

Abstract
More than 2,500 species of copepods (Class Maxillopoda; Subclass Copepoda) occur in the marine planktonic environment. The exceptional morphological conservation of the group, with numerous sibling species groups, makes the identification of species challenging, even for expert taxonomists. Molecular approaches to species identification have allowed rapid detection, discrimination, and identification of species based on DNA sequencing of single specimens and environmental samples. Despite the recent development of diverse genetic and genomic markers, the barcode region of the mitochondrial cytochrome c oxidase subunit I (COI) gene remains a useful and – in some cases – unequaled diagnostic character for species-level identification of copepods. This study reports 800 new barcode sequences for 63 copepod species not included in any previous study and examines the reliability and resolution of diverse statistical approaches to species identification based upon a dataset of 1,381 barcode sequences for 195 copepod species. We explore the impact of missing data (i.e., species not represented in the barcode database) on the accuracy and reliability of species identifications. Among the tested approaches, the best close match analysis resulted in accurate identification of all individuals to species, with no errors (false positives), and out-performed automated tree-based or BLAST based analyses. This comparative analysis yields new understanding of the strengths and weaknesses of DNA barcoding and confirms the value of DNA barcodes for species identification of copepods, including both individual specimens and bulk samples. Continued integrative morphological-molecular taxonomic analysis is needed to produce a taxonomically-comprehensive database of barcode sequences for all species of marine copepods.
Introduction
Marine copepods represent a predominant component of the zooplankton throughout the world oceans in both abundance and biomass 1 , 2. There are more than 2,500 described species of planktonic marine copepods, with species distributions ranging from shallow, brackish, estuarine waters to deep ocean (abysso- and hadopelagic) zones 3. Copepods exhibit a wide variety of biogeographical patterns, from very limited distributions to cosmopolitan and global-ocean ones.
Their high species diversity, together with their relatively small size and apparent similarity among different forms, has made the morphological identification and quantification of copepod species a challenging task 4. In addition, it is likely that there are large numbers of cryptic species within what are now considered recognized species, especially for geographically-widespread taxa 5 , 6 , 7.
Considerable effort has been focused on the development and use of genetic approaches to identifying and discriminating marine species in the past ~20 years (reviewed by Bucklin et al. 8). Use of a fragment of the cytochrome c oxidase subunit I (COI) gene for discrimination and identification of animal species, i.e., DNA barcoding 9 , 10, has moved rapidly from novelty to widespread use, although it has not been free of controversy. Objections have focused on uses of barcodes beyond the original intent as a species assignment tool, including DNA taxonomy 11 , 12, ecological assessment 13, and species discovery 14. Recent improvements in methods for statistical analysis of barcode data 13 , 15 , 16 , 17 and growing focus on the appropriate use and limitations of barcode analysis 18 are advancing the field of DNA barcoding.
Recent DNA barcoding studies of marine planktonic copepods have focused on examination of species-level diversity in particular regions of the ocean 19 , 20 , 21 , 22, and also on particular - usually problematical - taxa 23 , 24 , 25 , 26 , 27 , 28 , 29. Other studies have used DNA barcodes for biogeographical or phylogeographical analyses 30 , 31 , 32 , 33 , 34. A number of studies have revealed cryptic species 5 , 33 , 35 , 36 , 37.
This study provides 800 new barcode sequences for 63 copepod species not included in previous studies. These new barcoding records increase both the depth of sampling and also the geographical coverage of existing records, and continue progress toward a taxonomically-comprehensive database or library of DNA barcode sequences for all species of the groups or lineages of interest. Importantly, this study examines a variety of statistical and analytical approaches used for barcode data, and provides new information about the strengths, weaknesses and limitations of DNA barcodes for discrimination and identification of copepod species. A particular focus of this study is the impact of any missing data (i.e., species not represented in the barcode database) on the accuracy and reliability of species identifications. Finally, we offer new guidance and a conceptual framework for continued barcoding efforts to meet challenges of species identification of copepods, one of the most ecologically important and systematically complex groups of marine zooplankton.
Methods
Samples analyzed
Sequences of the COI barcoding region 38 were determined for identified individual specimens collected from various sources from 1992 to 2011, and archived at the University of New Hampshire (1992-2005) or the University of Connecticut (2005-2011). All specimen and collection metadata are included in the GenBank entries. Appropriate reference is made to previously published sequence data. Laboratory protocols (DNA purification, PCR amplification, and sequencing) are as described in previous publications by the authors 20 , 35.
DNA sequence data analysis
Sequences were analyzed using the Molecular Evolutionary Genetic Analysis (MEGA) Ver. 5 39. Sequences were aligned using ClustalW 40, as implemented in MEGA, using the corresponding amino acid translated version. This procedure allows better resolution by removing gap ambiguities, ensures designation of the correct codon reading frame, and minimizes risks of including nuclear pseudogenes with mitochondrial origins, known as numts 41 , 42. Initial tree runs were used to check for very divergent sequences (i.e., potential numts), which were removed prior to analysis. A total of eight individual sequences clustered in a single, highly supported, independent clade, that comprised a mixture of species from different orders (two) and families (six) of the Subclass Copepoda (five Calanoida and three Harpacticoida). When compared with the final number (1381 COI sequences) this value can be considered low, although it is necessary to recognize that these eight sequences were those that had passed all initial filters (for example, they did not code for a stop codon, or were extremely aberrant when translated into their correspondent amino-acid sequence).
Descriptive statistics
Three different alignments were prepared for analysis in order to study the influence and possible bias due to variation in sequence length and heterogeneity in levels of sequence divergence along the barcode region. The analyzed alignments will be referred to as follows: 1) original alignment, including all 1,381 sequences of any length; 2) standard barcode alignment, including only sequences of >500 bp (see Barcode of Life, http://www.barcodeoflife.org/); and 3) unique barcode alignment, considering only a 400 bp portion (positions 96 to 497) of the barcode region and including only a single copy of all the different haplotypes for each species (576 sequences in all). The unique alignment was subjected to a sliding window analysis of nucleotide diversity (π) using DnaSP Ver. 5 43. Two runs were performed with 10 bp step size and window lengths of 10 bp and 100 bp; results were compared to visualize differences in π along the analyzed region.
Genetic distances within species, genera, families, and orders and between orders were calculated in MEGA using the Kimura 2-parameter (K2P) model 44 for each of the three alignments previously described. Mann-Whitney U tests were carried out based upon the unique alignment distances matrix to compare distances within versus between species and between taxonomic levels. Although K2P was the second-best fit for the dataset (the best corresponded to GTR+I+ Γ), this model was used to allow direct comparison with previously published barcoding studies, which most frequently used this metric 8, despite growing criticism of this metric for barcode analysis 45 , 46. On the other hand, the same studies have shown that the choice of evolutionary model does not affect success rates of species identifications 45 , 46; uncorrected p-distances perform equally to any model; but see Fregin et al. 47.
Barcoding resolution
Initially, two automated statistical techniques for barcoding approaches to species identity assignment were evaluated: 1) automated identification of significant clades after tree reconstruction; and 2) genetic distance-based assignment by the Basic Local Alignment Search Tool (BLAST) method 48. Parallel analyses were carried out on the three alignments. In addition, a non-automated technique for species assignment was considered: the “best close match” 49 combines best match criteria and maximum within-species distance thresholds. Similar to the BLAST approach, this technique analyzes each query individually and identifies the closest sequence within a flexible threshold adapted to each dataset. Although computationally intensive and potentially time-consuming, this approach has been shown to out-perform automated and much more complicated methods 16, especially when the sequences are highly variable and many species are represented by one or a few sequences.
Neighbor-Joining (NJ) trees 50 were reconstructed in MEGA using the K2P evolutionary model for the standard and unique alignments. Maximum Likelihood (ML) phylogenetic tree analyses were done using RAxML Ver. 7.2.8 51 under the GTR+I+Γ model for the three datasets. This model-based method (ML) allows inclusion of non-overlapping sequences in the same analysis, which is not possible with distance-based methods, such as NJ. In addition, there is a growing concern about the validity and adequacy of both NJ and K2P for barcode analysis, especially when compared with methods like ML under the best fit evolutionary model 45 , 46 , 47. The NJ and ML trees were compared for the standard and unique alignments to evaluate consistency of the results. Confidence level was estimated for both methods as percentage recovery after 10,000 bootstraps. Putative species were inferred using the Poisson tree processes model (PTP) on the ML trees 52. These putative species are equivalent to molecular operational taxonomic units (MOTU 53) and were compared with the morphologically-identified species (OTUs).
For the BLAST approach, jMOTU 15 was used on the original, standard and unique alignments. The minimum alignment length (i.e., overlap between sequence pairs) for analysis of the original dataset was set at 100 bp. The standard alignment showed minimum overlap of ~350 bp between pairs of sequences; 400 bp was common to all sequences in the unique alignment. The BLAST filter was 85 for all analyses. The tree results, resolved MOTUs, and identified OTUs were compared for the three alignments.
Frequency distribution of the 1,381 sequences from the original dataset by length (in base pairs).
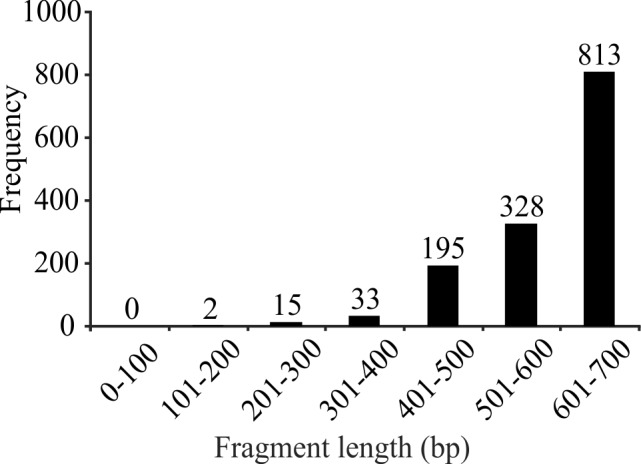
A total of 1,141 sequences (82%) fulfilled the minimum of 500 bp length definition of a gold standard barcode.
Species-by-species analyses
Taxa showing discrepancies between MOTUs and OTUs were selected for additional analysis, when possible based on available data, to examine possible reasons (e.g., variation among geographic areas or populations, cryptic speciation) for the observed disparities between morphological and molecular data. Analyses included parsimony haplotype networks (gene genealogies) using TCS Ver. 1.2.1 54 and calculation of F ST distances between samples or regions using Arlequin Ver. 3.5 55.
Results
This study reports a total of 800 new DNA barcode sequences for identified specimens of 63 species not included in previous studies. These new data were analyzed with 581 previously published sequences, yielding a total dataset of 1,381 sequences with an average length of 578.9 ± 84.3 bp (range: 105 – 658 bp); 82% of the sequences were > 500 bp (Fig. 1). The sequences originated from 195 different taxa or OTUs, including 71 genera, 37 families and 4 orders. Of the 1,381 total sequences, 1,354 belonged to the Order Calanoida (see Supplementary data S1.Alignment.fas at http://dx.doi.org/10.6084/m9.figshare.987095).
Descriptive statistics
The sliding window analysis of the unique alignment using the 100 bp window length showed that nucleotide diversity (π) was lower toward the 5’ end of the barcode region, but was relatively constant and higher in the half of the region toward the 3’ end (Fig. 2). For the analysis using the 10 bp window length, the results were markedly irregular, reflecting variation among different domains along the COI barcode region, with moderately conserved regions separated by highly variable ones.
Sliding window analyses of nucleotide diversity (π) along the “unique” alignment (see Methods).

The horizontal line indicates the average π for the fragment, 0.206. Analyses with window lengths of 10 bp and 100 bp were run with a 10 bp step size; both analyses showed lower variability on the 5’ end of the amplified fragment.
Based on the unique alignment, K2P distances between species were larger than those within species (p < 0.001), but some overlap was observed and no clear barcode gap 56 was identified. Some species showed high divergences between conspecific individuals, while in other cases there were no differences between individuals of different species (see Supplementary data S2.zip at http://dx.doi.org/10.6084/m9.figshare.987095). The range of variation of distances was reduced for the standard and original alignments, but there was some overlap of within- and between-species distances, which was more pronounced when comparing higher taxonomic levels (genus and above).
Analysis of the unique alignment revealed low densities of K2P distances between individuals from 0.05 to 0.15, and very low densities between 0.08 and 0.09 (Fig. 3). Overlap of within- and between-species distances was still observed (Fig. 3). Distances within and between higher-level groups also showed overlap, although these were significantly different when analyzed using multiple U tests (p < 0.001 in all cases).
Tree-based analysis of barcodes
The Maximum Likelihood trees based on the unique and standard alignments showed similar results to those of the NJ trees in terms of resolution and discrimination of clades, albeit with some differences in bootstrap values (see Supplementary data S3.zip at http://dx.doi.org/10.6084/m9.figshare.987095). Overall, the ML tree showed better grouping of closely-related taxa and higher recovery of deeper nodes than the NJ analysis.
Frequency distribution (in percentages) of Kimura-2-Parameter (K2P) distances by taxonomic level: species, genus, family, order and subclass.

Overlap of within- and between-species distances was still observed. Distances within and between higher-level groups also showed overlap.
Automated tree-based analyses of the unique alignment resolved 227 MOTUs for the ML tree and 241 for the NJ tree. Examining the tree by eye, these MOTUs could be reduced to 65 distinct species-specific clusters, each with more than one sequence separated by short internal branches. Bootstrap recovery was > 98% (100% in most cases; see Supplementary data S3.zip). A number of taxa showed fragmentation (i.e., separation of clusters within the species grouping), indicating geographic differentiation or cryptic speciation; these clusters were identified as different putative species by the PTP analysis (Supplementary data S3.zip). In contrast, there were highly supported clades comprising sequences from different species, including species of Calanus (C. helgolandicus Claus 1863 and C. euxinus Huselmann 1991; C. agulhensis De Decker, Kaczmaruk & Marska 1991 and C. sinicus Brodsky 1965); Centropages (C. typicus Kröyer 1849 and C. chierchiae Giesbrecht 1889); Acartia (A. tonsa Dana 1849 and A. hudsonica Pinhey 1926); Pleuromamma (P. gracilis Claus 1863 and P. piseki Farran 1929); as well as a Paracalanus Boeck 1964 species clade.
The standard alignment identified most species with 99 - 100% bootstrap confidence; the PTP automated method identified 222 putative species from the ML tree and 277 for the NJ tree. In general, the automated tree approach failed to group conspecific individuals when the depth of sampling in that taxon was low, even though the species were grouped in a single clade with high bootstrap support in other tree-based analyses. Inclusion of additional sequences allowed better resolution of MOTUs, especially for clades showing variable or complex results (e.g., Acartia tonsa / hudsonica, Pleuromamma gracilis / piseki, and Paracalanus species).
Number of MOTU inferred from the three alignments at a range of cut-offs (x-axis) expressed as percentage (relative to the mean length of each dataset) of differences between sequences.

The lower panel shows in detail the box from the upper panel.
ML analysis of the original alignment yielded results that were fully comparable to those for the standard and unique alignments; PTP identified 249 putative species. Deeper nodes showed better support when additional individuals were analyzed. Even the shortest sequences (17 sequences of 105 - 288 bp; Fig. 1), were placed in the correct species clade, with no decrease in the confidence value. The automated method again yielded variable results for taxa with small numbers of individuals. In some cases, analysis of additional individuals segregated closely-related sister species into different clades, although the monophyly of the morphological species was retained in all cases. When genetic distances between conspecific individuals were moderately high (5-7 %), PTP analysis failed to identify these as a single putative species (Supplementary data S3.zip).
BLAST analysis of barcodes
Results of the BLAST analyses carried out in jMOTU showed more sensitivity to sequence homogeneity than did the tree-based analyses (Fig. 4). For the unique and the standard alignments, there were marked shifts resulting in the attenuation of the slope indicating a within-species MOTU threshold of 2.5 – 3 % (Fig. 4). In contrast, the original alignment did not show this shift, instead showing progressive attenuation of the slope of the curve. For the sake of argument, if we apply a 3% sequence difference threshold level for species differentiation 9, all three alignments gave similar results: 225 clusters were identified from the unique alignment, 229 from the standard, and 228 from the original alignment, although there were differences in the taxa comprising the MOTUs between the analyses. For all three alignments, the number of MOTUs detected exceeded the number of OTUs. For the unique and standard alignments, each MOTU contained only one OTU (with the same exceptions indicated for the tree analyses). However, this analysis is not equivalent to a standard BLAST analysis of a single sequence, since the constraints (minimum length, percentage, etc) are based on averages calculated for the analyzed dataset. In a standard BLAST analysis, for which these thresholds are based on the query sequence length, even the shorter sequences are properly identified.
Best close match analysis of barcodes
Clades or MOTUS were analyzed individually using best close match, with a primary focus on those for which there were discrepancies between MOTUs and OTUs. The results indicated that, in nearly all cases when more than one individual of the same species was included in the analyzed dataset, the closest match was the same species. Exceptions to this outcome included closely-related species pairs of Calanus (C. helgolandicus and C. euxinus; C. agulhensis and C. sinicus); Centropages (C. typicus and C. chierchiae); and Acartia (A. tonsa and A. hudsonica). In sum, although the MOTU/OTU concordance can be improved in comparison with automated procedures in a flexible (but subjective) fashion, total agreement between morphological and molecular species assignment methods was not possible.
Species-by-species analyses
Taxa showing discrepancies between MOTUs and OTUs selected for additional analysis, when possible based on available data, to examine possible reasons (e.g., variation among geographic areas or populations, cryptic speciation) for the observed disparities between morphological and molecular data, are studied in detail in Appendix 1.
Discussion
The growing use of DNA barcodes to discriminate and identify marine animal species has included many studies on zooplankton and a number of studies of planktonic copepods (see Bucklin et al. 8 for a review). This study presents results of comparative analysis of a large dataset of 1,381 barcode sequences for 195 copepod species, including 800 new barcode sequences for 63 copepod species not included in any previous study. Evaluations include ML and NJ automated tree-based, BLAST, and “best close match” analyses of three different sequence alignments, varying the analyzed sequence domain and the numbers of individuals per species. We report here our conclusions regarding the reliability and resolution of diverse statistical approaches to species identification of planktonic marine copepods based on DNA barcodes.
The “best close match” 49 yielded the best results in terms of establishing a species threshold that avoids false positive results – even without a previously-identified and barcoded individual of the same species. Although individuals may fail to be assigned to a species, incorrect species assignments are avoided, unless the distance between two morphologically identified OTUs is zero. This analysis avoids a frequent error of NJ trees, for which individuals of different species may cluster together – albeit on long branches – with bootstrap support equal or very close to 100% when one or more of the species is missing from the analyzed barcode dataset 12 , 18.
The poor performance of the tree-based automated method compared to the others, can be attributed to the unbalanced dataset (large disparity in numbers of individuals among species) and with low coverage in many cases. This fact is known to limit the performance of PTP and other similar tree-based delimitation methods 52 . On the other hand, the failure of the PTP approach resulted in lack of power to identify the species, and not the much less desirable error of wrong species assignment.
The high levels of genetic diversity within species and the limited number of species for which DNA barcodes are available make character-based diagnosis 11 , 12 very unlikely to succeed. This approach may be appropriate for much-studied and well-defined groups of taxa, where much of variability has been characterized. Especially when a small number of nucleotide substitutions are used as taxon identifiers or as a step in a taxonomic key, accurate species identification will require complete knowledge of variability both among populations of a species and among species of the group of interest. We are still far away from this goal for marine copepods, due to their large effective population sizes (on the order of 108 57) and exceptional genetic diversity among eukaryotes 58.
Although the coincidence of these two concepts (large population sizes and exceptionally high genetic diversity) could seem counter-intuitive 59, this should be considered in light of factors inherent to the marine planktonic environment. The large distributional ranges of most species, in many cases across multiple ocean basins, might facilitate the isolation of lineages, while still allowing migration and continuous exchange of individuals across the distributional range. The short generation time for these species (usually weeks to several months, rarely multiple years) makes impossible the migration of individuals across the entire range of the species in a single or few generations. Thus, both oceanographic barriers and isolation by distance may results in population differentiation at large scales and among different ocean basins. However, if analyzed at fine scales, allele frequency differences would show continuous variation, with stronger differences at hydrographic or biogeographical barriers only 35 , 86 , 87 , 88.
One of the most powerful applications of barcoding for marine copepods is the analysis of the entire zooplankton community through high throughout DNA sequencing of environmental samples, out-performing the results obtained even by trained morphological analysts 60 . Recent technical advances allow determination of long sequences necessary for accurate identification of species in mixed assemblages. Limitations include inefficient amplification of the COI barcode region in samples containing diverse taxa resulting from variability in the amplification priming sites 61, which hinders annealing of consensus primers. However, higher affinity and amplification success rates of more conserved genes have the associated problem of under-estimating the real diversity of species in a community 62 , 63 , 64, due to low levels of sequence divergence and lack of discrimination between closely related species. The low affinity of the consensus COI barcoding primers by Folmer et al. 38 can be countered by design of suites of group-specific primers; copepod-specific primers have been designed for this purpose 20 , 65 . In the very near future, environmental barcoding approaches may employ nested sets of species- and group-specific amplification and sequencing primers and protocols to ensure reliable, accurate, cost-effective, and rapid assessments of species-level of diversity of pelagic communities, including the taxonomically complex and ecologically-important copepods.
OTUs vs MOTUs
Automated statistical analyses allow species identification and detection of species boundaries based on DNA barcodes 16 , 17. However, our results showed a large discrepancy between the numbers of OTUs and MOTUs (e.g., for the original alignment, 195 morphological species versus 249 / 228 putative species on the ML tree / jMOTU, respectively). Since these numbers are based on a 3% threshold for discrimination in jMOTUs, this may be due in some cases to unrecognized cryptic species. In other cases, the discrepancy may reflect strong population structuring of widely-distributed species, perhaps combined with incomplete sampling of populations across the geographic range (see discussion in Bergsten et al. , 66). Those errors could be corrected by non-automated approaches that would not be suited for larger dataset, such as the best-close match or examining the problematic clades on the tree by eye. It is not rare for marine copepods to show genetic differences over 5 % between individuals within and between populations 31 , 35. Although those cases may be easily resolved by considering the geographical reference (collection location) and/or closely-related species, detection by automated analysis is difficult without geographically and taxonomically dense and balanced sampling. In other cases, the putative species delimitation would be biased by over-sampled taxa 52. Marine planktonic copepods are excellent examples of the inherent challenges of sampling highly abundant, widely distributed populations: high spatial resolution and geographically-extensive sampling is needed for a perfect match between OTUs and MOTUs. But, despite under-sampling of intraspecific variation in the dataset analyzed here, there were no false positives (i.e., assignment of the wrong species to an individual) and the genetically closest individual to any specimen identified using a barcode almost always (with a few notable exceptions) belonged to the same species.
A criticism of metazoan barcoding is the reliance on a single gene, rather than multiple molecular markers. In fact, results obtained from additional genes do not always yield the same results, and caution is advised when using only one or few genetic markers. Additional sources of error include sample sizes, geographical coverage, and sampling bias. In sum, many problems associated with barcoding result not from the COI barcoding region, but from relying on a single molecular marker without necessary consideration for the inevitable limitations, since any gene – even very conserved ones – will have strengths and pitfalls 64. It is possible that there may be better regions for species assignment, and longer sequences do provide better accuracy and reliability 61, but our results confirm that even very short COI fragments (< 150 bp) show acceptable levels of accuracy for species identification. Further, although average COI divergence is significantly higher for deeper taxonomic levels, there is no consistent relationship between divergence and taxonomic level. COI shows marked saturation and erosion of the phylogenetic signal for deeper nodes 67.
A primary limitation of barcoding is the widespread problem of incorrect species identification in published datasets, which markedly reduces reliability and usefulness of the approach 18 , 49 , 68. This problem was detected in our dataset by comparison with data from GenBank and other public databases. In other cases, when the obvious morphological differences between the two species made misidentification unlikely, errors may result from laboratory procedures. Solutions include approaches that allow independent confirmation of identifications, e.g., inclusion of images, retention of voucher specimens for later examination, and ratings on the accuracy of taxonomists 69. Another solution is simply to continue to populate databases and increase taxon sampling densities both systematically and geographically, thus allowing recognition of errors at the time of data submission.
Conclusions
This study presents new DNA barcode data for marine copepods (800 sequences for 63 species not previously sequenced) and reports the results of new analyses of a larger dataset (1,381 sequences for 195 copepod species). Our conclusions include recommendations to improve the accuracy and feasibility of using DNA barcodes for species identification of marine planktonic copepods, including: 1) availability of PCR and sequencing primers suited to the targeted species; 2) availability of a taxonomically-comprehensive DNA barcode database linking DNA sequences to accurately identified specimens; 3) increased density of taxon sampling; and 4) near-complete coverage for the group of interest. In particular, comprehensive databases are needed for environmental barcoding efforts (i.e., barcoding of unsorted environmental samples) that seek to characterize species-level diversity of marine zooplankton assemblages and ecosystems.
Increasingly sophisticated approaches to statistical analysis of the barcode region of the mitochondrial cytochrome c oxidase subunit I (COI) gene have resulted in new appreciation for the strengths and weaknesses of this genetic marker for species assignment of planktonic copepods. An important result is that – for all analytical approaches – accurate identification requires inclusion in the analyzed dataset of a barcode sequence for that species. The lack of a complete DNA barcode library is thus the most limiting factor for accurate and reliable discrimination and identification of species of planktonic copepods. In fact, DNA barcodes are currently available for only ~ 400 copepod species, including many parasitic and freshwater taxa. In addition, extensive coverage of species diversity is especially critical for efficient resolution on large datasets using automated methods. Fortunately, many barcoding studies have focused on ecologically important, abundant, and/or geographically widespread species and species groups, making the available DNA barcode data particularly useful. Species that are rare or geographically restricted may remain unidentifiable using barcodes for the foreseeable future.
Acknowledgments
The Census of Marine Zooplankton (CMarZ) Steering Group members and Project Managers provided valuable assistance in the collection and taxonomic identification of planktonic copepods. We acknowledge in particular: J. Bradford-Grieve (National Institute of Water and Atmospheric Research, New Zealand), S. Schnack-Schiel (Alfred Wegener Institute, Germany), H. Verheye (Department of Environmental Affairs, South Africa), R. Escribano (Universidad de Concepción, Chile), R.R. Hopcroft (University of Alaska - Fairbanks, USA), and R. Machida (Academica Sinica, Taiwan). Technical assistance in determining DNA sequences during 1992-2005 was provided by Lisa D. Allen, Meredith A. Bailey, Jason G. Beaudet, Christopher Caudill and Christopher A. Manning (all then at the University of New Hampshire, USA). The UConn Bioinformatics Facility provided computing resources for some analyses performed for this study.
Appendix 1
Problematic taxa
Previous or ongoing publications have addressed some of the discordances between Operational Taxonomic Units (OTUs) and Molecular Operational Taxonomic Units (MOTUs) for various marine copepod taxa: Calanus agulhensis De Decker, Kaczmaruk & Marska 1991 / C. sinicus Brodsky 1965 34 ; Calanus helgolandicus Claus 1863 / C. euxinus Huselmann 1991 70 , 71 , 72 ; C. lividus Frost & Fleminger 1968 35 ; A. tonsa Dana 1849 / A. hudsonica Pinhey 1926 clades 73, Bucklin et al., unpublished; Paracalanus spp. 74 , 75; and Calanoides carinatus Krøyer 1848 (Viñas et al. unpublished).
Copepod taxa that are problematical for barcoding are most usually also problematical in classical morphological taxonomy (e.g., Acartia, Paracalanus, Nannocalanus). These groups show combined genetic, ecological, biogeographical and morphological complexities that must be resolved to define and delimit species boundaries, improve our understanding of marine zooplankton community ecology, and examine species-level responses to environmental change 13 . Several examples of such taxa are described here:
Nannocalanus minor Claus 1863
Nannocalanus minor barcodes form a cluster comprising three clades in both the tree-based (Supplementary data S3.zip) and TCS analysis (Fig. 1 in Appendix). The sequence variation did not show any geographical pattern; all three clades contained sequences from the NW Atlantic and in some cases were found in the same sample. Cluster A included a number of individuals with identical sequences collected from the NE and SE Atlantic; within each clade, individuals differed by < 1.5% on average; between clades, differences ranged from 9.2 - 14.3% (Table 1). In addition, ΦST (F ST distance under the K2P model) = 0.95 between clades A and B (P value < 0.0001 after 10,000 permutations); ΦST distances were not calculated for Clade C, which included a single sequence. Morphological analyses of two females from Clade A and B, respectively indicate different numbers of teeth on the inner edge of the basipodite 1 of P5: the Clade A individual showed 11 denticles, compared to 16 for Clade B. Individual variability is known to be high for this character, and either of the two described Nannocalanus species may exhibit 16 teeth. However, 11 teeth on the P5 B1 is outside the known range for this character for N. minor sensu stricto 3 , 76, suggesting potential reproductive isolation among the three clades, which may be sympatric, cryptic species 77.
Mesocalanus tenuicornis Dana 1849
Sequences for Mesocalanus tenuicornis clustered with low bootstrap support (Supplementary data S3.zip) and showed two clades, the SW Pacific and the NE Atlantic. Two other divergent sequences from SW Pacific and SE Atlantic were consistent with the jMOTU results that resolved four independent MOTUs. The divergence between these four potential clades comprised of two groups and two individuals ranged from 11.6 - 18.8% (average = 15.6 ± 3.2% S.D.), with individuals collected from the same location (e.g., AF332788 and AF462316) showing 16.7% COI divergence. Compared to another COI sequence for this species from GenBank (AB379998), genetic distances obtained here were in the same range (15.8 - 18.9%). Although some differences may result from geographic differentiation of regional populations, the presence of very divergent clades indicates the possible presence of cryptic species within this taxon.
Euterpina acutifrons Dana 1848
Tree-based and BLAST analysis of the harpacticoid copepod Euterpina acutifrons yielded two clusters corresponding to the NE (Bay of Biscay) and NW (Mid-Atlantic Bight) Atlantic. Within clades, differences ranged from 0.0 - 0.5%, while between-clade differences averaged 12% (range 11.8 – 12.6%; Supplementary data S2.zip and S3.zip). Although planktonic, E. acutifrons is rarely found in the open ocean 3 and naupliar stages are linked to the hyperbenthic environment 78, suggesting limited dispersal across and among ocean basins. Other harpacticoid copepods showed similarly large genetic divergences among geographic populations, including a species found in tidal pools and benthic environments 36, although open-ocean harpacticoid species showed lower divergences among ocean basin than in our study 31. These considerations, together with our small sample size, prevent conclusions of whether the observed differences reflect strong divergences among regional populations of a single species or the presence of a cryptic species complex within E. acutifrons.
Centropages typicus Kröyer 1849 and Centropages chierchiae Giesbrecht 1889
These two species co-occur throughout most of their distributions in the Atlantic Ocean 3. Subtle morphological differences discriminate the species 79 and many individuals exhibit ‘intermediate forms’ (L.B.B. unpubl. data.; Nieves Rodriguez Garcia, Oviedo, pers. comm.). Morphological variation has been widely described for C. typicus; comparative analyses showed no genetic basis for the morphological variation 32. In our study, genetic distances between individuals of both species from both NW and NE Atlantic (Bay of Biscay) were lower than 4%, and very similar within- and between-species (Table 2). In addition, an ‘intermediate-form’ individual showed the same range of variation as both species. In light of both morphological and molecular data, there is significant doubt about the validity of the distinction of C. chierchiae Giesbrecht, 1889 from C. typicus Kröyer, 1849.
Calocalanus contractus Farran 1926
Two very similar sequences (differing by < 1%) were found for Calocalanus contractus; a third sequence differed by 9% from these two; the corresponding amino acid sequence was identical for these three individuals. These three sequences differed from other Calocalanus species by 17.4% to 18.4%. The 9% difference is just slightly smaller to that between sister species within this genus 74, but some other copepod species show intra-species distances in this range 35. A detailed study combining morphology, taxonomy and barcoding is needed for the ~50 species described species 3, including many poorly-described species with small size and complex ornamentation.
Acartia tsuensis Ito 1956 and A. tonsa Dana 1849 / A. hudsonica Pinhey 1926
Three differentiated clades were detected within Acartia tsuensis. One was collected from Momoshima Island, Japan, while the other two were collected from fish ponds in the Philippines. Genetic distances within the clades were below 2%, while distances between the clades were equal or larger than 19% (average = 19.6%, SD = 0.4%), probably reflecting genetic isolation between the clades and indicating cryptic speciation.
For A. tonsa and A. hudsonica, individuals of both species were mixed within each clade; individuals from the NE Pacific samples (identified as two A. tonsa and one A. hudsonica individuals) clustered together. Cryptic speciation among geographically-close or sympatric populations has been described for other species of Acartia, likely driven by limited dispersal among populations and the effects of mesoscale and local environmental conditions. Alternatively, complex phylogeographic patterns might reflect high levels of genetic differentiation among conspecific populations 33 , 37 , 80 , 81.
Metridia lucens Boeck 1864
Metridia lucens is a cosmopolitan species that showed significant genetic differentiation among the six geographic regions analyzed: NW Atlantic (19 individuals), NE Atlantic (10), SW Pacific (4), NE Pacific (5) and Antarctic (1). No haplotypes were shared between ocean basins (Fig. 2 in Appendix), and genetic distances between regions were on average ten-times that obtained within regions (Table 3; the Antarctic individual was excluded from analysis). ΦST distances (under K2P model) indicated high and significant isolation between NW and NE Atlantic regions (Table 4), although this may result partly from small sample sizes bias (Fig. 2 in Appendix). It is possible that Metridia lucens, which exhibits strong diel vertical migration, may experience lower exchange among populations and show strong regional-scale genetic structure. The Antarctic sample showed a highly divergent sequence, ~8% different from other M. lucens individuals, which is similar to results obtained with larger sample sizes by Stupnikova et al. 82. As comparison, genetic distances between M. lucens and the closely related M. pacifica Brodsky 1950 ranged from 12.6% - 14.4%. Consequently, genetic variation among Antarctic M. lucens samples may reflect either recent speciation or strong regional isolation.
Pleuromamma gracilis Claus 1863 and P. piseki Farran 1929
The clade containing Pleuromamma gracilis and P. piseki was recovered with 100% bootstrap support, but the species formed a number of clusters and MOTUs. A detailed analysis of these two taxa showed a complex pattern, with strong regional-scale genetic structuring of P. gracilis, but no clear isolation from P. piseki (Fig. 3 in Appendix; Table 5). Pleuromamma gracilis from the SE Atlantic showed a number of highly divergent haplotypes, but still no amino acid substitutions (Fig. 3 in Appendix). Regional-scale genetic structure and the presence of highly-divergent rare haplotypes may be related to the large effective population size of cosmopolitan marine copepods 35 , 83. In contrast, P. piseki did not show strong spatial structure and lacked genetic divergence from P. gracilis (Table 5). Similar results have been recently 84 reported for the same species complex for other mitochondrial markers .Clearly, integrated molecular and morphological analysis of both species throughout their distributional ranges is needed to clarify both the status of the species and their population structure.
Clausocalanus Giesbrecht 1888 species
All 13 described species of Clausocalanus 85 are represented in the database, including multiple individuals from different ocean basins. All species clustered together in all analyses (Supplementary file S3.zip). Interestingly, two individuals collected from Sagami Bay (Japan) and identified as C. arcuicornis Dana 1849 (by L.B.B.) showed morphological oddities and were discriminated as a distinct clade with 12% or higher divergence from any of C. arcuicornis (or any other Clausocalanus) individuals. Since no intact individuals remained from the sample, a detailed morphological analysis cannot be done and we can only speculate that these individuals may represent either of two Clausocalanus that are incertae sedis (C. latipes T. Scott 1894 and C. dubius Brodsky 1950), or very divergent individuals of C. arcuicornis.
Figures and Tables
TCS analysis on the Nannocalanus minor sequences.
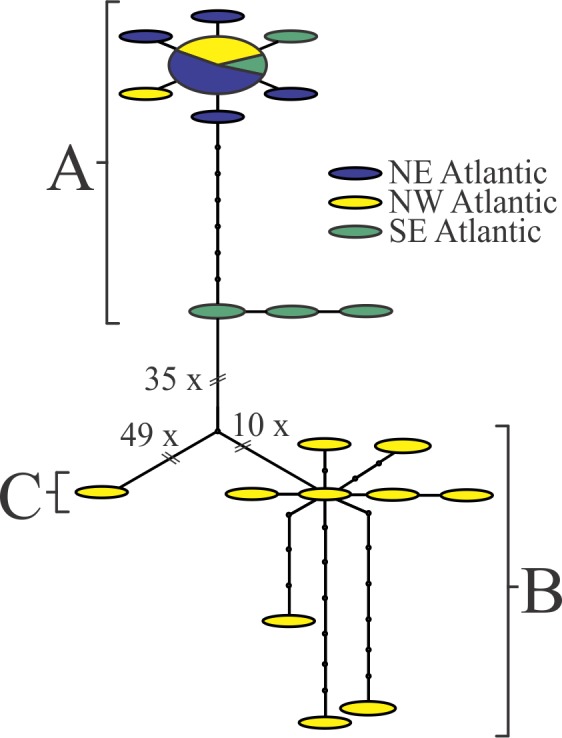
Each segment represents a mutational step, and small black circles, missing haplotypes needed to connect observed haplotypes. The size of the ovals is proportional to the number of sampled individuals with that haplotype. All sites containing ambiguous or missing data were excluded from the analysis (complete deletion; final length 485 bp). The three clusters reflect the clades identified in the tree analyses. No regional or distribution pattern could be related to these results.
TCS analysis on the Metridia lucens sequences.
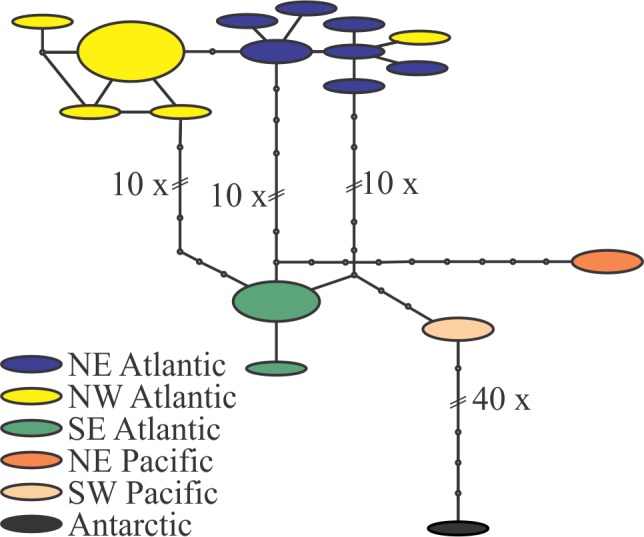
Each segment represents a mutational step, and small black circles are missing haplotypes needed to connect observed haplotypes. The size of the ovals is proportional to the number of sampled individuals with that haplotype. All sites containing ambiguous or missing data were excluded from the analysis (complete deletion; final length 502 bp). The resulting diagram reflects a strong regional pattern, with relative isolation between the different ocean basins especially in the North-South axis. The Antarctic region shows a strong isolation even from the close individuals from the Southern Hemisphere.
TCS analysis on the Pleuromamma piseki and Pleuromamma gracilis sequences.

Each segment represents a mutational step, and small black circles are missing haplotypes needed to connect observed haplotypes. The size of the ovals is proportional to the number of sampled individuals with that haplotype. All sites containing ambiguous or missing data were excluded from the analysis (complete deletion; final length 502 bp). The resulting diagram reflects a strong regional pattern, with relative isolation between the different ocean basins especially in the North-South axis. The Antarctic region shows a strong isolation even from the close individuals from the Southern Hemisphere.
Table 1. Average, standard deviation, maximum and minimum base differences per site (p-distance) within and between sequences of three clades of Nannocalanus minor.
| Within A | Within B | A vs B | B vs C | A vs C | |
|---|---|---|---|---|---|
| Average | 0.0044 | 0.0138 | 0.1170 | 0.1273 | 0.1403 |
| St. Dev. | 0.0060 | 0.0078 | 0.0074 | 0.0035 | 0.0016 |
| Max. | 0.0178 | 0.0258 | 0.1282 | 0.1322 | 0.1433 |
| Min. | 0.0000 | 0.0000 | 0.0928 | 0.1216 | 0.1367 |
Table 2. Average, standard deviation, maximum and minimum base differences per site (p-distance) within and between Centropages chierchiae and C. typicus, and between these species and the intermediate forma.
| C. chierchiae | C. typicus | ch. vs ty. | int. vs pure | |
|---|---|---|---|---|
| Average | 0.0127 | 0.0216 | 0.0160 | 0.0252 |
| St. Dev. | 0.0036 | 0.0123 | 0.0103 | 0.0067 |
| Max. | 0.0156 | 0.0362 | 0.0317 | 0.0362 |
| Min. | 0.0087 | 0.0035 | 0.0035 | 0.0173 |
Table 3. Average, standard deviation, maximum and minimum base differences per site (p-distance) between the Metridia lucens individuals within the locations, between all locations excluding the Antarctic, and those compared to the Antarctic individual.
| NE Atlantic | NW Atlantic | SE Atlantic | SW Pacific | NE Pacific | Between | Vs Antarctic | |
|---|---|---|---|---|---|---|---|
| Average | 0.0024 | 0.0016 | 0.0003 | 0.0000 | 0.0000 | 0.0236 | 0.0816 |
| St. Dev. | 0.0014 | 0.0028 | 0.0006 | 0.0000 | 0.0000 | 0.0132 | 0.0063 |
| Max. | 0.0047 | 0.0185 | 0.0016 | 0.0000 | 0.0000 | 0.0454 | 0.0965 |
| Min. | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0016 | 0.0717 |
Table 4. ΦST distances (K2P model, lower diagonal, and p-value (upper diagonal) between locations. Significant results after Bonferroni correction are indicated in bold.
| NE Atlantic | NW Atlantic | SE Atlantic | SW Pacific | NE Pacific | |
|---|---|---|---|---|---|
| NE Atlantic | <0.0001 | <0.0001 | 0.0006 | 0.0002 | |
| NW Atlantic | 0.763 | <0.0001 | <0.0001 | <0.0001 | |
| SE Atlantic | 0.978 | 0.969 | 0.001 | 0.0002 | |
| SW Pacific | 0.980 | 0.968 | 0.868 | 0.008 | |
| NE Pacific | 0.986 | 0.977 | 0.983 | 1.000 |
Table 5. Average, standard deviation, maximum and minimum base differences per site (p-distance) within and between the P. piseki and P. gracilis individuals, and within P. gracilis locations with N > 10 individuals.
| P. piseki | P. gracilis | Between | NE Atlantic | SE Atlantic | SW Pacific | |
|---|---|---|---|---|---|---|
| Average | 0.0321 | 0.0611 | 0.0662 | 0.0030 | 0.0401 | 0.0088 |
| St. Dev. | 0.0249 | 0.0359 | 0.0177 | 0.0019 | 0.0452 | 0.0059 |
| Max. | 0.0601 | 0.1030 | 0.0934 | 0.0064 | 0.1030 | 0.0218 |
| Min. | 0.0000 | 0.0000 | 0.0277 | 0.0000 | 0.0000 | 0.0016 |
Funding Statement
This study is a contribution from the Census of Marine Zooplankton (CMarZ, see www.CMarZ.org), an ocean realm field project of the Census of Marine Life. We gratefully acknowledge programmatic support from the Alfred P. Sloan Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors have declared that no competing interests exist.
Contributor Information
Leocadio Blanco-Bercial, University of Connecticut, Groton, Connecticut, USA.
Astrid Cornils, Alfred-Wegener-Institut Helmholtz-Zentrum für Polar- und Meeresforschung, Bremerhavn, Germany.
Nancy Copley, Woods Hole Oceanographic Institution, Woods Hole, Massachusetts, USA.
Ann Bucklin, University of Connecticut, Groton, Connecticut, USA.
References
- 1.Longhurst AR (1985) The structure and evolution of plankton communities. Prog Oceanogr 15: 1-35.
- 2.Mauchline J (1998) The biology of Calanoid Copepods: Academic Press. 710 pp.
- 3.Razouls C, de Bovée F, Kouwenberg J, Desreumaux N (2005-2012) Diversity and geographic distribution of marine planktonic copepods.
- 4.Turner JT (2004) The importance of small planktonic copepods and their roles in pelagic marine food webs. Zool Stud 43: 255-266.
- 5.Goetze E (2003) Cryptic speciation on the high seas; global phylogenetics of the copepod family Eucalanidae. Proc R Soc B: Biol Sci 270: 2321-2331. [DOI] [PMC free article] [PubMed]
- 6.Knowlton N (1993) Sibling Species in the Sea. Annu Rev Ecol Syst 24: 189-216.
- 7.Knowlton N (2000) Molecular genetic analyses of species boundaries in the sea. Hydrobiologia 420: 73-90.
- 8.Bucklin A, Steinke D, Blanco-Bercial L (2011) DNA Barcoding of marine Metazoa. Annu Rev Mar Sci 3: 471-508. [DOI] [PubMed]
- 9.Hebert PD, Cywinska A, Ball SL, deWaard JR (2003) Biological identifications through DNA barcodes. Proc R Soc B: Biol Sci 270: 313 - 321. [DOI] [PMC free article] [PubMed]
- 10.Hebert PD, Stoeckle MY, Zemlak TS, Francis CM (2004) Identification of birds through DNA barcodes. PLoS Biol 2: E312. [DOI] [PMC free article] [PubMed]
- 11.DeSalle R, Egan MG, Siddall M (2005) The Unholy Trinity: taxonomy, species delimitation and DNA Barcoding. Philos Trans Biol Sci 360: 1905-1916. [DOI] [PMC free article] [PubMed]
- 12.Goldstein PZ, DeSalle R (2011) Integrating DNA barcode data and taxonomic practice: Determination, discovery, and description. BioEssays 33: 135-147. [DOI] [PubMed]
- 13.Köhler F (2007) From DNA taxonomy to barcoding - how a vague idea evolved into a biosystematic tool. Mitteilungen aus dem Museum für Naturkunde in Berlin - Zoologische Reihe 83: 44-51.
- 14.Heimeier D, Lavery S, Sewell MA (2010) Using DNA barcoding and phylogenetics to identify Antarctic invertebrate larvae: Lessons from a large scale study. Mar Genomics 3: 165-177. [DOI] [PubMed]
- 15.Jones M, Ghoorah A, Blaxter M (2011) jMOTU and Taxonerator: Turning DNA Barcode sequences into annotated Operational Taxonomic Units. PLoS ONE 6: e19259. [DOI] [PMC free article] [PubMed]
- 16.Zhang AB, Muster C, Liang HB, Zhu CD, Crozier R, et al. (2012) A fuzzy-set-theory-based approach to analyse species membership in DNA barcoding. Mol Ecol 21: 1848-1863. [DOI] [PubMed]
- 17.Puillandre N, Modica MV, Zhang Y, Sirovich L, Boisselier MC, et al. (2012) Large-scale species delimitation method for hyperdiverse groups. Mol Ecol 21: 2671-2691. [DOI] [PubMed]
- 18.Collins RA, Cruickshank RH (2013) The seven deadly sins of DNA barcoding. Mol Ecol Res 13: 969-975. [DOI] [PubMed]
- 19.Bucklin A, Hopcroft RR, Kosobokova KN, Nigro LM, Ortman BD, et al. (2010) DNA barcoding of Arctic Ocean holozooplankton for species identification and recognition. Deep-Sea Res II 57: 40-48.
- 20.Bucklin A, Ortman BD, Jennings RM, Nigro LM, Sweetman CJ, et al. (2010) A "Rosetta Stone" for metazoan zooplankton: DNA barcode analysis of species diversity of the Sargasso Sea (Northwest Atlantic Ocean). Deep-Sea Res II 57: 2234-2247.
- 21.Machida R, Hashiguchi Y, Nishida M, Nishida S (2009) Zooplankton diversity analysis through single-gene sequencing of a community sample. BMC Genomics 10: 438. [DOI] [PMC free article] [PubMed]
- 22.Laakmann S, Gerdts G, Erler R, Knebelsberger T, Martínez Arbizu P, et al. (2013) Comparison of molecular species identification for North Sea calanoid copepods (Crustacea) using proteome fingerprints and DNA sequences. Mol Ecol Res 13: 862-876. [DOI] [PubMed]
- 23.Böttger-Schnack R, Machida R (2011) Comparison of morphological and molecular traits for species identification and taxonomic grouping of oncaeid copepods. Hydrobiologia 666: 111-125.
- 24.Bucklin A, Frost BW (2009) Morphological and molecular phylogenetic analysis of evolutionary lineages within Clausocalanus (Copepoda: Calanoida). J Crust Biol 29: 111-120.
- 25.Machida RJ, Tsuda A (2010) Dissimilarity of species and forms of planktonic Neocalanus copepods using mitochondrial COI, 12S, nuclear ITS, and 28S gene sequences. PLoS ONE 5: e10278. [DOI] [PMC free article] [PubMed]
- 26.Goetze E, Bradford-Grieve J (2005) Genetic and morphological description of Eucalanus spinifer T. Scott, 1894 (Calanoida: Eucalanidae), a circumglobal sister species of the copepod E. hyalinus s.s. (Claus, 1866). Prog Oceanogr 65: 55-87.
- 27.Ueda H, Bucklin A (2006) Acartia (Odontacartia) ohtsukai, a new brackish-water calanoid copepod from Ariake Bay, Japan, with a redescription of the closely related A. pacifica from the Seto Inland Sea. Hydrobiologia 560: 77-91.
- 28.Ueda H, Yamaguchi A, Saitoh S-i, Sakaguchi SO, Tachihara K (2011) Speciation of two salinity-associated size forms of Oithona dissimilis (Copepoda: Cyclopoida) in estuaries. J Nat Hist 45: 2069-2079.
- 29.Hill RS, Allen LD, Bucklin A (2001) Multiplexed species-specific PCR protocol to discriminate four N. Atlantic Calanus species, with an mtCOI gene tree for ten Calanus species. Mar Biol 139: 279-287.
- 30.Laakmann S, Auel H, Kochzius M (2012) Evolution in the deep sea: Biological traits, ecology and phylogenetics of pelagic copepods. Mol Phylogenet Evol 65: 535-546. [DOI] [PubMed]
- 31.Eberl R, Cohen S, Cipriano F, Carpenter EJ (2007) Genetic diversity of the pelagic harpacticoid copepod Macrosetella gracilis on colonies of the cyanobacterium Trichodesmium spp. Aquat Biol 1: 33-43.
- 32.Castellani C, Lindley AJ, Wootton M, Lee CM, Kirby RR (2012) Morphological and genetic variation in the North Atlantic copepod, Centropages typicus. J Mar Biol Assoc UK 92: 99-106.
- 33.Chen G, Hare MP (2011) Cryptic diversity and comparative phylogeography of the estuarine copepod Acartia tonsa on the US Atlantic coast. Mol Ecol 20: 2425-2441. [DOI] [PubMed]
- 34.Kozol R, Blanco-Bercial L, Bucklin A (2012) Multi-Gene analysis reveals a lack of genetic divergence between Calanus agulhensis and C. sinicus (Copepoda; Calanoida). PLoS ONE 7: e45710. [DOI] [PMC free article] [PubMed]
- 35.Blanco-Bercial L, Álvarez-Marqués F, Bucklin A (2011) Comparative phylogeography and connectivity of sibling species of the marine copepod Clausocalanus (Calanoida). J Exp Mar Biol Ecol 404: 108-115.
- 36.Garlitska L, Neretina T, Schepetov D, Mugue N, De Troch M, et al. (2012) Cryptic diversity of the ‘cosmopolitan’ harpacticoid copepod Nannopus palustris: genetic and morphological evidence. Mol Ecol 21: 5336-5347. [DOI] [PubMed]
- 37.Caudill CC, Bucklin A (2004) Molecular phylogeography and evolutionary history of the estuarine copepod, Acartia tonsa, on the Northwest Atlantic coast. Hydrobiologia 511: 91-102.
- 38.Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3: 294-299. [PubMed]
- 39.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731-2739. [DOI] [PMC free article] [PubMed]
- 40.Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673-4680. [DOI] [PMC free article] [PubMed]
- 41.Lopez JV, Yuhki N, Masuda R, Modi W, O'Brien SJ (1994) Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J Mol Evol 39: 174-190. [DOI] [PubMed]
- 42.Song H, Buhay JE, Whiting MF, Crandall KA (2008) Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc Natl Acad Sci USA 105: 13486-13491. [DOI] [PMC free article] [PubMed]
- 43.Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451-1452. [DOI] [PubMed]
- 44.Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16: 111 - 120. [DOI] [PubMed]
- 45.Collins RA, Boykin LM, Cruickshank RH, Armstrong KF (2012) Barcoding's next top model: an evaluation of nucleotide substitution models for specimen identification. Methods Ecol Evol 3: 457-465.
- 46.Srivathsan A, Meier R (2012) On the inappropriate use of Kimura-2-parameter (K2P) divergences in the DNA-barcoding literature. Cladistics 28: 190-194. [DOI] [PubMed]
- 47.Fregin S, Haase M, Olsson U, Alström P (2012) Pitfalls in comparisons of genetic distances: A case study of the avian family Acrocephalidae. Mol Phylogenet Evol 62: 319-328. [DOI] [PubMed]
- 48.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403-410. [DOI] [PubMed]
- 49.Meier R, Shiyang K, Vaidya G, Ng PKL (2006) DNA barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Syst Biol 55: 715-728. [DOI] [PubMed]
- 50.Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406 - 425. [DOI] [PubMed]
- 51.Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688-2690. [DOI] [PubMed]
- 52.Zhang J, Kapli P, Pavlidis P, Stamatakis A (2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29: 2869-2876. [DOI] [PMC free article] [PubMed]
- 53.Floyd R, Abebe E, Papert A, Blaxter M (2002) Molecular barcodes for soil nematode identification. Mol Ecol 11: 839-850. [DOI] [PubMed]
- 54.Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9: 1657-1659. [DOI] [PubMed]
- 55.Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10: 564-567. [DOI] [PubMed]
- 56.Meyer CP, Paulay G (2005) DNA Barcoding: error rates based on comprehensive sampling. PLoS Biol 3: e422. [DOI] [PMC free article] [PubMed]
- 57.Bucklin A, Wiebe P (1998) Low mitochondrial diversity and small effective population sizes of the copepods Calanus finmarchicus and Nannocalanus minor: possible impact of climatic variation during recent glaciation. J Hered 89: 383-392. [DOI] [PubMed]
- 58.Bron J, Frisch D, Goetze E, Johnson S, Lee C, et al. (2011) Observing copepods through a genomic lens. Frontiers in Zoology 8: 22. [DOI] [PMC free article] [PubMed]
- 59.Lanfear R, Kokko H, Eyre-Walker A (2014) Population size and the rate of evolution. Trends Ecol Evol 29: 33-41. [DOI] [PubMed]
- 60.Lindeque PK, Parry HE, Harmer RA, Somerfield PJ, Atkinson A (2013) Next Generation Sequencing reveals the hidden diversity of zooplankton assemblages. PLoS ONE 8: e81327. [DOI] [PMC free article] [PubMed]
- 61.Roe AD, Sperling FAH (2007) Patterns of evolution of mitochondrial cytochrome c oxidase I and II DNA and implications for DNA barcoding. Mol Phylogenet Evol 44: 325-345. [DOI] [PubMed]
- 62.Hirai J, Shimode S, Tsuda A (2013) Evaluation of ITS2-28S as a molecular marker for identification of calanoid copepods in the subtropical western North Pacific. J Plankton Res 35: 644-656.
- 63.Creer S, Fonseca VG, Porazinska DL, Giblin-Davis RM, Sung W, et al. (2010) Ultrasequencing of the meiofaunal biosphere: practice, pitfalls and promises. Mol Ecol 19: 4-20. [DOI] [PubMed]
- 64.Tang CQ, Leasi F, Obertegger U, Kieneke A, Barraclough TG, et al. (2012) The widely used small subunit 18S rDNA molecule greatly underestimates true diversity in biodiversity surveys of the meiofauna. Proc Natl Acad Sci USA 109: 16208-16212. [DOI] [PMC free article] [PubMed]
- 65.Hoareau TB, Boissin E (2010) Design of phylum-specific hybrid primers for DNA barcoding: addressing the need for efficient COI amplification in the Echinodermata. Mol Ecol Res 10: 960-967. [DOI] [PubMed]
- 66.Bergsten J, Bilton DT, Fujisawa T, Elliott M, Monaghan MT, et al. (2012) The effect of geographical scale of sampling on DNA barcoding. Syst Biol 61: 851-869. [DOI] [PMC free article] [PubMed]
- 67.Wägele J-W (1999) Major sources of errors in phylogenese systematics. Zool Anz 238: 329-337.
- 68.Valentini A, Pompanon F, Taberlet P (2009) DNA barcoding for ecologists. Trends Ecol Evol 24: 110-117. [DOI] [PubMed]
- 69.Steinke D, Hanner R (2011) The FISH-BOL collaborators' protocol. Mitochondrial DNA 22: 10-14. [DOI] [PubMed]
- 70.Unal E, Frost BW, Armbrust V, Kideys AE (2006) Phylogeography of Calanus helgolandicus and the Black Sea copepod Calanus euxinus, with notes on Pseudocalanus elongatus (Copepoda, Calanoida). Deep-Sea Res II 53: 1961-1975.
- 71.Papadopoulos LN, Peijnenburg KTCA, Luttikhuizen PC (2005) Phylogeography of the calanoid copepods Calanus helgolandicus and C. euxinus suggests Pleistocene divergences between Atlantic, Mediterranean, and Black Sea populations. Mar Biol 147: 1353-1365.
- 72.Yebra L, Bonnet D, Harris R, Lindeque P, Peijnenburg K (2011) Barriers in the pelagic: population structuring of Calanus helgolandicus and C. euxinus in European waters. Mar Ecol Prog Ser 428: 135-149.
- 73.Hill RS (2004) Genetic diversity and structure of calanoid copepods: molecular evolutionary patterns in coastal estuaries (Acartia tonsa) and the open ocean (Calanus spp.) [Ph.D. Thesis]. Durham: University of New Hampshire.
- 74.Cornils A, Blanco-Bercial L (2013) Phylogeny of the Paracalanidae Giesbrecht, 1888 (Crustacea: Copepoda: Calanoida). Mol Phylogenet Evol 69: 861-872. [DOI] [PubMed]
- 75.Cornils A, Held C (2014) Evidence of cryptic and pseudocryptic speciation in the Paracalanus parvus species complex (Crustacea, Copepoda, Calanoida). Front Zool 11: 19. [DOI] [PMC free article] [PubMed]
- 76.Andronov VN (2001) On the taxonomy of the genus Nannocalanus Sars, 1925 (Crustacea, Copepoda: Calanidae). Zoosystem Ross 9: 277-283.
- 77.Bucklin A, LaJeunesse TC, Curry E, Wallinga J, Garrison K (1996) Molecular diversity of the copepod, Nannocalanus minor: Genetic evidence of species and population structure in the North Atlantic Ocean. J Mar Res 54: 285-310.
- 78.Haq SM (1965) Development of the copepod Euterpina acutifrons with special reference to dimorphism in the male. Proc Zool Soc Lond 144: 175-201.
- 79.Rose M (1933) Copépodes pélagiques. Faune de France 26: 374 pp.
- 80.Milligan P, Stahl E, Schizas N, Turner J (2011) Phylogeography of the copepod Acartia hudsonica in estuaries of the northeastern United States. Hydrobiologia 666: 155-165.
- 81.Soh HY, Moon SY, Park EO, Maran BV (2013) A new species of Acartia subgenus Euacartia (Copepoda: Calanoida: Acartiidae) from Korean estuaries based on morphological and molecular evidence. J Crust Biol 33: 718-729.
- 82.Stupnikova AN, Molodtsova TN, Mugue NS, Neretina TV (2013) Genetic variability of the Metridia lucens complex (Copepoda) in the Southern Ocean. J Mar Syst 128: 175-184.
- 83.Goetze E (2010) Species discovery in marine planktonic invertebrates through global molecular screening. Mol Ecol 19: 952-967. [DOI] [PubMed]
- 84.Halbert KMK, Goetze E, Carlon DB (2013) High cryptic diversity across the global range of the migratory planktonic copepods Pleuromamma piseki and P. gracilis. PLoS ONE 8: e77011. [DOI] [PMC free article] [PubMed]
- 85.Frost BW, Fleminger A (1968) A revision of the genus Clausocalanus (Copepoda: Calanoida) with remarks on distributional patterns in diagnostic characters. Bull Scripps Inst Oceanogr Univ Calif 12: 1-235.
- 86.Nelson RJ, Carmack EC, McLaughlin FA, Cooper GA (2009) Penetration of Pacific zooplankton into the western Arctic Ocean tracked with molecular population genetics. Mar Ecol Prog Ser 381: 129-138.
- 87.Unal E, Bucklin A (2010) Basin-scale population genetic structure of the planktonic copepod Calanus finmarchicus in the North Atlantic Ocean. Prog Oceanogr 87: 175-185.
- 88.Goetze E (2011) Population differentiation in the Open Sea: insights from the pelagic copepod Pleuromamma xiphias. Integr Comp Biol 51: 580-597. [DOI] [PubMed]