neurobiol Aging. 2007a doi: 10.1016/j.neurobiolaging.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Tan EK, Shen H, Tan LC, Farrer M, Yew K, Chua E, Jamora RD, Puvan K, Puong KY, Zhao Y, Pavanni R, Wong MC, Yih Y, Skipper L, Liu JJ. The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson’s disease patients. Neurosci Lett. 2005;384(3):327–329. doi: 10.1016/j.neulet.2005.04.103. [DOI] [PubMed] [Google Scholar]
- Tan EK, Tan LC, Lim HQ, Li R, Tang M, Yih Y, Pavanni R, Prakash KM, Fook-Chong S, Zhao Y. LRRK2 R1628P increases risk of Parkinson’s disease: replication evidence. Hum Genet. 2008 doi: 10.1007/s00439-008-0544-2. [DOI] [PubMed] [Google Scholar]
- Tan EK, Zhao Y, Skipper L, Tan MG, Di Fonzo A, Sun L, Fook-Chong S, Tang S, Chua E, Yuen Y, Tan L, Pavanni R, Wong MC, Kolatkar P, Lu CS, Bonifati V, Liu JJ. The LRRK2 Gly2385Arg variant is associated with Parkinson’s disease: genetic and functional evidence. Hum Genet. 2007b;120(6):857–863. doi: 10.1007/s00439-006-0268-0. [DOI] [PubMed] [Google Scholar]
- Taylor JPMI, Farrer MJ. LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol Med. 2006;12(2):76–82. doi: 10.1016/j.molmed.2005.12.004. Epub 2006 Jan 2010. [DOI] [PubMed] [Google Scholar]
- Taymans JM, Van den Haute C, Baekelandt V. Distribution of PINK1 and LRRK2 in rat and mouse brain. J Neurochem. 2006;98(3):951–961. doi: 10.1111/j.1471-4159.2006.03919.x. [DOI] [PubMed] [Google Scholar]
- Toft M, Sando SB, Melquist S, Ross OA, White LR, Aasly JO, Farrer MJ. LRRK2 mutations are not common in Alzheimer’s disease. Mech Ageing Dev. 2005;126(11):1201–1205. doi: 10.1016/j.mad.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Tomiyama H, Li Y, Funayama M, Hasegawa K, Yoshino H, Kubo S, Sato K, Hattori T, Lu CS, Inzelberg R, Djaldetti R, Melamed E, Amouri R, Gouider-Khouja N, Hentati F, Hatano Y, Wang M, Imamichi Y, Mizoguchi K, Miyajima H, Obata F, Toda T, Farrer MJ, Mizuno Y, Hattori N. Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson’s disease patients from 18 countries. Mov Disord. 2006;21(8):1102–1108. doi: 10.1002/mds.20886. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Wang D, Tang B, Zhao G, Pan Q, Xia K, Bodmer R, Zhang Z. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol Neurodegener. 2008a;3:3. doi: 10.1186/1750-1326-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Xie C, Greggio E, Parisiadou L, Shim H, Sun L, Chandran J, Lin X, Lai C, Yang WJ, Moore DJ, Dawson TM, Dawson VL, Chiosis G, Cookson MR, Cai H. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J Neurosci. 2008b;28(13):3384–3391. doi: 10.1523/JNEUROSCI.0185-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102(46):16842–16847. doi: 10.1073/pnas.0507360102. Epub 12005 Nov 16843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16(2):223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- White LR, Toft M, Kvam SN, Farrer MJ, Aasly JO. MAPK-pathway activity, Lrrk2 G2019S, and Parkinson’s disease. J Neurosci Res. 2007;85(6):1288–1294. doi: 10.1002/jnr.21240. [DOI] [PubMed] [Google Scholar]
- Williams-Gray CH, Goris A, Foltynie T, Brown J, Maranian M, Walton A, Compston DA, Sawcer SJ, Barker RA. Prevalence of the LRRK2 G2019S mutation in a UK community based idiopathic Parkinson’s disease cohort. J Neurol Neurosurg Psychiatry. 2006;77(5):665–667. doi: 10.1136/jnnp.2005.085019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, Saha S, Guillily M, Ferree A, Riley M. Investigating convergent actions of genes linked to familial Parkinson’s disease. Neurodegener Dis. 2008;5(3–4):182–185. doi: 10.1159/000113697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiromerisiou G, Hadjigeorgiou GM, Gourbali V, Johnson J, Papakonstantinou I, Papadimitriou A, Singleton AB. Screening for SNCA and LRRK2 mutations in Greek sporadic and autosomal dominant Parkinson’s disease: identification of two novel LRRK2 variants. Eur J Neurol. 2007;14(1):7–11. doi: 10.1111/j.1468-1331.2006.01551.x. [DOI] [PubMed] [Google Scholar]
- Zabetian CP, Hutter CM, Yearout D, Lopez AN, Factor SA, Griffith A, Leis BC, Bird TD, Nutt JG, Higgins DS, Roberts JW, Kay DM, Edwards KL, Samii A, Payami H. LRRK2 G2019S in families with Parkinson disease who originated from Europe and the Middle East: evidence of two distinct founding events beginning two millennia ago. Am J Hum Genet. 2006a;79(4):752–758. doi: 10.1086/508025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabetian CP, Morino H, Ujike H, Yamamoto M, Oda M, Maruyama H, Izumi Y, Kaji R, Griffith A, Leis BC, Roberts JW, Yearout D, Samii A, Kawakami H. Identification and haplotype analysis of LRRK2 G2019S in Japanese patients with Parkinson disease. Neurology. 2006b;67(4):697–699. doi: 10.1212/01.wnl.0000227732.37801.d4. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wu W, Du Y, Santos SJ, Conrad SE, Watson JT, Grammatikakis N, Gallo KA. Hsp90/p50cdc37 is required for mixed-lineage kinase (MLK) 3 signaling. J Biol Chem. 2004;279(19):19457–19463. doi: 10.1074/jbc.M311377200. [DOI] [PubMed] [Google Scholar]
- Zhu X, Babar A, Siedlak SL, Yang Q, Ito G, Iwatsubo T, Smith MA, Perry G, Chen SG. LRRK2 in Parkinson’s disease and dementia with Lewy bodies. Mol Neurodegener. 2006a;1:17. doi: 10.1186/1750-1326-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Siedlak SL, Smith MA, Perry G, Chen SG. LRRK2 protein is a component of lewy bodies. Ann Neurol. 2006b;59(2):388–393. doi: 10.1002/ana.20928. [DOI] [PubMed] [Google Scholar]
- Zimprich ABS, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]