

Abstract
Many globular and natively disordered proteins can convert into amyloid fibers. These fibers are associated with numerous pathologies1 as well as with normal cellular functions2,3, and frequently form during protein denaturation4,5. Inhibitors of pathological amyloid fibers could serve as leads for therapeutics, provided the inhibitors were specific enough to avoid interfering with normal processes. Here we show that computer-aided, structure-based design can yield highly specific peptide inhibitors of amyloid formation. Using known atomic structures of segments of amyloid fibers as templates, we have designed and characterized an all D-amino acid inhibitor of fibrillation of the tau protein found in Alzheimer’s disease, and a non-natural L-amino acid inhibitor of an amyloid fiber that enhances sexual transmission of HIV. Our results indicate that peptides from structure-based designs can disrupt the fibrillation of full-length proteins, including those like tau that lack fully ordered native structures.
The finding that dozens of devastating pathologies, including Alzheimer’s disease, are associated with amyloid fibers has stimulated research on fiber inhibition. One approach employs the self-associating property of proteins that form fibers to poison fibrillation with short peptide segments6–11. A second approach is based on screening for molecules that can disrupt fiber formation12,13. Here we take a third approach to fiber inhibition: structure-based design of non-natural peptides targeted to block the ends of fibers. With advanced sampling techniques and minimizing an appropriate energy function, we computationally identify novel candidate inhibitors from a vast peptide space that interact favorably with our template structure. This approach has become possible following the determination of several dozen fiber-like atomic structures of segments from amyloid-forming proteins14–16.
These structures reveal a common motif termed a steric zipper, in which a pair of β-sheets is held together by the interdigitation of their side-chains14. Using the steric-zipper structures formed by segments of two pathological proteins as templates, here we design inhibitors that cap fiber ends. As we show, the inhibitors greatly slow fibrillation of their parent proteins, offering a route to designed chemical interventions, and also supporting the hypothesis that steric zippers are the principal structural element of these fibers.
One of the two fiber-like steric zippers that we have chosen as a target for inhibitor design is the hexapeptide 306VQIVYK311 from tau, a protein that forms intracellular amyloid fibers in Alzheimer’s disease17. This segment has been shown to be important for fibrillation of the full-length protein and itself forms fibers with biophysical properties similar to full-length tau fibers15,18,19. Our second template for inhibitor design, identified by the 3D Profile algorithm20,21, is the steric-zipper structure of the peptide segment GGVLVN from the amyloid fiber formed by 248PAP286, a proteolytic fragment of prostatic acid phosphatase (PAP), a protein abundant in semen. 248PAP286 fibers (also termed SEVI, or Semen derived Enhancer of Virus Infection) enhance HIV infection by orders of magnitude in cell culture studies, while the monomeric peptide is inactive22.
Our computational approach to designing non-natural peptides that inhibit fibrillation is summarized in Fig. 1 for the VQIVYK segment of tau; the same general strategy is used for the GGVLVN segment of 248PAP286. In both systems, we design a tight interface between the inhibiting peptide and the end of the steric zipper to block additional segments from joining the fiber. By sampling L- or D- amino acids, or commercially available non-natural amino acids, we can design candidate inhibitors with side chains that maximize hydrogen bonding and apolar interactions across the interface.
Figure 1. Scheme for the design and characterization of peptide inhibitors of amyloid fibrillation.

Tau constructs form fibers in vitro (top left)24. The VQIVYK segment in isolation forms fibers and microcrystals (bottom left). The atomic structure of the fiber-like VQIVYK segment reveals a characteristic steric zipper motif15, comprising a pair of interacting β-sheets running along the fiber axis (grey arrow), in purple and grey (bottom right). We designed a D-amino acid peptide to bind to the end of the steric zipper template and prevent fiber elongation (middle right). The D-peptide, in red, is designed to satisfy hydrogen bonds and make favorable apolar interactions with the molecule below, while preventing the addition of other molecules above and on the opposite β-sheet. As shown in vitro, the designed D-peptide prevents the formation of fibers when incubated with tau K19 (upper right). Scale bars are 100 μm and 200 nm on the microcrystal image and electron micrographs, respectively.
We hypothesize that the steric-zipper structures of the VQIVYK and GGVLVN segments represent the spines of the fibers formed by their parent proteins. Supporting our hypothesis are our results that D-amino acid inhibitors designed on the VQIVYK steric zipper template inhibit fiber formation not only of the VQIVYK segment, but also of two tau constructs, K12 and K1923,24 (Fig. 2a). Similarly, the peptide composed of non-natural amino acids designed on the GGVLVN template inhibits the fibrillation of 248PAP286 and greatly inhibits the HIV infectivity of human cells in culture.
Figure 2. Designed D-peptide delays tau K12 fibrillation in a sequence-specific manner.

a, Tau construct composition23. The largest human tau isoform hTau40 contains four microtubule-binding repeats, R1 – R4, while K12 and K19 lack R2. The black bars at the N-termini of R2 and R3 represent the fibrillogenic segments VQIINK and VQIVYK, respectively. b, The inhibitor D-TLKIVW is designed to interact with atoms on both β-strands of the VQIVYK steric zipper primarily through hydrophobic packing and hydrogen bonding interactions. c, The inhibitor (red) interacts with the VQIVYK structure below (grey). The transparent spheres show where the two molecules interact favorably. Black and red dashes indicate main chain and side chain hydrogen bonds, respectively. d, The seeded fibrillation of 50 μM K12 in the presence and absence of ten-fold molar excess peptide was monitored by Thioflavin S fluorescence. In the presence of the scrambled peptide D-TIWKVL (dark green) or alone (black), seeded K12 fibrillation occurs with almost no lag time. However, D-TLKIVW prevents fibrillation for days (maroon). e, At equimolar concentrations, D-TLKIVW (red) inhibits the fibrillation of 50μM K12. D-TIKWVL (blue) with only three residues scrambled shows weak inhibition. However, no inhibition is observed for either D-TIWKVL (green) or D-LKTWIV (cyan). f, The replacement of D-Leu2, designed to clash with VQIVYK on the opposite sheet, with D-Ala eliminates the inhibition of fibrillation.
To design a D-amino acid hexapeptide sequence that interacts favorably with the VQIVYK steric zipper15, and also prevents further addition of tau molecules to the fiber, we used the RosettaDesign software25. This led to the identification of four D-amino acid peptides: D-TLKIVW, D-TWKLVL, D-DYYFEF, and D-YVIIER, in which D- signifies that all α-carbon atoms are in the D-configuration (Fig. 2b,c, Supplementary Figs. 1, 2 and Supplementary Table 1). In the D-TLKIVW design model (Fig. 2b, c and Supplementary Fig. 1), the inhibitor packs tightly across the top of the VQIYVK steric-zipper structure, maintaining all main chain hydrogen bonds. The side-chain hydrogen bonding between layers of stacked Gln307 residues is replaced in the designed interface by an interaction with D-Lys3. Several hydrophobic interactions between D-TLKIVW and the two VQIVYK β-strands contribute to the favorable binding energy (Supplementary Table 1). In the design, the D-peptide blocks the addition of another layer of VQIVYK, both above the D-peptide and across on the mating β-sheet (Supplementary Fig. 3). D-Leu2 of the designed inhibitor prevents the addition of a VQIVYK molecule above it through a steric clash with Ile308 of VQIVYK and on the mating sheet through a clash with Val306 and Ile308 (Supplementary Fig. 3). These steric clashes involving D-Leu2 are intended to block fiber growth.
We used fluorescence spectroscopy and electron microscopy to assess whether the designed D-peptides inhibit the fibrillation of the tau segment VQIVYK, and the K12 and K19 tau constructs. Among our designed inhibitors, D-TLKIVW is the most effective (Supplementary Fig. 4). Electron microscopy verified that incubation with D-TLKIVW prevents K19 fibrillation, which would otherwise have occurred within the elapsed time (Fig. 1 upper right). D-TLKIVW delays fiber formation even at sub-equimolar concentration relative to VQIVYK, K12, and K19 (Supplementary Fig. 5). Five-fold molar excess of D-TLKIVW delays K12 fibrillation for more than two weeks in some experimental replicates (Supplementary Fig. 5c, d). In ten-fold molar excess, D-TLKIVW prevents the fibrillation of K12 in the presence of preformed K12 fiber seeds, suggesting that the peptide interacts with fibers (Fig. 2d). Also, kinetic analysis shows that the fiber elongation rate decreases in the presence of increasing concentrations of inhibitor peptide (Supplementary Fig. 6). The large increase in lag time in un-seeded reactions may be due to interactions with small aggregates on pathway to fiber formation.
To investigate the specificity of the designed inhibitor, we tested scrambled sequence variants of D-TLKIVW that have poor calculated energy scores and unfavorable packing (Supplementary Table 1). The scrambled peptides D-TIKWVL, D-TIWKVL, and D-LKTWIV exhibit little inhibitory effect at an equimolar ratio with VQIVYK, K12, and K19 (Fig. 2e and Supplementary Fig 7), showing that the inhibition is sequence-specific. Also, the diastereomer, L-TLKIVW, is less effective than D-TLKIVW (Supplementary Fig 8). As a further test of the specificity of our design, we confirmed that D-TLKIVW is unable to block the fibrillation of amyloid beta, also associated with Alzheimer’s disease. This suggests that the D-peptide inhibitor is not general for amyloid systems, but is specific for the VQIVYK interface in tau (Supplementary Fig. 9). Such specificity for designed inhibitors is essential if they are not to interfere with proteins that natively function in an amyloid state3.
To confirm that the designed D-peptide inhibits in accord with the design model (Fig. 2b,c and Supplementary Fig. 1), we performed several additional tests. First, we visualized the position of the inhibitor D-TLKIVW relative to fibers of the tau construct K19 using electron microscopy. We covalently linked maleimido-nanogold particles to both the inhibitor and, separately, to a scrambled hexapeptide, D-LKTWIV. We used a blind counting assay and found that D-TLKIVW shows a significant binding preference for the end of fibers compared to nanogold alone, in contrast to the scrambled control peptide, D-LKTWIV (Fig. 3a and Supplementary Fig. 10).
Figure 3. Mechanism of interaction.

a, Nanogold covalently bound to D-TLKIVW localizes at the ends of two tau K19 fibers. Scale bar represents 50 nm. b, The inhibitor D-TLKIVW binds to fibers with an estimated affinity constant in the low micromolar range, as shown by the indole proton region of the 500 MHz 1H NMR spectra of D-TLKIVW (9.83 ppm) and D-LKTWIV (9.98 ppm) in the presence of increasing concentrations of VQIVYK fibers. The resonance of the D-TLKIVW indole proton is reduced in the presence of increasing concentrations of VQIVYK fibers, whereas the indole proton signal for the scrambled control peptide D-LKTWIV is only slightly affected. Fiber solutions contain 0 to 1500 μM of VQIVYK monomers, as indicated.
As a further test of the model, we used nuclear magnetic resonance (NMR) to characterize the binding affinity of D-TLKIVW for tau fibers. The 1H NMR spectra for D-TLKIVW were collected in the presence of increasing concentrations of VQIVYK or K19 fibers. Because neither K19 nor VQIVYK contains tryptophan, we were able to monitor the 1H resonance of the indole proton of the tryptophan in our inhibitor. When bound to a fiber, the inhibitor, D-TLKIVW, is removed from the soluble phase and the 1H resonance is diminished (Fig. 3b and Supplemental Fig. 11)26. As a control, both D-TLKIVW and the non-inhibiting peptide D-LKTWIV were included in the same binding reaction mixture. As shown in Fig. 3b, the D-TLKIVW indole resonance is reduced greatly, whereas the D-LKTWIV indole resonance is only slightly affected. Spectra of the two peptides are shown in Supplementary Fig. 12. By monitoring the D-TLKIVW indole resonance over a range of VQIVYK fiber concentrations, we estimate the apparent Kd value of the interaction between D-TLKIVW and VQIVYK fibers to be ~2 μM (Supplementary Fig. 11a and Methods). This value corresponds to a standard free binding energy of ~7.4 kcal/mol, with ~2.5 kcal/mol from apolar interactions, and ~4.9 kcal/mol from six hydrogen bonds (see Methods). Repeating the NMR binding experiment with K19 fibers yields a similar trend (Supplementary Fig 11b). To determine whether D-TLKIVW has affinity for soluble VQIVYK, we measured 1H NMR spectra of D-TLKIVW and D-LKTWIV in the presence of increasing amounts of soluble VQIVYK. Only a slight change in the chemical shifts of the indole proton peaks of D-TLKIVW and D-LKTWIV is observed, even at 70-fold molar excess of VQIVYK (Supplementary Fig. 13). This, together with the ability of the peptide to prevent seeded fibrillation, suggests that D-TLKIVW does not interact with monomers, but rather with a structured, fiber-like species.
For another test of our design model, we replaced the D-Leu residue with D-Ala in D-TLKIVW. Our structural model suggests that D-Leu2 of D-TLKIVW is important for preventing tau fibrillation because of its favorable interaction with the Ile residue of the VQIVYK molecule below and with Ile and the first Val of VQIVYK across the steric zipper (Fig. 2b,c and Supplementary Fig. 1). The D-Ala replacement eliminates these interactions and, further, removes a steric clash that would occur were another VQIVYK molecule placed across from the inhibitor (Supplementary Fig. 3 and Supplementary Table 1). When the D-Ala variant is incubated with VQIVYK and the tau constructs, it shows no inhibitory effect on fibrillation (Fig. 2f and Supplementary Fig. 14). This confirms that D-Leu2 is critical for the efficacy of D-TLKIVW, consistent with our model.
In summary, while our electron microscopy, NMR, and D-Ala replacement results support a model in which the designed D-TLKIVW peptide binds to the ends of tau fibers, they do not constitute proof that the inhibitor binds exactly as anticipated in the design (Supplementary Fig. 15).
To expand on our design methodology, we computationally designed an inhibitor of 248PAP286 fibrillation containing non-natural L-amino acids (Fig. 4b, Supplementary Fig. 16) using the GGVLVN structure as a template (Fig. 4a and Supplementary Table 2). This peptide, W-H-K-chAla-W-hydroxyTic (WW61), contains an alanine derivative, β-cyclohexyl-L-alanine (chAla) and a tyrosine/proline derivative, 7-hydroxy-(S)-1,2,3,4- tetrahydroisoquinoline-3-carboxylic acid (hydroxyTic), both of which increase contact area with the GGVLVN template. The non-natural chAla forms hydrophobic interactions with the leucine residue in the steric zipper interface, and hydroxyTic supports the favorable placement of chAla through hydrophobic packing (Fig. 4b and Supplementary Fig. 16b). Moreover, we hypothesize that the bulky side-chains and steric constraints of hydroxyTic provide hindrance to further fibril growth.
Figure 4. A designed non-natural peptide inhibits 248PAP286 fibrillation.
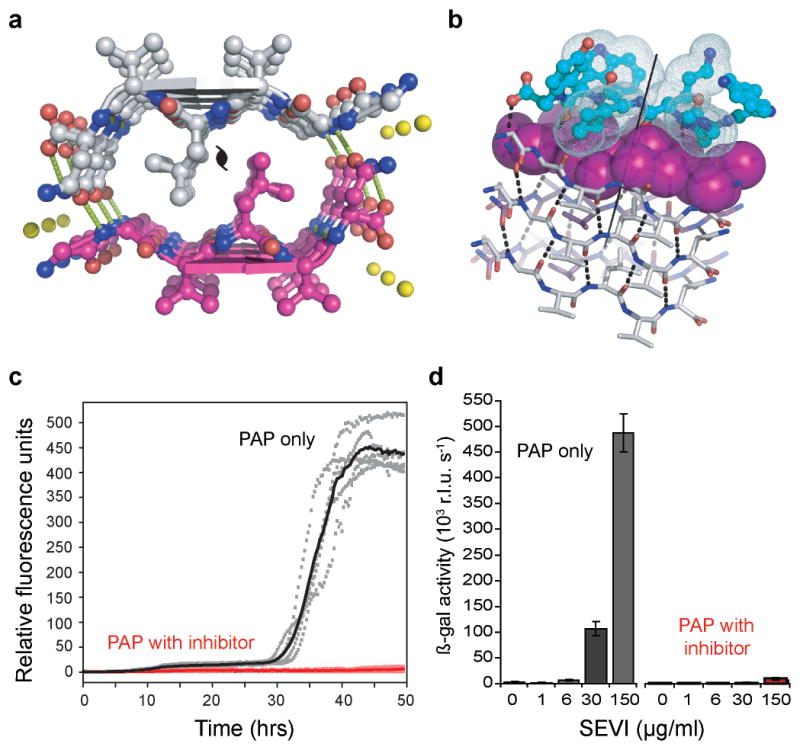
a, The view down the fiber axis of the crystal structure of the GGVLVN steric zipper reveals two mating β-sheets with parallel, in-register β-strands (hydrogen bonds depicted as green dashed lines; water molecules as yellow spheres). b, View roughly perpendicular to a fiber of 3 layers, with the atoms of the side-chains of the top layer shown as purple spheres. On top, in aqua, is a designed non-natural peptide inhibitor, W-H-K-chAla-W-hydroxyTic (see Supplementary Fig. 13). c, The inhibitor blocks 248PAP286 fibrillation, as shown by monitoring Thioflavin T fluorescence. With two-fold molar excess of the inhibitor (red), the fluorescence remains low over the course of the experiment for all five replicates, in contrast to 248PAP286 alone (black). d, HIV infection rates were determined by monitoring β-galactosidase activity. Agitated 248PAP286 (SEVI) alone efficiently increases viral infection, whereas 248PAP286 mixtures incubated with inhibitor were unable to enhance HIV infection. Peptide concentrations during virion treatment are indicated on the x-axis. Error bars show the s.d. of 3 measurements per sample.
This designed peptide, WW61, effectively delays both seeded and unseeded fibrillation of 248PAP286 in vitro (Fig. 4c, Supplementary Figs. 17,18). In the presence of twofold molar excess of this inhibitor, seeded fibrillation is efficiently blocked for more than two days (Fig. 4c). Further, we see that increasing the concentration of this inhibitor extends the fibrillation lag time (Fig. 19). These inhibition assay results were further confirmed by electron microscopy (Supplementary Fig 20). As a control for specificity, we tested the effect of GIHKQK, from the N-terminus of 248PAP286, and PYKLWN, a peptide with the same charge as WW61. Neither peptide affected fibrillation kinetics, indicating that the inhibitory activity of the designed peptide is sequence specific (Supplementary Fig. 21).
Because 248PAP286 fibers (SEVI) have been shown to enhance HIV infection22, we tested whether WW61 is able to prevent this enhancement using a functional assay. In this experiment, we treated HIV particles with 248PAP286 solutions that had been agitated for 20 hours (to allow fiber formation) in the presence or absence of WW61, and infected TZM-bl indicator cells. As previously observed, SEVI efficiently enhanced HIV infection22. However, 248PAP286 incubated with the designed inhibitor prevented HIV infection (Fig. 4d).
We performed several control experiments to verify that the lack of infectivity observed in the assay is indeed due to the inhibition of SEVI formation. First, we confirmed that in the absence of SEVI, the designed inhibitor WW61 does not affect HIV infectivity (Supplementary Fig. 22a). We also found that the control peptides GIHKQK and PYKLWN, which do not inhibit 248PAP286 fibrillation, fail to decrease HIV infectivity (Supplementary Fig 22b). Additionally, we observed that WW61 has no inhibitory effect on poly-lysine mediated HIV infectivity27, further ruling out a non-specific electrostatic interaction mechanism (Supplementary Fig. 22a). Together, these results demonstrate that a peptide capable of preventing 248PAP286 fibrillation also inhibits the generation of virus-enhancing material.
Structure-based design of inhibitors of amyloid fibrillation has been challenging in the absence of detailed information about the atomic-level interactions that form the fiber spine. To date, one of the most successful structure-based approaches to preventing fibrillation has been to stabilize the native tetrameric structure of transthyretin28. While that approach is well-suited to prevent fibrillation of proteins with known native structures, other proteins involved in amyloid-related diseases, such as tau, amyloid beta, and 248PAP286 lack fully ordered native structures29. Our structure-based approach designs inhibitors independent of native structure. Instead, the templates are atomic-level structures of short fiber-forming segments14,15. Using these fiber-like templates, and adopting computational methods successful in designing novel proteins and protein-protein interfaces25,30, we have created specific inhibitors of proteins that normally fibrillate. These results support the hypothesis that the steric zipper is a principal feature of tau-related and SEVI fibers, and suggest that with current computational methods and steric-zipper structures, we have the tools to design specific inhibitors to prevent the formation of other amyloid fibers.
Full Methods
Computational design
Computational designs were carried out using the RosettaDesign software (www.rosettacommons.org)25. This algorithm involves building side-chain rotamers of all amino acids onto a fixed protein backbone. The optimal set of rotamers is then identified as those which minimize an energy function containing a Lennard-Jones potential, an orientation-dependent hydrogen bond potential, a solvation term, amino acid-dependent reference energies, and a statistical torsional potential that depends on the backbone and side-chain dihedral angles.
D-amino acid tau inhibitors
The crystal structure of VQIVYK was used as a starting scaffold for computational design (PDB accession code 2on9)15. To take full advantage of the statistical nature of the rotamer library and some terms in the Rosetta energy function, the stereochemistry of the fiber scaffold was inverted so that design would take place using L-amino acids. An extended L-peptide was aligned with the N, C, and O backbone atoms of the D-fiber scaffold. This L-peptide was subsequently redesigned, keeping all atoms of the D-fiber fixed. The stereochemistry of the final design model was then inverted, yielding a D-peptide designed to cap an L-fiber. We inspected the finished models to confirm that inversion of the stereochemistry at the Thr and Ile Cβ atoms did not make the designs energetically unfavorable. Energetic consequences of incorporating a D-blocker peptide in the middle of an L-fiber were subsequently evaluated to ensure that fiber propagation could not continue after association of a blocker. Area buried and shape complementarity calculations were performed with areaimol and sc, respectively31,32.
L-peptide 248PAP286 inhibitors
The crystal structure of GGVLVN (PDB accession code 3PPD) was used as a template for the following design procedure: An extended L-peptide was aligned according to crystal symmetry. Small random perturbations to the L-peptide were performed to optimize the rigid body arrangement between the fiber template and peptide inhibitor. Full sequence optimization of the blocker was performed using RosettaDesign, allowing residues directly contacting the blocker to repack; other scaffold residues remained fixed. Because the design calculations employ a discrete rotamer representation of the side-chains, we next performed simultaneous quasi-Newton optimization of the blocker rigid body orientation, the side-chain torsion angles, and in some cases, the backbone torsion angles using the full-atom Rosetta energy function. This optimization was essential to the subsequent assessment of the inhibition of the design. Several iterative runs of small perturbation of blocker placement, interface design and refinement were performed to improve hydrogen bonding and packing interactions. The top ranked designs based on the total binding energy of the blocker to the fiber scaffold and the interfacial shape complementarity32 were subsequently synthesized and tested.
For each initial active L-peptide design, the non-natural L-amino acids were incorporated using a growth strategy. Non-natural amino acids, structurally similar to those of initial active designs, were selected based on their solubility, side-chain shape, and commercial availability. Side-chain conformations were approximately sampled by adopting side-chain torsion angles from those in their natural counterparts. Sequence optimization of the blocker was performed and the optimal set of rotamers identified using Monte Carlo Simulated Annealing with the full-atom energy function described above. The resulting designs were ranked based on the total binding energy of the blocker to the fiber scaffold.
Tau construct expression and purification
pNG2 expression vectors (derived from pET-3b33) containing either the K12 or K19 gene, were generously provided by E. Mandelkow34. Expression in BL21 (DE3) E. coli33 was induced with 1mM isopropyl thiogalactoside when the Abs600 was between 0.8 and 1.0 and cells were harvested after 3–4 hours. K12 and K19 were purified based on previously described methods35. Cells were pelleted for 20 minutes at 4700 × g, and resuspended in 20 mM MES pH 6.8, 1 mM EDTA, 0.2 mM MgCl2, 5 mM DTT, 1 mM PMSF, and a protease inhibitor cocktail. The cells were sonicated for 2.5 minutes and, following addition of NaCl to bring cell lysate to 0.5 M NaCl, the lysate was boiled for 20 minutes. The lysate was sedimented at 30,000 × g for 20 min and dialyzed twice against 20 mM MES pH 6.8, 50 mM NaCl, 1 mM EDTA, 1 mM MgCl2, 2 mM DTT, and 0.1 mM PMSF at 4°C. The dialysate was pelleted for 20 minutes at 30,000 × g and filtered before cation exchange chromatography on an AKTA Explorer (GE – Pharmacia) with a HighTrap HP SP 5mL column (GE Healthcare). The sample was eluted with a linear gradient of up to 60% Buffer B (20 mM MES pH 6.8, 1 M NaCl, 1 mM EDTA, 1 mM MgCl2, 2 mM DTT, and 0.1 mM PMSF). Size exclusion chromatography was optionally performed with a Superdex™ 75 10/300 GL column (GE Healthcare) in PBS buffer (137 mM NaCl, 3 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) with 1 mM DTT on the AKTA Explorer depending on preparation purity as assessed by SDS-PAGE.
Tau construct inhibition assays
150 μL reactions containing 50 μM tau K12 or K19, as determined by the micro BCA Protein Assay Kit (Pierce), were incubated in 250 mM sodium phosphate buffer pH 7.4 with 1 mM DTT, 12.5 μM of heparin (6000 average molecular weight) from Sigma, and 10 μM Thioflavine S (ThS), from MP Bio, Inc.36–38. Inhibitor peptides (CS Bio, Inc., Celtek Biosciences, LLC) were dissolved in 250 mM phosphate buffer pH 7.4 to 0.5 mM and added at specified molar ratios. Reactions were split into a minimum of 3 replicates in black 96-well optically-clear plates (Nunc), sealed with Corning pressure seal tape and monitored using a Varioskan plate reader (Thermo Scientific, Inc) for K12 and a SpectraMax M5 for K19. The fluorescence signal was measured every 15 minutes with excitation and emission wavelengths of 440 and 510 nm, respectively, at 37°C with continuous shaking at 900 rpm with a diameter of 1 mm for K12, and quiescent incubation, with shaking 2 seconds prior to each reading for K19. Plots showing the fluorescence trace of the replicate with median lag time for each sample were created using R39. Lag time plots depict the mean time value at which each replicate crossed an arbitrary fluorescence value above the noise (values were selected per experiment and applied to all samples). Error bars represent the standard deviation of the replicate lag times for each sample.
Seeded K12 fiber formation assays
Seeds were produced by incubating 50 μM K12 as above, but without ThS present, and added at 0.25% (v/v). Peptide stock concentrations were 0.75 mM and added at a final concentration of 10-fold molar excess relative to soluble K12. Reaction mixtures were otherwise prepared and monitored as above.
VQIVYK inhibition assays
The VQIVYK fibrillation assay was modified from a previously published protocol40. Buffers and plates were kept on ice to delay VQIVYK fibrillation while the reaction mixtures were prepared. Replicate solutions of 180 μL of 25 mM MOPS pH 7.2, 100 μM ThS, and inhibitor peptides were added to black, clear-bottom, 96-well Nunc plates with 1/8″ PTFE beads (Orange Products, Inc.). Acetylated and amidated VQIVYK (Genscript, Inc.) was dissolved in H2O to 1.3 mM and filtered through a Millipore microcon 100-kDa filter device at 14,000 rpm for 5 minutes at 4°C to remove large aggregates (final concentration ~1mM). 20 μL of filtered VQIVYK were added to each reaction well. ThS fluorescence was monitored at room temperature every 2 minutes using a SpectraMax M5 fluorimeter with 2 seconds of mixing before each reading.
Amyloid beta fibrillation assay
Lyophilized amyloid beta(1-42) was diluted to 0.2 mg/ml in 50 mM NH4OH and filtered with a 0.2 μm filter. The reaction mixture contained a final concentration of 11.5 μM Aβ1-42, 10 μM Thioflavin T (ThT), 23 mM NH4OH in 100 mM Bicine pH 9.1 and 11.5 μM D-TLKIVW in reactions with peptide present. Reactions were split into 4 replicates and the ThT fluorescence signal was measured every minute (Ex. 444/Em. 482) at 37°C with continuous shaking at 960 rpm with a 1 mm diameter in a Varioskan fluorimeter.
Electron microscopy
5 μL of sample were applied to glow-discharged, 400-mesh carbon-coated, formvar films on copper grids (Ted Pella, Inc.) for 3 minutes. Grids were rinsed twice with distilled water and stained with 1% uranyl acetate for 90 seconds. Grids were examined in a Hitachi H-7000 transmission electron microscope at 75 kEv or a JEOL JEM1200-EX operating at 80 kEv.
Tau fibrillation kinetic analysis
The nucleation (k1) and propagation (k2) rates were determined by fitting the form of the Finke-Watzky 2-step mechanism41. Plateau values were determined and the remaining parameters were fit using the leasqr non-linear least-squares regression function (http://fly.isti.cnr.it/pub/software/octave/leasqr/) through the octave software package (http://www.gnu.org/software/octave/).
Preparation of peptide-gold conjugates
Peptide-nanogold conjugates were prepared as described earlier for similarly sized peptides42. Briefly, 60 nmol of the peptides CGGG-(D)-TLKIVW and CGGG-(D)-LKTWIV (CS Bio) were dissolved in 110 μl of phosphate-buffered saline (20 mM, pH 6.5, 0.15 M NaCl), added to 6 nmol of monomaleimido nanogold (Nanoprobes, Inc.), dissolved in 200 μl H2O, and incubated for 1 hour at room temperature with constant rotation. Peptide-nanogold conjugates were separated from excess unbound peptides by membrane centrifugation (Microcon-10 system; Amicon) using a molecular weight cut-off of 10 kDa. Peptide-nanogold conjugates were then diluted into phosphate-buffered saline, aliquoted, and stored at -20°C for no longer than 1 month.
Preparation of K19 fibers
K19 fibers were generated by incubating 100 μM of soluble K19 with 25 μM of 6-kDa heparin overnight at 37°C in phosphate buffer (50 mM, pH 7.4). K19 fibers were sonicated for 15 s, using a microtip set to 35% amplitude. Residual heparin and small oligomers were removed by centrifuging the mixture through a 100-kDa Microcon concentrator for 10 minutes at 14,000 × g, washing the retentate with phosphate buffer, and repeating three times; the retentate was restored to its original volume with phosphate buffer. These short fiber segments were stored at 4°C for no longer than 1 week. For NMR studies, fiber samples were similarly prepared, but washed in H2O and concentrated to 2mM K19 (by monomer).
Preparation of samples for nanogold binding experiments
10 nM of the nanogold conjugate (or control) was incubated with 1.67 μM (by monomer) of K19 fibers in MOPS buffer (25 mM, pH 7.2) for 1 hour. 5 μL was applied to a glow-discharged 400 mesh carbon-stabilized copper grid (Ted Pella) for 3 minutes. The grids were washed twice with H2O and 10 μl of the Goldenhance reagent was applied for 10 seconds. The grids were washed 5 times with H2O and negatively stained with 2% uranyl acetate.
Quantification and localization of nanogold binding
For each sample, 75 nanogold particles ≤ 15 nm in diameter were counted and classified as bound or unbound. The 15 nm cutoff was to exclude unbound, but adjacent, particles enlarged by Goldenhance that only apparently bind fibers. To establish the localization of the binding observed, individual nanogold particles bound to fibers were categorized as bound to the fiber end or side. In both of these experiments, sample identities were concealed from the microscopist to ensure unbiased counting. Grids were examined with a JEOL JEM1200-EX and images were recorded using Digital Micrograph (Gatan, Inc.).
Statistical analysis of nanogold binding
We compared counts of nanogold-conjugated peptides and unconjugated nanogold bound to fibers or localizing to fiber ends. 21 unconjugated nanogold particles out of 75 counted bound to fibers. We modeled nanogold particles bound to fibers using a binomial distribution with parameters n = 75 (sample size) observations and p = 0.28 (probability of success). In a separate experiment, 22 unconjugated nanogold particles bound to fibers that localized to fiber ends, following a binomial distribution with n = 105 and p = 0.21.
Because the number of counts is fairly large, we assumed a normal distribution, and used a standard z-test to compare the number of bound nanogold-peptide conjugates to the expected distribution based on the number of bound unconjugated nanogold particles. We used analogous analysis to determine the significant of localization to fiber ends.
The number of nanogold-D-TLKIVW conjugates bound to fibers (xbound = 43, n = 75) and bound nanogold-D-TLKIVW localizing to the end of fibers (xend = 49, n = 86) was significantly different from nanogold alone, whereas the number of nanogold-D-LKTWIV conjugates bound (xbound = 15, n = 75) or the number localized to fiber ends (xend = 17, n = 100) does not differ significantly from nanogold alone.
VQIVYK preparation for binding studies
Acetylated and amidated VQIVYK peptide (Genscript, Inc) was dissolved to 1 mM in 25 mM MOPS pH 7.2 and incubated at room temperature for at least 24 hours. Fibers were washed with H2O, concentrated using a 3-kDa molecular weight cutoff Amicon ultracentrifugal filter, and resuspended in H2O to a final concentration (by monomer) of 4 mM. Soluble VQIVYK was prepared by dissolving VQIVYK peptide (CS Bio, Inc.) with free N- and C- termini in H2O.
1H nuclear magnetic resonance sample preparation and measurements
NMR samples were prepared with 5% D2O and 10mM NaOAc pH 5.0. D-Peptides were added from 1 mM stocks in H2O to a final concentration of 100 μM. Soluble and fibrillar VQIVYK and tau were added at indicated concentrations to make a final volume of 550 μL. 500 MHz 1H NMR spectra were collected on a Bruker DRX500 at 283 K. H2O resonance was suppressed through presaturation. Spectra were processed with XWINNMR 3.6.
Binding constant estimations
NMR data were analyzed to estimate a binding constant for the interaction between D-TLKIVW and VQIVYK fibers. At about 1000 μM VQIVYK (concentration as monomer), 50% of D-TLKIVW is bound (Supplementary Fig. 11). The steric zipper model suggests that there are 2 monomers per 4.7 Å (0.47 nm) layer in a fiber14:
and we estimate the fiber concentration using the monomer concentration:
If we assume one binding site and estimate from electron microscopy an average length of ~140 nm per fiber, then there are about 600 monomers per fiber, and the apparent Kd is about 2 μM.
Hydrogen bonding energy calculation
We used areaimol31 to calculate the apolar and polar area buried by the interaction D-TLKIVW with the VQIVYK steric zipper (Supplementary Fig. 1). We calculate 201, 24, and 102 Å2 buried for carbon, nitrogen, and oxygen atoms, respectively. Using the atomic solvation parameters of Eisenberg and coworkers43, we estimate that the ΔGsolvation is approximately 2.5 kcal/mol. Based on an apparent Kd of 2 μM, we estimate ΔGbinding to be 7.4 kcal/mol. From the interaction model (Fig. 2c and Supplementary Fig. 1), we maintain 6 hydrogen bonds between D-TLKIVW and VQIVYK, and if
GGVLVN Crystallization and Structure determination
The GGVLVN peptide was dissolved in 10mM Tris, pH 9 at 1.8 mg/ml and crystallized in 10% (w/v) PEG-8000, 0.1 M MES pH 6.0, 0.2 M Zn(OAc)2. X-ray diffraction data was collected at APS beamline 24-IDE. Phases were determined by molecular replacement using an idealized β-strand in Phaser44. Crystallographic refinement was performed using Refmac45. Model building was performed with Coot46 and illustrated with PyMOL47.
248PAP286 fibrillation and inhibition
Fmoc-β-cyclohexyl-L-alanine and Fmoc-7-hydroxy-(S)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid were purchased from AnaSpec, Inc and the inhibitor peptide H-[W-H-K-chAla-W-hydroxyTic]-OH (WW61) was synthesized by Celtek Biosciences LLC. 248PAP286 and WW61 were dissolved as 1.25X and 5X stocks in PBS, respectively, and filtered with a 0.1 μm filter. 248PAP286 was diluted with PBS to 0.66 mM and ThT was added to 10μM final. Samples were optionally mixed with 1.32 mM WW61 and vortexed. 5 replicates of 150 μL were immediately dispensed into a 96-well plate. In dose-response experiments, WW61 final concentrations were 0.33 mM, 0.66 mM, and 1.32 mM. Plates were continuously agitated at 960 rpm at 37°C, and ThT fluoresence readings were recorded (Ex. 440/Em. 482) in 15-minute intervals with a Varioskan Flash fluorimeter. Lag time was determined when fluorescence crossed an arbitrary value (3 RFU) above background.
Effect of fibrillation inhibitors on 248PAP286 fiber-mediated enhancement of HIV infection
The CCR5 tropic molecular HIV-1 clone NL4_3/92TH014-248 was generated by transient transfection of 293T cells with proviral DNA. Supernatants were collected 48 hours later and p24 concentrations determined by ELISA. TZM-bl reporter cells encoding a lacZ gene under the control of the viral LTR promoter were obtained through the NIH AIDS Research and Reference Reagent Program and provided by Dr. John C. Kappes, Dr. Xiaoyun Wu and Tranzyme Inc49. 40 μl HIV-1 containing 0.1 ng p24 antigen was incubated with 40 μl dilutions of mixtures of 248PAP286 and inhibitory peptide that was either freshly prepared or had been agitated for 23 hours. Peptide concentrations and experimental conditions during agitation were similar to those described above. Thereafter, 20 μl of the mixtures were used to infect 180 μl TZM-bl cells seeded the day before (1×105/well). 2 days later infection rates were determined by quantifying β-galactosidase activities in cellular lysates using the Gal-Screen assay (Applied Biosystem, T1027). Luminescence was recorded on an Orion microplate luminometer as relative light units per second (RLU/s).
Effect of WW61 on poly-lysine mediated enhancement of HIV-1 infection
50 μl of poly-lysine (Sigma Aldrich) were mixed with equal volumes of WW61. Thereafter, 35 μl of 5-fold dilutions of poly-lysine/WW61 mixtures or poly-lysine alone were incubated with the same volume of virus and incubated for 5 minutes at room temperature. Poly-lysine/WW61 concentrations were 100, 20, 4, 0.8, 0.16, 0.032, 0.064 and 0 μg/ml during preincubation with virus stocks. Thereafter, 20 μl of each mixture were added to 180 μl TZM-bl cells. The infection rate was determined 2 days later as described above.
Effect of WW61, GIHKQK and PYKLWN on HIV-1 infection
40 μl of each peptide was incubated with an equal volume of virus containing 1 ng of p24 antigens for 5 minutes at room temperature. Peptide concentrations were 150, 30, 6, 1.2 and 0 μg/ml during preincubation with virus stocks. Thereafter, 20 μl of mixtures were added to 180 μl TZM-bl cells (10-fold dilution) and the infection rate was determined as above.
Supplementary Material
Acknowledgments
We thank M.I. Ivanova, J. Corn, T. Kortemme, D. Anderson, M.R. Sawaya, M. Phillips, S. Sambashivan, J. Park, M. Landau, Q. Zhang, R. Clubb, F. Guo, T. Yeates, J. Nowick, J. Zheng, and M.J. Thompson for discussions, HHMI, NIH, NSF, the GATES foundation, and the Joint Center for Translational Medicine for support, R. Peterson for help with NMR experiments, E. Mandelkow for providing tau constructs, R. Riek for providing amyloid beta, J. Stroud for amyloid beta preparation. Support for JK was from the Damon Runyon Cancer Research Foundation, for HWC by the Ruth L. Kirschstein National Research Service Award, for JM from the programme for junior-professors by the ministry of science, Baden-Württemberg, and for SAS by a UCLA-IGERT bioinformatics traineeship.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions S.A.S., J.K., D.B., J.M. and D.E. designed the project. J.K. and S.A.S. created design protocol. J.K. designed D-peptides. L.J. expanded design methodology and designed non-natural amino acid peptides. S.A.S, H.W.C., and A.Z. performed fluorescence experiments and electron microscopy. A.Z. determined the structure of GGVLVN. O.Z. performed HIV infectivity experiments. J.T.S. performed kinetic data analysis. S.A.S performed NMR experiments. S.A.S, J.K., and D.E. wrote the manuscript coordinating contributions by other authors.
Atomic coordinates and structure factors for the reported GGVLVN structure have been deposited in the Protein Data Bank with accession code 3PPD.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Westermark P, et al. A primer of amyloid nomenclature. Amyloid. 2007;14:179–183. doi: 10.1080/13506120701460923. [DOI] [PubMed] [Google Scholar]
- 2.Maji SK, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science (New York, NY) 2009;325:328. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid--from bacteria to humans. Trends Biochem Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Astbury WT, Dickinson S. The X-ray interpretation of denaturation and the structure of the seed globulins. The Biochemical Journal. 1935;29:2351–2360. doi: 10.1042/bj0292351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calamai M, Chiti F, Dobson CM. Amyloid fibril formation can proceed from different conformations of a partially unfolded protein. Biophysical Journal. 2005;89:4201. doi: 10.1529/biophysj.105.068726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tjernberg LO, et al. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J Biol Chem. 1996;271:8545–8548. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- 7.Findeis MA. Peptide inhibitors of beta amyloid aggregation. Curr Top Med Chem. 2002;2:417–423. doi: 10.2174/1568026024607508. [DOI] [PubMed] [Google Scholar]
- 8.Sciarretta KL, Gordon DJ, Meredith SC. Peptide-based inhibitors of amyloid assembly. Methods Enzymol. 2006;413:273–312. doi: 10.1016/S0076-6879(06)13015-3. [DOI] [PubMed] [Google Scholar]
- 9.Soto C, Kindy MS, Baumann M, Frangione B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent beta-sheet conformation. Biochem Biophys Res Commun. 1996;226:672–680. doi: 10.1006/bbrc.1996.1413. [DOI] [PubMed] [Google Scholar]
- 10.Kokkoni N, Stott K, Amijee H, Mason JM, Doig AJ. N-Methylated Peptide Inhibitors of Amyloid Aggregation and Toxicity. Optimization of the Inhibitor Structure. Biochemistry. 2006;45:9906–9918. doi: 10.1021/bi060837s. [DOI] [PubMed] [Google Scholar]
- 11.Sato T, et al. Inhibitors of amyloid toxicity based on beta-sheet packing of Abeta40 and Abeta42. Biochemistry. 2006;45:5503–5516. doi: 10.1021/bi052485f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larbig G, Pickhardt M, Lloyd DG, Schmidt B, Mandelkow E. Screening for inhibitors of tau protein aggregation into Alzheimer paired helical filaments: a ligand based approach results in successful scaffold hopping. Curr Alzheimer Res. 2007;4:315–323. doi: 10.2174/156720507781077250. [DOI] [PubMed] [Google Scholar]
- 13.Wiesehan K, et al. Selection of D-amino-acid peptides that bind to Alzheimer’s disease amyloid peptide abeta1–42 by mirror image phage display. Chembiochem. 2003;4:748–753. doi: 10.1002/cbic.200300631. [DOI] [PubMed] [Google Scholar]
- 14.Nelson R, et al. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawaya MR, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 16.Wiltzius JJ, et al. Molecular mechanisms for protein-encoded inheritance. Nat Struct Mol Biol. 2009;16:973–978. doi: 10.1038/nsmb.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 18.Goux WJ, et al. The formation of straight and twisted filaments from short tau peptides. J Biol Chem. 2004;279:26868–26875. doi: 10.1074/jbc.M402379200. [DOI] [PubMed] [Google Scholar]
- 19.von Bergen M, et al. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci U S A. 2000;97:5129–5134. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci U S A. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson MJ, et al. The 3D profile method for identifying fibril-forming segments of proteins. Proc Natl Acad Sci U S A. 2006;103:4074–4078. doi: 10.1073/pnas.0511295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Münch J, et al. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell. 2007;131:1059. doi: 10.1016/j.cell.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 23.Friedhoff P, von Bergen M, Mandelkow EM, Davies P, Mandelkow E. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci U S A. 1998;95:15712–15717. doi: 10.1073/pnas.95.26.15712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wille H, Drewes G, Biernat J, Mandelkow EM, Mandelkow E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J Cell Biol. 1992;118:573–584. doi: 10.1083/jcb.118.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z, Krause G, Reif B. Structure and orientation of peptide inhibitors bound to beta-amyloid fibrils. J Mol Biol. 2005;354:760–776. doi: 10.1016/j.jmb.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 27.Roan NR, et al. The cationic properties of SEVI underlie its ability to enhance human immunodeficiency virus infection. J Virol. 2009;83:73–80. doi: 10.1128/JVI.01366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petrassi HM, Klabunde T, Sacchettini J, Kelly JW. Structure-Based Design of N-Phenyl Phenoxazine Transthyretin Amyloid Fibril Inhibitors. J Am Chem Soc. 2000;122:2178–2192. [Google Scholar]
- 29.Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- 30.Kortemme T, et al. Computational redesign of protein-protein interaction specificity. Nat Struct Mol Biol. 2004;11:371–379. doi: 10.1038/nsmb749. [DOI] [PubMed] [Google Scholar]
- 31.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 33.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 34.Biernat J, et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. Embo J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barghorn S, Biernat J, Mandelkow E. Purification of recombinant tau protein and preparation of Alzheimer-paired helical filaments in vitro. Methods Mol Biol. 2005;299:35–51. doi: 10.1385/1-59259-874-9:035. [DOI] [PubMed] [Google Scholar]
- 36.Friedhoff P, Schneider A, Mandelkow EM, Mandelkow E. Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein tau monitored by fluorescence in solution. Biochemistry. 1998;37:10223–10230. doi: 10.1021/bi980537d. [DOI] [PubMed] [Google Scholar]
- 37.Perez M, Valpuesta JM, Medina M, Montejo de Garcini E, Avila J. Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. J Neurochem. 1996;67:1183–1190. doi: 10.1046/j.1471-4159.1996.67031183.x. [DOI] [PubMed] [Google Scholar]
- 38.Schweers O, Mandelkow EM, Biernat J, Mandelkow E. Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc Natl Acad Sci U S A. 1995;92:8463–8467. doi: 10.1073/pnas.92.18.8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Team, R. D. C. R Foundation for Statistical Computing. 2008. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 40.Rojas Quijano FA, Morrow D, Wise BM, Brancia FL, Goux WJ. Prediction of nucleating sequences from amyloidogenic propensities of tau-related peptides. Biochemistry. 2006;45:4638–4652. doi: 10.1021/bi052226q. [DOI] [PubMed] [Google Scholar]
- 41.Morris AM, Watzky MA, Agar JN, Finke RG. Fitting neurological protein aggregation kinetic data via a 2-step, minimal/“Ockham’s razor” model: the Finke-Watzky mechanism of nucleation followed by autocatalytic surface growth. Biochemistry. 2008;47:2413–2427. doi: 10.1021/bi701899y. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt K, Segond von Banchet G, Heppelmann B. Labelling of peptides with 1.4-nm gold particles to demonstrate their binding sites in the rat spinal cord. J Neurosci Methods. 1999;87:195–200. doi: 10.1016/s0165-0270(99)00003-5. [DOI] [PubMed] [Google Scholar]
- 43.Eisenberg D, Wesson M, Yamashita M. Interpretation of Protein Folding and Binding with Atomic Solvation Parameters. Chemica Scripta. 1989;29A:217–221. [Google Scholar]
- 44.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 46.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.DeLano WL. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 48.Papkalla A, Munch J, Otto C, Kirchhoff F. Nef enhances human immunodeficiency virus type 1 infectivity and replication independently of viral coreceptor tropism. J Virol. 2002;76:8455–8459. doi: 10.1128/JVI.76.16.8455-8459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.