

Abstract
Strongly asymmetric exchange-coupled biradicals, like the trityl-nitroxides (TN), possess particular magnetic properties opening new possibilities for their application in biophysical, physicochemical and biological studies. In the present work, we investigated the effect of the linker length on the spin-spin coupling interaction in TN biradicals using the newly synthesized biradicals CT02-GT, CT02-AT, CT02-VT and CT02-PPT as well as the previously reported biradicals TNN14 and TN1. Results show that the magnitude of the spin-spin interaction (J) can be easily tuned from ~ 4 G (conformer 1 in CT02-PPT) to over 1200 G (in TNN14) using various linkers separating the two radical moieties and with varying temperature. Computer simulation of EPR spectra was carried out to directly estimate J values of the TN biradicals. In addition to the spin-spin coupling interaction of TN biradicals, their g, hyperfine splitting and zero-field splitting interactions were explored at low temperature (220 K). Our present study clearly shows that the spin-spin interaction variation as a function of linker distance and temperature provides an effective strategy to develop new TN biradicals which can find wide applications in relevant fields.
Introduction
Exchange-coupled biradicals have found applications as polarizing agents in solid state dynamic nuclear polarization (DNP),1–6 building blocks in molecular magnetic materials,7–13 polymerization initiators,14–18 spin labels for structural investigation of biomolecules using inter-spin distance determination19–22 and as molecular probes.23–25 The magnitude and sign of the spin-spin coupling interaction exerts a crucial effect on the physiochemical properties of biradicals. The spin-spin coupling interaction can be through-bonded and/or through-space26 and its value varies by many orders of magnitude.10–12 Several factors such as nature of the linkers between two spins, conformations, substituents and environment (e.g., temperature, solvent, etc) control the exchange coupling magnitude in biradicals.27 Through conformational constraint into the co-planarity of two radical moieties with m-phenylene10,28,29 or simple direct linkage,30 stable trimethylenemethane (TMM)-type biradicals with large positive exchange interactions have been recently obtained which show great potential as building blocks for robust magnetic materials. In contrast, biradicals with rigid geometries holding two nitroxide moieties approximately orthogonal to one another have weak exchange coupling but show enhanced DNP properties.3–6 Therefore, fundamental understanding of the factors controlling exchange coupling could allow development of new biradicals with improved properties.
Most of the reported biradicals are based on two homogenous radical parts, rather than mixed radical moieties.9 The latter case allows the combination of two different heterospin properties into unique molecules. Recently, it has been proposed31–33 that mixed biradicals based on tetrathiatriarylmethyl (trityl) and nitroxide radicals are potentially the best biradical candidate so far to attain the maximal DNP enhancement owing to: 1) ideal electron paramagnetic resonance (EPR) frequency separation between the nitroxide gyy component and the isotropic g-value of trityl radicals and; 2) optimal relaxation times allowing simultaneous microwave saturation and polarization turnover. The moderately weak coupling interaction in which J is typically smaller than the 14N hyperfine splitting in the case of nitroxide biradicals34 does not perturb the EPR frequency matching between the two spins and is a requisite to maximal enhancement. On the other hand, these trityl-nitroxide (TN) biradicals are well suitable for simultaneous measurement of oxygenation and redox status as well as thiol concentration by EPR spectroscopy and imaging but the strong coupling interaction (typically, J > ten times of the 14N hyperfine splitting) is preferred in order to minimize the background signal.35–37 Moreover, TN biradicals with strong ferromagnetic exchange coupling may offer potential as building blocks for molecule-based magnetic materials.7,13 Therefore, in order to expand the application of TN biradicals, there is a great need for fine tuning the coupling interaction between trityl and nitroxide moieties.
Our previous studies showed that TNN14 with a direct and short linkage between two radical moieties has a large J value of 800 G at room temperature,36 whereas TN1 and TSSN has much smaller J values of 300 and 100 G, respectively, due to the longer linker groups.35,37 Note, the J value in our previous and present studies denotes the triplet-singlet separation, its sign was not determined, and its units are presented in Gauss (G) and can be converted into MHz with multiplication by 2.8. These studies led us to the idea of fine tuning the spin-spin coupling interaction in TN biradicals by varying the linker group length. In the present manuscript, we synthesized new TN biradicals CT02-GT, CT02-AT, CT02-VT and CT02-PPT (Chart 1). While CT02-GT, CT02-AT and CT02-VT have flexible linkers with various lengths between two radical parts, CT02-PPT has a rigid linker group. EPR spectroscopy coupled with computer simulation was used to investigate temperature- and linker length- dependent spin-spin coupling interactions in these new biradicals along with TN1 and TNN14. A survey of the EPR spectral profiles of TN biradicals with various coupling magnitudes in the range of 0–1000 G was obtained using computer simulation. In addition, the g, hyperfine splitting and the zero-field splitting tensors were determined from the solid-state EPR spectra.
Chart 1.

Molecular structure of TN biradicals
Results and discussion
Computer simulation of EPR spectra
Computer simulation of the EPR spectra in liquid and frozen solutions was carried out using the EPR simulation program (ROKI\EPR) developed by Prof. Rockenbauer.38 The fitting routine to determine the J values of the trityl-nitroxide biradicals was similar to the method described in our previous studies.35–37 In liquid state, the following parameters were optimized: g1, g2, AN, the relaxation or line width variation parameters: α, β, and γ, and J and its standard deviation ΔJ. The impact of J distribution gives contribution to the line width variation. All the 8 parameters including also ΔJ are simultaneously optimized until the square deviation between experimental and calculated spectra will achieve its minimum. When the best J value is significantly larger than the hyperfine constant, we check the limit of J, when its further increase cannot improve significantly the fit. The criterion is given by the noise of the experimental spectrum. We found for the TN radicals that the J exceeds the hyperfine constant of the nitrogen atom by an order of magnitude but the optimized value is still reliable, while for the various nitroxide-nitroxide biradicals reliable J can be obtained only when it is maximum three times larger than the hyperfine constant. Occasionally a superimposed signal of two conformers is observed. In this case the two component spectra are described by the same parameters except for the J value, which is supposed to vary significantly with the molecular geometry.
Comparatively, the EPR spectral simulation in the solid state is more difficult since more parameters such as dipolar electron-electron interaction and anisotropic g-tensor and hyperfine splitting interaction were included. The Hamiltonian of biradicals can be written:
| (1) |
We restrict the treatment for the case where the J exchange is stronger than the dipolar and hyperfine interactions and the Zeeman level separation for the two radical components. Then the S=1 triplet state will determine the EPR resonance and the mixing of S=1 and S=0 levels will not mix significantly. The effective S=1 Hamiltonian:
| (2) |
Here the components of effective g-tensor can be given as: with i = x, y, z. Similarly the tensor elements of hyperfine splitting constants are also the arithmetical means of two components: . The principal directions of g, zero-field and hf tensors are supposed to be parallel. We still apply the above approach, when the exchange coupling is comparable with the hyperfine interaction. This reduces the reliability of optimized data, but the trend of dipolar parameters characterizing the distance between the two centers of unpaired electron is still correct, when the impact of different linkers is compared.
The simulation requires the optimization of three tensors with altogether 8 elements (three-three for the g- and hyperfine tensor, and two for the zero-field or dipolar tensor, respectively). Furthermore the line width should be also optimized. To obtain the best fit for the 9 non-linear parameters a combination of various strategies is necessary. As great number of local minima exists on the nine-dimensional error surface, the setting of initial parameters set has a great importance. Typically around 100 different starting parameter sets were examined before the automatic adjustment of 9 parameters offered the best available fit.
Experimental studies on the effect of linker distance on the spin-spin coupling interaction in TN biradicals
Figure 1 shows the EPR spectra of TN biradicals at 357 K in which CT02-VT, CT02-AT, CT02-GT and CT02-PPT were newly synthesized and, TNN14 and TN1 were previously reported.35,36 Due to variations in the magnitude of spin-spin coupling interaction, these biradicals exhibit markedly different EPR spectral profiles. TNN14 and TN1 have triplet EPR signals with J = 1230 and 430 G, respectively, which are higher than the reported values of ~160 G for TN1 and ~400 G for TNN14 at room temperature. 35,36 Due to the stronger spin-spin coupling interaction, TNN14 has a symmetric triplet as compared to the asymmetric signal with a relatively weak low-field peak observed for TN1. Despite the direct linkage through an amide bond between the two radical moieties, the use of pyrrolidinyl nitroxide in TNN14 instead of the piperidinyl nitroxide in TN1 leads to a shorter distance between the two spins and therefore exhibits stronger spin-spin coupling interaction. The large J value for TNN14 indicates that the five-membered ring has an almost planar geometry that makes the plane of C2NO parallel to the central plane of trityl moiety. In this case, the dominant spin-spin mechanism has through-bond character. While the biradicals CT02-GT, CT02-AT and CT02-VT have relatively flexible linkers with various lengths, CT02-PPT has a rigid linker. The increase in linker distance in CT02-GT results in a significant decrease of the J value (J = 91G) as evidenced by the splitting of the low-field peak into the doublet (See the experimental EPR spectrum in Figure 1 and the calculated spectral pattern for J = 100 G in Figure 3). However, further increasing the linker lengths does not lead to the decrease of the J values. For instance, CT02-AT and CT02-VT have one and three more methylene groups than CT02-GT between the two radical moieties, respectively, but they have similar J values (i.e., 91 G for CT02-GT, 110 G for CT02-AT and 105 G for CT02-VT). For the latter two biradicals, the flexible linker leads to the large scattering of J values (ΔJ = 7.5 G for CT02-GT, 17 G for CT02-AT and 20 G for CT02-VT) and broadens their low-field doublets to the less resolved patterns (Figure 1). Previously, a similar result was observed for TSSN which has a J value of 82 G but affords an unresolved low-field doublet due to its long and flexible cystamine linker between nitroxide and trityl moieties.37 The linker length-independent J values for CT02-GT, CT02-AT, CT02-VT and TSSN are most likely due to fast folding of the flexible linkers which leads to averaging of different conformations and produces the geometries where the distances between the two radical centers are almost the same for four biradicals. Therefore, the through-space spin-spin interactions are dominant for CT02-GT, CT02-AT, CT02-VT and TSSN. The through-space exchange coupling interaction was similarly observed between the diagonal nitroxides in calix[4]arene nitroxide tetraradicals and diradicals.26 Interestingly, CT02-PPT with the long but rigid linker affords a complicated EPR spectrum with two separated doublets at low-field area possibly due to the presence of two stable conformers (Conformers 1 and 2) whose inter-conversion is relatively slow and their characteristic EPR peaks are distinguishable even at the relatively high temperature (i.e., 357 K). Figure 2 shows the structure of two possible stable conformers in CT02-PPT. Conformer 1 featuring the wing doublet pattern (Figure 1) with a small J value of 3.7 G has a relatively long interspin distance (13.2 Å) (Figure 2). In contrast, the inner doublet can be assigned to Conformer 2 with the interspin distance of 11.1 Å which affords an intermediate J value of 41 G.
Figure 1.

Experimental and simulated EPR spectra of TN biradicals at 357K.
Figure 3.

Simulated EPR spectra of TN biradicals with various J values. The simulation input standard deviation of J is zero, and no relaxation effect is considered. T and N indicate the trityl and nitroxide signal, respectively; (*) shows forbidden transitions of biradicals.
Figure 2.
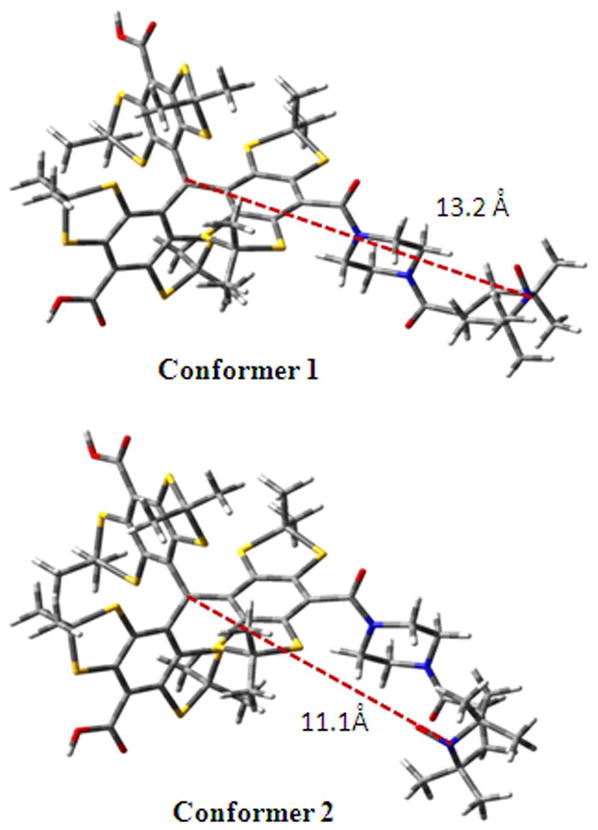
Two possible stable conformations of CT02-PPT showing different interspin distances. The structures were obtained on the basis of the most stable and unique conformers using Spartan 04 at the MMFF level.
Theoretical investigation of the effect of spin-spin exchange coupling on EPR spectra of TN biradicals
Since fast molecular tumbling averages the anisotropic g, the hyperfine and the dipolar interactions at a relatively high temperature (> 293K), only the spin-spin coupling interaction of TN biradicals exerts a significant effect on their EPR spectra as shown above. Thus, computer calculation was carried out to quantitatively describe the effect of J values on EPR spectra of TN biradicals. Figure 3 shows the simulated EPR spectra of TN biradicals as a function of J values. When the trityl and nitroxide moieties are far away from each other and uncoupled (J = 0), the superimposed trityl (a single line denoted by T) and nitroxide signals (a triplet denoted by N with a line separation of AN = 17 G) can be observed. With the increase in J values but smaller than the hyperfine coupling (AN) (i.e., J = 2 and 5 G), each line from trityl and nitroxide signals splits into doublet, affording eight lines in total. If J is comparable to AN (i.e., J = 10 and 25 G) the forbidden transitions (*) appear besides the doublet lines. When J is moderately larger than AN, a doublet pattern can be seen at the low-field part of the spectra (i.e., J = 25, 50, 100 G). The separation of this doublet is inversely proportional to the J value. With further increase of J value, this doublet merges into singlet line (See J = 200 G), thus affording an EPR triplet pattern with the separation of approximately 8.5 G (AN/2). When J is very large (see J = 1000 G), the EPR spectrum is almost symmetric with the same width and amplitude for the triplet.
Temperature-dependent effect on J coupling
As mentioned above, the predominant exchange mechanism has the through-space character since the number of σ-bonds in the linker does not play a decisive role in the magnitude of spin-spin exchange in the TN biradicals. Therefore, it can be expected that the increase of temperature can lead to different J values because of the varying distances possibly resulting from the conformational changes.26 As shown in Figure 4, for the TN biradicals (TNN14, TN1 and CT02-GT) with relatively short and rigid linkers, their J couplings increase with temperature up to 357 K which can be explained by the interconversion of two envelope or twist conformers of the five-membered nitroxide ring for TNN14 or two chair conformers of six-membered nitroxide ring for TN1 and CT02-GT. Their ground-state conformers correspond to the ring geometries with the relatively longer distances between the nitroxide and trityl moieties, thus resulting in smaller J couplings. Due to the large J value for TNN14, the tendency of J to increase as a function of temperature is not that favorable. For CT02-AT and CT02-VT, the J has a maximal value at around 330–340 K. This behavior probably shows the activation of intramolecular rotation around the Csp3-Csp3 bonds of the linker. Much higher J maxima for CT02-AT and CT02-VT than CT02-GT further verify that the spin-spin interaction of these TN biradicals is through-spaced except for TNN14 where it is through-bonded.
Figure 4.
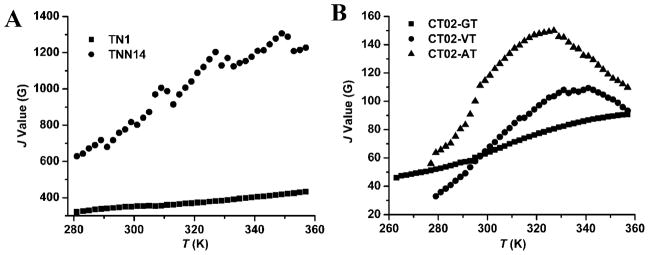
J values of (A) trityl-nitroxide biradicals TN1 and TNN14 as well as (B) CT02-GT, CT02-AT and CT02-GT as a function of temperature obtained by simulating the corresponding EPR spectra at various temperatures.
On the other hand, the substituted piperazine represents a rigid linker where the exchange interaction between radical centers is rather weak. In this case, the transition between the two chair conformers is slow which allows for the detection of lines from the two conformers (i.e., Conformers 1 and 2 of CT02-PPT in Figure 1). In Conformer 2 with a relatively large J (41 G at 357 K), the biradical adopts a geometry where the separation of paramagnetic centers is relatively small. The increase of its J coupling with temperature can be attributed to the enhanced amplitude of oscillation of the ground chair conformation (Figure 5A). Since the oscillation can reduce the non-planarity of the nitroxide moiety and accordingly decrease AN coupling, we examined the variation of AN with temperature. As expected, the AN value decreased from 17.75 G at 279 K to 16.92 G at 357 K (Figure 5B). Further inspection shows that the relative population of Conformer 2 significantly increases with temperature although it diminishes at high temperature indicating that Conformer 1 has lower energy and is the ground-state conformer (Figure 5C).
Figure 5.

(A) J values of two conformers, (B) hyperfine splitting (AN) and (C) population of Conformer 2 of CT02-VT as a function of temperature which were obtained by simulating EPR spectra at various temperatures.
Solid-state EPR studies of TN biradicals
The magnitude of the dipolar interaction and the anisotropic parameters for g- and hyperfine tensors are also important for the application of biradicals in structural studies, and for this reason, estimation of these parameters from the frozen solution spectra was attempted. One critical step in this analysis is to compare the J exchange coupling with the hyperfine interaction. According to the EPR measurements in solution, the J exchange couplings of both TNN14 and TN1 are stronger than the dipolar and hyperfine interactions, and the Zeeman level separation of the two radical components and therefore, no significant triplet-singlet mixing can occur, that is, J is large enough to fulfill the restrictions of solid-state EPR simulation as mentioned above in the description of simulation program. Figure 6 shows the EPR spectra of TNN14 and TN1 at 220 K in ethylene glycol/H2O (1:1, v/v) glass-forming solution, which can be analyzed well to give tensorial elements for g, hyperfine splitting and zero-field splitting interactions. Since trityl radical is a stable carbon-centered radical with a small spin orbit coupling, the very small g-anisotropy was observed at solid state with all g values of approximately 2.0030. In contrast, the gzz value of nitroxide radical is close to 2.0023 and its gxx and gyy are typically 2.008–2.010 due to the significant spin orbit coupling. While the z-direction of nitroxide monoradical is mainly determined by the π-lobe of unpaired electron, the direction of principal axes for the above biradicals is determined by the large zero-field (or dipolar) interaction. Since gxx’s of both biradicals (i.e., 2.0025 for TNN14 and 2.0028 for TN1) are nearly equal to the gzz of mono-nitroxide (2.0023), the x-direction of biradicals agrees with the direction of π-lobes of the nitroxide moiety where the unpaired electron is localized. The same assignment follows from the hfsc values (i.e., for trityl radicals Axx=Ayy=Azz=0, while for nitroxides Azz = 34–36 G, Axx=Ayy=5–8 G) which are in excellent agreement with the values in Table 1. Since the rhombic zero-field parameter E is small, the z-direction of biradical is determined by the axis connecting the two radical centers. According to the point-dipole approach, the R distance between the radical centers can be given by the D zero-field term as R3 = 27810/D. This relation gives R = 10.6 Å for TN1 and R = 9.7 Å for TNN14. The g-, D- and A tensors of either biradical have nearly the same values at different temperatures except for the different line widths.
Figure 6.
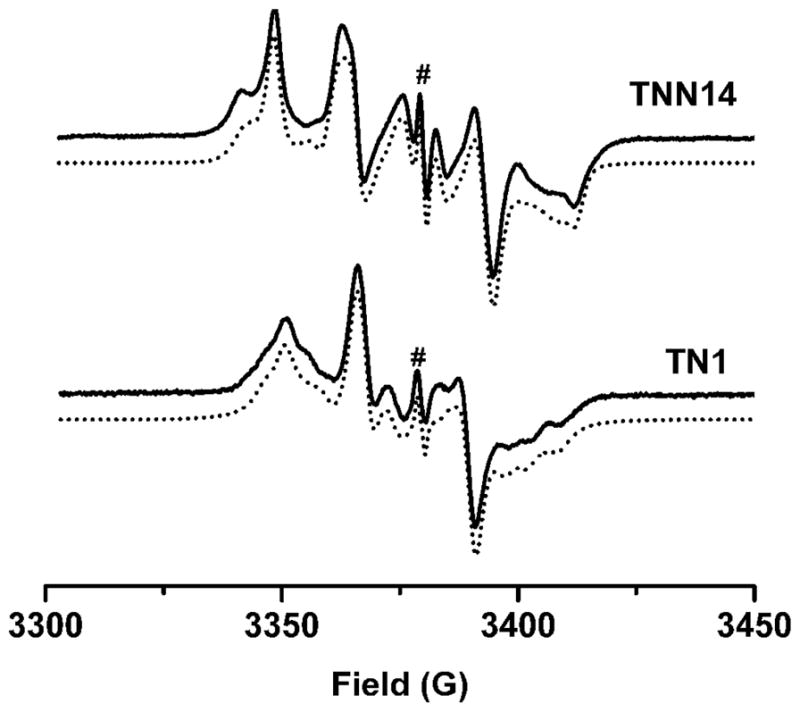
Experimental and simulated EPR spectra of TNN14 and TN1 at 220 K in ethylene glycol/H2O(1:1, v/v) glass-forming solution. #, the signal of trityl monoradical.
Table 1.
Anisotropic values of g, hyperfine splitting (AN) and zero-field splitting constant (D) of biradicals.
| Biradicals | G | AN (G) | D (G) | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| gxx | gyy | gzz | Axx | Ayy | Azz | Dxx | Dyy | Dzz | |
| TNN14 | 2.0025 | 2.0048 | 2.0057 | 18.0 | 3.9 | 2.8 | −10.0 | −10.4 | 20.4 |
| TN1 | 2.0028 | 2.0040 | 2.0063 | 17.9 | 5.8 | 3.8 | −7.8 | −7.8 | 15.6 |
| CT02-GT | 2.0039 | 2.0070 | 2.0028 | 1.1 | 0 | 18.5 | −3.3 | −9.0 | 12.3 |
| CT02-AT | 2.0016 | 2.0088 | 2.0040 | 0 | 7.3 | 15.9 | −3.0 | −3.7 | 6.6 |
| CT02-VT | 2.0103 | 2.0057 | 2.0027 | 0 | 4.4 | 38 | −1.9 | −0.8 | 2.7 |
| CT02-PPT | 2.0046 | 2.0049 | 2.0027 | 0.5 | 1.0 | 40 | 1 | −3 | 2 |
The small gzz (2.0028, Table 1) and large Azz (18.5, Table 1), which is close to the half of a typical Azz (34–36 G) in nitroxide radical, observed for CT02-GT indicate that the lobe of unpaired electron on the nitroxide moiety is parallel to the direction connecting the two radical centers of this biradical. The very different g- and hyperfine tensors for CT02-GT compared to the mono nitroxides indicate that the J exchange coupling is indeed larger than the hyperfine interaction. In the case of CT02-AT, the Axx value is very small (~ 0 G, Table 1) perhaps because the J coupling has a comparable value with the hyperfine coupling resulting in the significant triplet-singlet mixing and uncertain tensorial elements. Similar to CT02-GT, CT02-AT has an Azz value close to the half of the value for nitroxide monoradical, indicating that the direction of π-lobes of nitroxide moiety is almost parallel to the direction interconnecting the two radical centers.
As for CT02-VT, both the g- and hyperfine values are typical for nitroxide monoradical, implying that the J exchange coupling is comparable or less than the hyperfine coupling. Thus, the D value and the calculated R distance between the radical centers are very approximate and the same is also true for CT02-PPT. The negligible D values are in accordance with the extrapolated small J values at low temperature, since D and J should have the same order of magnitude.
Conclusion
In summary, we investigated the effects of linker distance and temperature on the spin-spin coupling interaction in TN biradicals. The coupling magnitude can be easily tuned from ~ 4 to 1200 G by changing the nature and length of the tether group. Further tuning of the coupling interaction can be also achieved by varying the temperature. Depending on the coupling magnitude, these TN radicals could find various applications in redox sensing, magnetic materials and DNP enhancement. The biradical CT02-PPT with a relatively small exchange interaction will be a good candidate as DNP agent and the DNP enhancement induced by TN biradicals can be further expanded by replacing the piperazine linker with a more rigid and bulky linker.4–6 Overall, our present work provides fundamental understanding of the spin-spin interaction in TN or other biradicals and could shed new lights on the design of new TN biradicals with desirable properties.
Experimental section
Synthesis of TEMPO derivatives GT, AT, VT and PPT
To a solution of Boc-glycine (201 mg, 1.15 mmol) in dry CH2Cl2 (25 mL) at 0 °C were added successively 4-amino-TEMPO (216 mg, 1.26 mmol), N,N-diisopropylethylamine (DIPEA, 300 μL), 1-hydroxybenzotriazole (HOBt, 80%, 213 mg, 1.26 mmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbo-diimide hydrochloride (EDCI, 265 mg, 1.38 mmol). The resulting orange solution was stirred at room temperature for 14 h, and then washed with saturated aqueous NH4Cl solution. The aqueous phase was separated and extracted once with CH2Cl2, and the combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. Chromatography of the residue on SiO2 (1% to 6% methanol in CH2Cl2) afforded 313 mg (83%) of GT as a red solid. HRMS (ESI) m/z = 351.2134 calcd for C16H30N3NaO4• [M+Na]+, found: 351.2151. The product was used in the next step without further characterization. Similar procedure was utilized for the synthesis of AT and VT using Boc-β-alanine and 5-(Boc-amino)valeric acid as starting materials, respectively, instead of Boc-glycine. In addition, PPT was synthesized from 4-carboxy-TEMPO and 1-Boc-piperazine using the similar procedure. AT (250 mg, 86%): HRMS (ESI) m/z = 365.2291 calcd for C17H32N3NaO4• [M+Na]+, found: 365.2295. VT (230 mg, 78%): HRMS (ESI) m/z = 393.2604 calcd for C19H36N3NaO4• [M+Na]+, found: 393.2620. PPT (183mg, 75%): HRMS (ESI) m/z = 391.2447 calcd for C19H34N3NaO4•+ [M+Na]+, found: 391.2442.
Synthesis of trityl-nitroxide diradicals CT02-GT, CT02-AT, CT02-VT and CT02-PPT
To a solution of GT (3.0 mg, 9.1 μmol) in DCM (1 mL) was added TFA (1 mL). The reaction mixture was stirred for 3 h at room temperature and evaporated to dryness under vacuum. The residue was redissolved in 1 mL of DMF and added dropwise to the solution of CT-03 (10 mg, 10 μmol), HOBt (80%, 4.6 mg, 27 μmol), (benzotriazol-1-yloxy) tris(dimethylamino)phosphonium hexafluorophosphate (BOP, 4.4 mg, 10 μmol), and DIPEA (15 μL) in dry DMF (4 mL). The resulting reaction mixture was continuously stirred for 18 h at room temperature. Solvent was removed under vacuum, and the residue was dissolved in phosphate buffer (0.1 M, pH 7.4) and purified by column chromatography on reversed phase C-18 using water followed by 0–25% acetonitrile in water as eluents to give CT02-GT as a green solid (6.4 mg, 58%). HRMS (MALDI-TOF, DHB as the matrix) m/z calcd for C51H59N3O7S12•• ([M]+) 1209.100, found 1209.049; calcd for C51H59N3NaO7S12••+ ([M+Na]+) 1232.089, found 1232.051;
Similar procedure was utilized for the synthesis of CT02-AT, CT02-VT and CT02-PPT using AT, VT and PPT as starting materials, respectively. CT02-AT (5.4 mg, 60%) HRMS (MALDI-TOF, DHB as the matrix) m/z calcd for C52H61N3O7S12•• ([M]+) 1223.116, found 1223.049; calcd for C52H61N3NaO7S12••+ ([M+Na]+) 1246.105, found 1246.058. CT02-VT (4.1 mg, 52%) HRMS (MALDI-TOF, DHB as the matrix) m/z calcd for C54H66N3O7S12••+ ([M+H]+) 1252.154, found 1252.158; calcd for C54H65N3NaO7S12••+ ([M+Na]+) 1274.136, found 1274.177. CT02-PPT (6.8 mg, 38%): HRMS (ESI) m/z = 1272.1212 calcd for C54H63N3NaO7S12••+, found: 1272.1179.
Supplementary Material
Acknowledgments
This work was supported by NIH grants HL38324, EB0890, EB4900 (J.L.Z.), and HL81248 (F.A.V.).
Footnotes
Supporting Information Available: Spectroscopic characterization of new biradicals. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Song CS, Hu KN, Joo CG, Swager TM, Griffin RG. J Am Chem Soc. 2006;128:11385–11390. doi: 10.1021/ja061284b. [DOI] [PubMed] [Google Scholar]
- 2.Dane EL, Maly T, Debelouchina GT, Griffin RG, Swager TM. Org Lett. 2009;11:1871–1874. doi: 10.1021/ol9001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dane EL, Corzilius B, Rizzato E, Stocker P, Maly T, Smith AA, Griffin RG, Ouari O, Tordo P, Swagert TM. J Org Chem. 2012;77:1789–1797. doi: 10.1021/jo202349j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zagdoun A, Casano G, Ouari O, Lapadula G, Rossini AJ, Lelli M, Baffert M, Gajan D, Veyre L, Maas WE, Rosay M, Weber RT, Thieuleux C, Coperet C, Lesage A, Tordo P, Emsley L. J Am Chem Soc. 2012;134:2284–2291. doi: 10.1021/ja210177v. [DOI] [PubMed] [Google Scholar]
- 5.Kiesewetter MK, Corzilius B, Smith AA, Griffin RG, Swager TM. J Am Chem Soc. 2012;134:4537–4540. doi: 10.1021/ja212054e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuki Y, Maly T, Ouari O, Karoui H, Le Moigne F, Rizzato E, Lyubenova S, Herzfeld J, Prisner T, Tordo P, Griffin RG. Angew Chem-Int Edit. 2009;48:4996–5000. doi: 10.1002/anie.200805940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller JS, Drillon M, editors. Magnetism: Molecules to Materials. I–IV Wiley-VCH; Weinheim: 2001–2003. [Google Scholar]
- 8.Iwamura H, Koga N. Accounts Chem Res. 1993;26:346–351. [Google Scholar]
- 9.Rajca A. Chem Rev. 1994;94:871–893. [Google Scholar]
- 10.Rajca A, Olankitwanit A, Rajca S. J Am Chem Soc. 2011;133:4750–4753. doi: 10.1021/ja200708b. [DOI] [PubMed] [Google Scholar]
- 11.Shultz DA, Fico RM, Lee H, Kampf JW, Kirschbaum K, Pinkerton AA, Boyle PD. J Am Chem Soc. 2003;125:15426–15432. doi: 10.1021/ja037787o. [DOI] [PubMed] [Google Scholar]
- 12.Shultz DA, Boal AK, Farmer GT. J Am Chem Soc. 1997;119:3846–3847. [Google Scholar]
- 13.Lahti PM, editor. Magnetic Properties of Organic Materials. Marcel Dekker; New York: 1999. [Google Scholar]
- 14.Yoshida E, Takeda K. Polym J. 2001;33:590–596. [Google Scholar]
- 15.Bothe M, Schmidt-Naake G. Macromol Chem Phys. 2004;205:208–216. [Google Scholar]
- 16.Hill NL, Braslau R. Macromolecules. 2005;38:9066–9074. [Google Scholar]
- 17.Ruehl J, Hill NL, Walter ED, Milihauser G, Braslau R. Macromolecules. 2008;41:1972–1982. doi: 10.1021/ma702358c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaim A, Pietrasik K, Stoklosa T. Eur Polym J. 2010;46:519–527. [Google Scholar]
- 19.Hubbell WL, Cafiso DS, Altenbach C. Nat Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 20.Hagelueken G, Ingledew WJ, Huang H, Petrovic-Stojanovska B, Whitfield C, ElMkami H, Schiemann O, Naismith JH. Angew Chem-Int Edit. 2009;48:2904–2906. doi: 10.1002/anie.200805758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berliner LJ, Eaton GR, Eaton SS, editors. Distance Measurements in Biological Systems by EPR. Vol. 19 Kluwer; New York: 2000. [Google Scholar]
- 22.Yang ZY, Liu YP, Borbat P, Zweier JL, Freed JH, Hubbell WL. J Am Chem Soc. 2012;134:9950–9952. doi: 10.1021/ja303791p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marx L, Rassat A. Angew Chem-Int Edit. 2000;39:4494–4496. doi: 10.1002/1521-3773(20001215)39:24<4494::aid-anie4494>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 24.Ishiguro K, Ozaki M, Sekine N, Sawaki Y. J Am Chem Soc. 1997;119:3625–3626. [Google Scholar]
- 25.Roshchupkina GI, Bobko AA, Bratasz A, Reznikov VA, Kuppusamy P, Khramtsov VV. Free Radic Biol Med. 2008;45:312–320. doi: 10.1016/j.freeradbiomed.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajca A, Mukherjee S, Pink M, Rajca S. J Am Chem Soc. 2006;128:13497–13507. doi: 10.1021/ja063567+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forbes MDE, Dukes KE, Avdievich NI, Harbron EJ, DeSimone JM. J Phys Chem A. 2006;110:1767–1774. doi: 10.1021/jp053183q. [DOI] [PubMed] [Google Scholar]
- 28.Rajca A, Takahashi M, Pink M, Spagnol G, Rajca S. J Am Chem Soc. 2007;129:10159–10170. doi: 10.1021/ja0712017. [DOI] [PubMed] [Google Scholar]
- 29.Rajca A, Shiraishi K, Pink M, Rajca S. J Am Chem Soc. 2007;129:7232–7233. doi: 10.1021/ja071881d. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki S, Furui T, Kuratsu M, Kozaki M, Shiomi D, Sato K, Takui T, Okada K. J Am Chem Soc. 2010;132:15908–15910. doi: 10.1021/ja107769z. [DOI] [PubMed] [Google Scholar]
- 31.Hu KN, Bajaj VS, Rosay M, Griffin RG. J Chem Phys. 2007;126:044512. doi: 10.1063/1.2429658. [DOI] [PubMed] [Google Scholar]
- 32.Maly T, Debelouchina GT, Bajaj VS, Hu KN, Joo CG, Mak-Jurkauskas ML, Sirigiri JR, van der Wel PCA, Herzfeld J, Temkin RJ, Griffin RG. J Chem Phys. 2008;128:052211. doi: 10.1063/1.2833582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu KN. Solid State Nucl Magn Reson. 2011;40:31–41. doi: 10.1016/j.ssnmr.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu KN, Debelouchina GT, Smith AA, Griffin RG. J Chem Phys. 2011;134:125105. doi: 10.1063/1.3564920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu YP, Villamena FA, Rockenbauer A, Zweier JL. Chem Commun. 2010;46:628–630. doi: 10.1039/b919279d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu YP, Villamena FA, Song YG, Sun J, Rockenbauer A, Zweier JL. J Org Chem. 2010;75:7796–7802. doi: 10.1021/jo1016844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu YP, Song YG, Rockenbauer A, Sun J, Hemann C, Villamena FA, Zweier JL. J Org Chem. 2011;76:3853–3860. doi: 10.1021/jo200265u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rockenbauer A, Korecz L. Appl Magn Reson. 1996;10:29–43. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.