

Abstract
Modification at the glycerol side chain of sialic acid in sialosides modulate their recognition by sialic acid-binding proteins and sialidases. However, limited work has been focused on the synthesis and functional studies of sialosides with C7-modified sialic acids. Here we report chemical synthesis of C4-modified ManNAc and mannose and their application as sialic acid precursors in a highly efficient one-pot three-enzyme system for chemoenzymatic synthesis of α2–3- and α2–6-linked sialyl para-nitrophenyl galactosides in which the C7-hydroxyl group in sialic acid (N-acetylneuraminic acid, Neu5Ac, or 2-keto-3-deoxynonulosonic acid, Kdn) was systematically substituted by -F, -OMe, -H, and -N3 groups. Substrate specificity study of bacterial and human sialidases using the obtained sialoside library containing C7-modified sialic acids showed that sialosides containing C7-deoxy Neu5Ac were selective substrates for all bacterial sialidases tested but not for human NEU2. The information obtained from sialidase substrate specificity can be used to guide the design of new inhibitors that are selective against bacterial sialidases.
Keywords: Chemoenzymatic synthesis, C7-modified sialic acid, Sialidase, Sialoside, Sialyltransferase, Substrate specificity
1. Introduction
Sialic acids are common terminal monosaccharides on the carbohydrate moieties of surface glycoconjugates on mammalian cells. They are directly involved in numerous molecular recognition events involved in immune regulation, cell-cell interaction, inflammation, bacterial and viral infection. Three basic forms of sialic acids are N-acetylneuraminic acid (Neu5Ac), N-glycolylneuraminic acid (Neu5Gc), and 2-keto-3-deoxynonulosonic acid (Kdn) (Figure 1). In nature, sialic acids can undergo additional post-glycosylational modifications (modifications after the formation of glycosidic linkages)1 including mono- and multiple O-acetylation at any hydroxyl groups, O-methylation and O-sulfation at C-8, as well as O-lactylation and O-phosphorylation at C-9. So far, more than 50 structurally distinct sialic acid forms have been identified in nature.2–4 Although the functions of some sialic acid modifications have been elucidated,2,3 the roles of the majority of modified sialic acid residues remain unknown.
Figure 1.
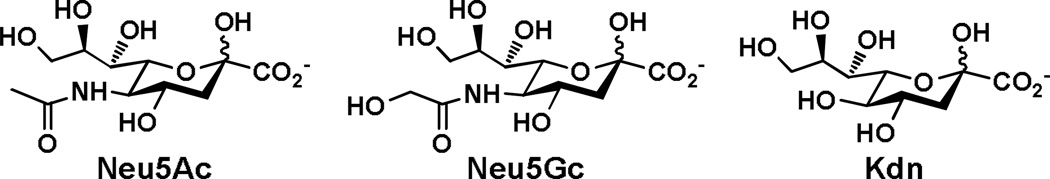
Three basic forms of sialic acid.
In order to facilitate the understanding of the significance of sialic acid modifications in nature and develop sialic acid-based therapeutics, a highly efficient one-pot multienzyme chemoenzymatic sialylation system has been developed for the synthesis of sialosides containing naturally occurring sialic acids and their non-natural derivatives.5–7 In this system, N-acetylmannosamine (ManNAc) and mannose derivatives are chemically or enzymatically synthesized as sialic acid precursors. ManNAc, mannose, and their derivatives are converted by a sialic acid aldolase (NanA)-catalyzed reaction to form sialic acids and their derivatives, which are activated by a CMP-sialic acid synthetase (CSS), and transferred to proper acceptors by suitable sialyltransferases (SiaTs) to form targeted sialosides with desired sialic acid forms, sialyl linkages, and underlying glycans.5 The enzymatic reactions containing three enzymes can be carried out in one-pot in a buffered aqueous solution at room temperature or 37 °C without the isolation of intermediates. It has been shown that all enzymes used including recombinant bacterial NanAs, CSSs, and SiaTs have quite good tolerance towards substrate modifications. Sialosides with natural and non-natural modifications at C-5, C-9, and C-8 of sialic acid have been successfully synthesized.6–12 α2–3- and α2–6-Linked sialosides Siaα2–3/6GalβpNP containing C-5 and/or C-9 modified sialic acids have been used to elucidate the substrate specificities of bacterial, human, and viral sialidases.9–11,13 Selective sialidase inhibitors against human NEU2 or bacterial sialidases have been designed and synthesized based on the structural features of the sialic acid modification obtained from sialidase substrate specificity studies.14,15 Nevertheless, the method has not been used for the synthesis of sialosides containing C7-modified sialic acids.
Compared to sialosides containing C5- and C9-modified sialic acids which are more commonly investigated, sialosides containing C7-modified sialic acids are underexplored. Neu5Ac analogs containing C7-modification have been chemically synthesized16,17 and tested as substrates for CMP-sialic acid synthetase (CSS).17 C4-Modified ManNAc derivatives have also been synthesized18,19 and used as substrates for sialic acid aldolase for the synthesis of Neu5Ac analogs20–22 as well as Neu5Ac2en-derivatives as transition state analog inhibitors19 or prodrugs23 against influenza virus sialidases. N-Acetyl-4-fluoro-4-deoxy-D-mannosamine (4-F-ManNAc or ManNAc4F) has been synthesized and used as a starting material for one-pot two-enzyme synthesis of CMP-ManNAc4F.24 Recently, peracetylated N-acetyl-4-azido-4-deoxymannosamine (peracetylated 4-azido-ManNAc) was used successfully for metabolic engineering of mammalian cell surface glycoproteins with 7-azido-Neu5Ac, indicating the tolerance of 4-azido-modified ManNAc by mammalian cellular enzymes involved in sialic acid biosynthesis, activation, and transfer to glycoproteins.25 Unlike metabolic engineering of cultured mammalian cells with peracetylated N-azidoacetylmannosamine (peracetylated ManNAz) which introduced Neu5Az to both N- and O-glycans on cell surface glycoproteins, incubating UDP-N-acetylglucosamine 2-epimerase/ManNAc kinase (GNE) deficient HEK293 cells with peracetylated 4-azido-ManNAc introduced 7-azido-Neu5Ac only to the O-glycans of cell surface glycoproteins. Success in labelling O-glycosylated proteins in live zebrafish embryos during development was also achieved.25 Nevertheless, there are only a few reports on the chemical synthesis of sialosides containing 7-deoxy Neu5Ac26,27 and testing their binding to sialoside-binding proteins.27 Enzymatic synthesis of sialosides containing C7-modified sialic acid and their applications in investigating sialidase activities have not been achieved.
Here we describe the highly efficient one-pot three-enzyme chemoenzymatic synthesis of α2–3-and α2–6-linked sialosides containing C7-modified sialic acids with the C7-OH group of Neu5Ac and Kdn being systematically replaced by -F, -OMe, -H, and -N3 from the corresponding C4-modified ManNAc and mannose as sialic acid precursors. Good to excellent (53–98%) yields have been obtained which illustrates again the high promiscuity of bacterial sialoside biosynthetic enzymes in tolerating substrate modifications. Sialidase substrate specificity studies using this set of sialosides clearly show selective cleavage of sialosides containing 7-deoxy-Neu5Ac by bacterial enzymes but not by human cytosolic sialidase NEU2. Therefore, 7-deoxy-Neu5Ac2en is a potential selective inhibitor against bacterial sialidases but not human NEU2. We are in the process of synthesizing sialidase inhibitors based on 7-deoxy-Neu5Ac and the results will be reported in due course.
2. Results and discussion
2.1 Chemical synthesis of sialic acid precursors
In order to synthesize desired α2–3- and α2–6-linked sialosides containing C7-modified sialic acids with the C7-OH group of Neu5Ac and Kdn being systematically replaced by -F, -OMe, -H, and -N3, we designed and synthesized eight ManNAc and mannose derivatives 1a–8a in which the C4-OH is replaced with -F, -OMe, -H, and -N3, respectively (Figure 2).
Figure 2.

Structures of C4-modified sialic acid precursors that are synthesized.
The synthesis of 4-F-ManNAc (1a) was achieved by following a previously reported method.24 As shown in Scheme 1, benzyl glycopyranoside 9 readily prepared from D-glucose28 was selectively protected at the hydroxyl groups of both C4 and C6 with benzylidene to form 10. The C2-OH of 10 was triflated and converted to azide 11 with concomitant reversal of the stereochemistry by an SN2 reaction using sodium azide in DMF. Benzylation of the C3-OH of compound 11 formed 12 followed by hydrolysis of the benzylidene acetal under acidic conditions yielded 4,6-diol, which was selectively protected by benzoate at C6-OH to form 13. Reacting the C4-OH of 13 with DAST in dichloromethane produced fluorinated hexose 14. Converting the C2-azido group in 14 to NHAc group formed 15 which was deprotected by catalytic hydrogenation and the Zemplén reaction to form the desired 4-F-ManNAc 1a.
Scheme 1.

Synthesis of 4-F-ManNAc 1a.
The preparation of 4-O-methyl-ManNAc 2a and 4-deoxy-ManNAc 3a is shown in Scheme 2. Selectively benzoylation of ManNAc provided tri-benzoate 16 with a free hydroxyl group at the C-4. It was treated with iodomethane and silver oxide to produce the methylation product. After removing the benzoyl protection groups by treating with KOH in methanol, the desired 4-O-methyl ManNAc 2a was obtained in a reasonable yield. On the other hand, treatment of benzoate 16 with 1,1'-thiocarbonyldiimidazole, followed by reaction with tri-n-butylstannane and azobisisobutyronitrile (AIBN) provided a deoxy intermediate. Subsequent removal of benzoyl groups by the Zemplén reaction produced the target compound 4-deoxy-ManNAc 3a.
Scheme 2.

Synthesis of 4-O-methyl ManNAc 2a and 4-deoxy ManNAc 3a.
4-N3-ManNAc 4a was prepared via a modified 1,6-anhydro sugar similar to that described in a report by von Itzstein et al.18 Briefly, as shown in Scheme 3, tosylation of ManNAc and subsequent reaction with DBU produced 1,6-anhydro compound 17. Compound 17 was treated with triphenylphosphine and diisopropyl azodicarboxylate (DIAD) to yield 3,4-epoxide 18. Treating 11 with sodium azide, followed by acetylation using acetic anhydride and acetolysis using Ac2O and TMSOTf, yielded the 4-azido product 21. Subsequent removal of the acetyl groups using NaOMe in methanol produced the target compound 4-N3-ManNAc 4a.
Scheme 3.

Synthesis of 4-N3-ManNAc 4a.
The preparation of mannose derivatives 5a–8a (Scheme 4) started from mannose. Selectively benzoylation of mannose29 produced the key intermediate tetra-benzoate 22. The remaining hydroxyl group of 22 was then transformed into a triflate, followed by the treatment of Bu4NNO2 to produce 23. Fluorination of the 23 with DAST in CH2Cl2 gave 4-F-mannose 5a after deprotection. For the synthesis of 4-O-methyl-mannose 6a, benzoate 22 was treated with iodomethane and silver oxide followed by removal of the benzoyl groups to produce the desired 6a. 4-Dexoy-mannose 7a was obtained via activation of 22 with 1,1'-thiocarbonyldiimidazole followed by treatment with tributyltin hydride and azobisisobutyronitrile, and then de-benzoylation. In order to synthesize 4-N3-mannose 8a, compound 23 was subjected to triflation and concomitant treatment with TBAN3, the target compound 8a was obtained after the Zemplén debenzoylation.
Scheme 4.

Synthesis of C4-modified mannose derivatives 5a–8a.
2.2. Enzymatic synthesis of C7-modified sialosides
A highly efficient one-pot three-enzyme chemoenzymatic sialylation approach5 was used to synthesize a library of α2–3- and α2–6-linked sialyl para-nitrophenyl galactosides in which the C7-hydroxyl group in the sialic acid was systematically substituted with -F, -OMe, -H, and -N3 groups. The enzymatic system (Scheme 5) contained p-nitrophenyl β-galactopyranoside as the sialyltransferase acceptor, a C4-mdofied ManNAc or mannose chosen from 1a–8a as the sialic acid precursor, sodium pyruvate, CTP, and three enzymes including Pasteurella multocida sialic acid aldolase (PmNanA), Neisseria meningitidis CMP-sialic acid synthetase (NmCSS), and Pasteurella multocida sialyltransferase 1(PmST1) or Photobacterium damselae α2–6-sialyltransferase (Pd2,6ST) for the synthesis of α2–3-linked sialosides or α2–6-linked sialosides, respectively.
Scheme 5.

One-pot three-enzyme synthesis of sialosides containing C7-modified sialic acids.
As shown in Table 1, the α2–3-linked sialosides containing C7-modified Neu5Ac (1b–4b) or C7-modified Kdn (5b–8b ) were obtained in excellent yields (80–98%). Except for Neu5Ac7OMeα2–6GalβpNP 2c (57%), other α2–6-linked sialosides containing C7-modified Neu5Ac (1c and 3c–4c) were obtained in excellent yields (88–98%). Except for Kdn7OMeα2–6GalpNP, 6c (53%) and Kdn7deoxyα2–6GalpNP, 7c (58%), other α2–6-linked sialosides containing C7-modified Kdn including Kdn7Fα2–6GalpNP, 5c (95%) and Kdn7N3α2–6GalpNP, 8c (89%) were also obtained in excellent yields. The higher yields obtained for α2–3-linked sialosides containing Neu5Ac7OMe (95%) and Kdn7OMe (89%) compared to that for α2–6-linked counterparts (57% and 53%, respectively) indicates that the 7-O-methylation on CMP-sialic acid is better tolerated by PmST1 compared to Pd2,6ST. Similarly, replacing C7-OH in the Kdn of CMP-Kdn is also better tolerated by PmST1 compared to Pd2,6ST leading to a significant difference in the yields obtained (90% for Kdn7deoxyα2–3GalpNP 7b and 58% for Kdn7deoxyα2–6GalpNP 7c).
Table 1.
One-pot three-enzyme synthesis of α2–3/6-linked sialosides containing C7-modified sialic acids.
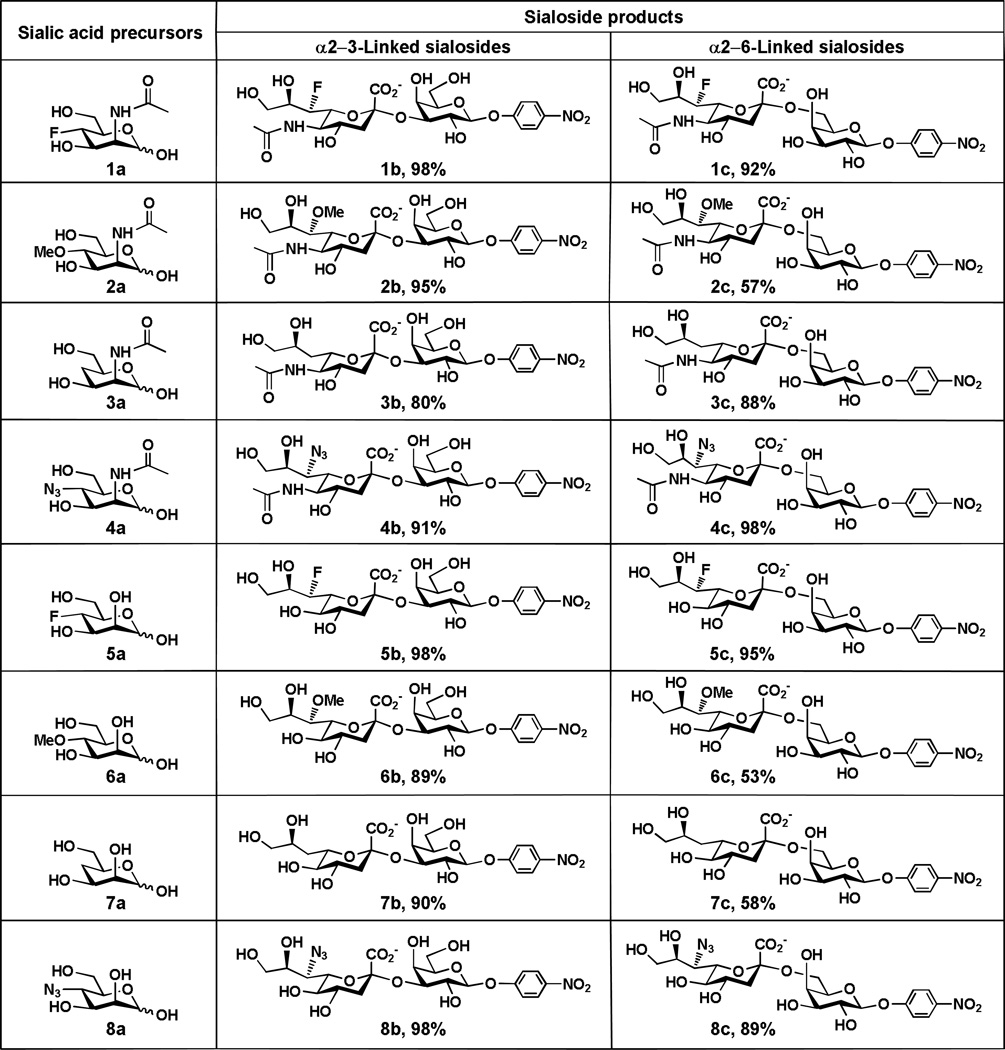 |
2.3. Sialidase substrate specificity studies
The synthesized sialosides 1b–8b and 1c–8c were used together with Neu5Acα2–3/6GalβpNP and Kdnα2–3/6GalβpNP to test the substrate specificity of a recombinant human cytosolic sialidase NEU2 and several bacterial sialidases including the α2–3-sialidase function of PmST1 and four commercially available bacterial sialidases such as Vibrio cholerae sialidase, Salmonella typhimurium sialidase, Streptococcus pneumoniae sialidase, and Clostridium perfringens sialidase. As shown in Figure 3, while sialosides containing C7-deoxy Neu5Ac are generally better or similarly good substrates compared to those containing non-modified Neu5Ac for bacterial sialidases, substitution of the C7-OH of Neu5Ac in sialosides by hydrogen diminishes the activity of human NEU2.
Figure 3.

Sialidase substrate specificity studies using α2–3- (1b–8b) and α2–6-linked (1c–8c) sialyl GalβpNP containing C7-modified sialic acids. Neu5Acα2-3/6Galβ3pNP and Kdnα2–3/6GalβpNP are used as controls. Results using α2–3-linked sialosides as substrates are shown in white bars and results using α2–6-linked sialosides as substrates are shown in black bars.
Previously we have shown that replacing one of the methyl hydrogen atoms in the C5-N-acetyl group of Neu5Ac in sialosides by a fluorine atom does not affect the activities of either bacterial sialidases tested10 or human NEU2.14 We have also shown that replacing the C9-OH group of Neu5Ac in sialosides by a fluorine atom, a hydrogen atom (deoxy-Neu5Ac), or an azido group does not significantly change the activities of all bacterial sialidases tested although human NEU2 is quite sensitive to C9-modifications on Neu5Ac11 Here we show that both Streptococcus pneumoniae sialidase (Figure 3D) and Clostridium perfringens sialidase (Figure 3E) as well as human NEU2 (Figure 3F) are quite sensitive to Neu5Ac C7-fluorine substitution which diminishes their activities significantly. Azido-substitution at C7-OH of Neu5Ac is also not well tolerated by either human NEU2 or bacterial sialidases tested except for the α2–6-sialidase activity of Clostridium perfringens sialidase which remains a reasonable activity (50%) compared to non-modified Neu5Acα2–6GalβpNP substrate (Figure 3E). Neu5Ac C7-OMe modification is not tolerated well by any of the enzymes tested.
3. Conclusion
In conclusion, we report here that the one-pot three-enzyme chemoenzymatic sialylation system is highly efficient (yields vary from 53% to 98%) in producing sialosides containing C7-modified sialic acids from a library of C4-modified ManNAc and mannose derivatives containing various functional groups (-F, -OMe, -H, and -N3). We demonstrate again the substrate promiscuity of bacterial sialoside biosynthetic enzymes and their application in the synthesis of diverse sialosides. Substrate specificity studies of five bacterial sialidases and a human cytosolic sialidase NEU2 indicate that in general, substituting C7-OH of sialic acids in sialosides by -OMe or -N3 diminishes the activity of sialidases tested significantly. Quite interestingly, sialosides containing C7-deoxy Neu5Ac are good substrates for all bacterial sialidases tested but not for human NEU2. Sialidase inhibitors based on C7-deoxy Neu5Ac could be selective inhibitors against bacterial sialidases without affecting the activity of human NEU2 significantly. We are in the process of testing this hypothesis.
4. Experimental section
4.1. General
All chemicals were obtained from commercial suppliers and used without further purification unless otherwise noted. Anhydrous solvents were used to carry out chemical reactions under inert argon or nitrogen environment. 1H NMR (600 MHz) and 13C NMR (150 MHz) spectra were recorded on a Varian Inova-600 spectrometer. High resolution electrospray ionization (ESI) mass spectra were obtained at the Mass Spectrometry Facility in the University of California, Davis. Silica gel 60 Å (200–425 mesh, Fisher Chemical) was used for flash column chromatography. Thin-layer chromatography (TLC) was performed on silica gel plates 60 GF254 (Sorbent technologies) using p-anisaldehyde sugar stain. Gel filtration chromatography was performed using a column (100 cm × 2.5 cm) packed with Bio-gel P-2 fine resins (Bio-Rad, Hercules, CA).
4.2. Chemical synthesis of ManNAc and Man derivatives as sialoside precursors 1a–8a
4.2.1. 2-Acetamido-2,4-dideoxy-4-fluoro -d-mannopyranoside (4-F-ManNAc, 1a)
Benzyl glucopyranoside 928 (7.2 g, 26.64 mmol) was dissolved in 30 mL of anhydrous DMF. PhCH(OMe)2 (6.0 mL, 1.5 equiv.) and p-toluenesulfonic acid (100 mg) were added. The reaction was carried out at 50 °C for 1 h using rotavap at 70 mbar. Reaction was monitored by TLC (CH2Cl2:MeOH = 10:0.2 by volume). The solution mixture was concentrated, co-evaporated with toluene, and the residue was dissolved in CH2Cl2. The sample was loaded to a silica gel column and washed with hexanes, CH2Cl2, and a mixture of CH2Cl2 and MeOH (100:1–20:1) to produce 1-benzyl-4,6-benzylidene-d-glucopyranoside 10 (8.27 g, 87% yield). 1H NMR (600 MHz, CDCl3) δ 7.91–7.26 (m, 10H), 5.48 (s, 1H), 4.93 (d, J = 4.2 Hz, 1H, H-1), 4.72 (d, J = 12.0 Hz, 1H), 4.53 (d, J = 11.4 Hz, 1H), 4.19 (dd, J = 5.4 and 11.2 Hz, 1H), 3.91 (t, J = 11.2 Hz, 1H), 3.82–3.35 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 137.37, 137.14, 128.78, 128.72, 128.51, 128.50, 128.36, 128.32, 126.60, 126.59, 102.06, 98.56, 81.22, 73.07, 71.65, 70.28, 69.10, 62.96.
Compound 10 (4.23 g, 11.8 mmol) was dissolved in anhydrous CH2Cl2 (50 mL) and pyridine (20 mL) was added. The solution mixture was placed in acetone-dry ice (–20 °C), and trifluoromethanesulfonic acid anhydride (Tf2O) (2.35 mL, 14.16 mmol) was added drop-wisely. The mixture was stirred for 2 h at −20 °C. The reaction was monitored by TLC (Hexanes:EtOAc = 3:1, by volume). Upon completion, the reaction was quenched and washed with brine three times. The organic solution was concentrated, re-dissolved in CH2Cl2, dried over MgSO4, and filtered. The filtrate was concentrated, co-evaporated with toluene, and the residue was dried under vacuum. Without purification, the residue was dissolved in anhydrous CH2Cl2 (30 mL) under nitrogen. Sodium azide (7.68 g, 118 mmol) was added and the suspension was stirred at 50 °C for 24 h. the reaction was monitored by TLC and terminated by adding water (50 mL). The solution mixture was extracted with ethyl acetate, dried over anhydrous MgSO4, filtered, concentrated, and purified by silica gel column to produce 2-azido-2-deoxy-4,6-benzylidene-1,3-dibenzylmannpyranoside 11 (2.11 g, 47% yield). 1H NMR (600 MHz, CDCl3) δ 7.51–7.32 (m, 10H), 5.58 (s, 1H), 4.88 (d, J = 1.2 Hz, 1H, H-1), 4.72 (d, J = 11.4 Hz, 1H), 4.53 (d, J = 12.0 Hz, 1H), 4.31–4.29 (m, 1H), 4.24 (dd, J = 3.6 and 9.0 Hz, 1H), 3.96 (d, J = 3.6 Hz, 1H), 3.93 (t, J = 9.0 Hz, 1H), 3.87–3.79 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 137.24, 136.72, 129.58, 128.85, 128.64, 128.47, 128.31, 126.50, 102.50, 98.46, 79.25, 69.80, 69.16, 68.86, 63.97, 63.87.
Compound 11 (2.0g, 5.22 mmol) was dissolved in anhydrous DMF (20 mL) under nitrogen and the reaction was placed in ice bath at 0 °C. Sodium anhydride (0.19 g, 7.83 mmol), tetrabutylammonium iodide (0.12 g, 0.33 mmol), and benzyl bromide (1.25 mL, 10.5 mmol) were added. The solution mixture was stirred at 0 °C for 2 h then at room temperature for overnight. The reaction was terminated by adding methanol (2 mL) followed by water (50 mL) and the mixture was extracted with ethyl acetate. The organic solution was dried over anhydrous MgSO4, filtered, concentrated and purified by silica gel column (Hexanes:EtOAc = 10:1 to 5:1, by volume) to produce compound 12 (2.23 g, 90% yield).
Compound 12 (2.20 g, 4.65 mmol) was dissolved in methanol (40 mL). p-Toluenesulfonic acid (0.30g, 1.58 mmol) was added and the reaction mixture was stirred at room temperature for 3 h. The reaction was monitored by TLC (Hexanes:EtOAc = 3:1, by volume). Triethylamine (0.25 mL) was added and stirred for 10 min to neutralize the reaction mixture. The reaction mixture was concentrated, co-evaporated with toluene, and dried under vacuum for overnight. The residue was dissolved in CH2Cl2 (30 mL) under nitrogen atmosphere, placed in ice-bath at 0 °C. 2,6-Lutidine (0.81 mL, 6.95 mmol) was added followed by benzoyl chloride (0.64 mL, 5.55 mmol). The reaction mixture was stirred at 0 °C for 3 h, washed with water (2 × 30 mL), and then once with brine. The organic solution was dried over anhydrous magnesium sulfate and concentrated. The residue was dissolved in CH2Cl2 and purified by silica gel column (Hexanes:EtOAc = 10:1 to 5:1, by volume) to produce compound 13 (1.94 g, 85%). 1H NMR (600 MHz, CDCl3) δ 8.09–7.32 (m, 15H), 4.92 (s, 1H, H-1), 4.77 (d, J = 11.4 Hz, 1H), 4.73 (d, J = 12.0 Hz, 1H), 4.67 (d, J = 11.4 Hz, 1H), 4.65 (dd, J = 4.8 and 12.0 Hz, 1H), 4.56 (dd, J = 1.8 and 12.0 Hz, 1H), 4.52 (d, J = 12.0 Hz, 1H), 4.02–3.91 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 167.06, 137.60, 136.77, 133.40, 128.90, 128.82, 128.64, 128.44, 128.43, 128.37, 128.30, 97.68, 79.54, 72.88, 71.21, 69.57, 66.89, 64.00, 60.77.
In a 50 mL centrifuge tube, DAST (3.0 mL, 24.5 mmol) was added to anhydrous CH2Cl2 (10 mL). A solution containing compound 13 (1.5 g, 3.06 mmol) in anhydrous CH2Cl2 was slowly added at −78 °C, and the mixture was stirred at room temperature for 3 d. The reaction was checked by TLC (Hexanes:EtOAc = 3:1, by volume), cooled down to −20 °C, and terminated by adding methanol (2 mL). The mixture was transferred into a flask, concentrated, re-dissolved in CH2Cl2, and washed with water. The organic solution was dried over anhydrous MgSO4, filtered, concentrated, and purified by silica gel column (Hexanes:EtOAc = 12:1 to 6:1, by volume) to produce compound 14 (0.53 g, 35% yield).
Compound 14 (0.49 g, 1.00 mmol) was dissolved in pyridine (4 mL), and thioacetic acid (8 mL) was added. The reaction was allowed to stir at rt for overnight and monitored by TLC (Hexanes:EtOAc = 1:1, by volume). The reaction mixture was concentrated and the residue was purified by flash chromatography (Hexanes:EtOAc = 4:1 to 1:1, by volume) to produce compound 15 (0.43 g, 85% yield). 1H NMR (600 MHz, CDCl3) δ 8.06–7.28 (m, 15H), 5.89 (d, J = 8.4 Hz, 1H, NH), 5.00 (s, 1H, H-1), 4.79–4.46 (m, 8H), 4.20–4.15 (m, 2H).
To a solution of 15 (204 mg, 0.40 mmol) in MeOH (10 mL) was added 10% Pd-C (100 mg). The resulting mixture was hydrogenated on a hydrogenation apparatus for 18 h under hydrogen atmosphere. The resulting suspension was filtered and concentrated. The residue was dissolved in dry MeOH (10 mL) containing a catalytic amount of NaOMe. The mixture was stirred at room temperature for overnight. The reaction was neutralized using Dowex (H+) resin. The resulting suspension was filtrated and concentrated. Silica gel column purification (EtOAc:MeOH = 9:1, by volume) produced the desired product 4-F-ManNAc 1a (60 mg, 67% yield). 1H NMR (600 MHz, D2O) δ 5.13 (s, 0.7H), 4.49 (d, J = 1.8 Hz, 0.3H), 3.71–3.91 (m, 3H), 3.66–3.45 (m, 3H), 3.39 (s, 1H) 3.38 (s, 2H); 13C NMR (150 MHz, D2O) δ 94.1, 93.8, 74.7, 73.1, 71.6, 71.3, 70.9, 70.8, 70.3, 67.1, 66.8, 58.6, 58.5.
4.2.2. 2-Acetamido-2-deoxy-4-O-methyl-d-mannopyranoside (4-O-Me-ManNAc, 2a)
To a solution of pre-dried ManNAc (6.0 g, 27.1 mmol) in anhydrous pyridine (120 mL) was added benzoyl chloride (13.5 mL, 108.0 mmol) at −40 °C drop-wisely for 20 min. The reaction solution was stirred at −40 °C for 2 h. The reaction was stopped by adding MeOH and the reaction mixture was concentrated. The concentrated residue was dissolved in CH2Cl2 and washed sequentially with HCl (100 mL, 2 M), saturated NaHCO3 (60 mL), and water (100 mL). The product was purified by silica gel column (Hexanes:EtOAc = 6:4) to give 16 (12.3 g, 85%). 1H NMR (600 MHz, CDCl3) δ 7.99 (d, J = 7.8 Hz, 4H), 7.96 (d, J = 7.2 Hz, 2H), 7.55–7.51 (m, 3H), 7.38 (t, J = 7.2 Hz, 6 H), 6.18 (s, 1H), 6.11 (d, J = 9.0 Hz, 1H), 5.34 (dd, J = 8.4 and 4.2 Hz, 1H), 5.01 (dd, J = 9.0 and 3.0 Hz, 1H), 4.69–4.61 (m, 2H), 3.97 (d, J = 6.0 Hz, 2H), 2.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 170.73, 167.04, 166.88, 164.40, 134.01, 133.66, 133.54, 130.24, 130.14, 129.99, 129.79, 129.60, 128.99, 128.75, 128.69, 128.65, 91.89, 75.74, 74.68, 66.60, 63.75, 50.00, 23.44.
Compound 16 (0.30 g, 0.56 mmol) and Ag2O (1.30 g, 5.6 mmol) were dissolved in anhydrous CH2Cl2 (15 mL) containing 4 Å molecular sieves (0.6 g). CH3I (0.35 mL, 5.6 mmol) was added drop-wisely at 0 °C and the suspension was stirred for 48 h at room temperature. The mixture was filtered over Celite and concentrated. Silica gel column purification (Hexanes:EtOAc = 3:2, by volume) produced the benzoyl-protected methylated intermediate (0.27 g, 90%). 1H NMR (600 MHz, CDCl3) δ 8.06–7.97 (m, 6H), 7.59–7.55 (m, 3H), 7.46–7.39 (m, 6H), 6.24 (s, 1H), 5.99 (d, J = 9.0 Hz, 1H), 5.43 (dd, J = 8.4 and 4.2 Hz, 1H), 5.09–5.07 (m, 1H), 4.66–4.65 (m, 2H), 4.06–4.04 (m, 1H), 3.70 (t, J = 8.4 Hz, 1H), 3.52 (s, 3H), 2.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 170.17, 166.09, 165.57, 164.28, 133.84, 133.33, 130.07, 129.70, 128.57, 91.27, 74.93, 74.13, 73.64, 63.34, 60.47, 49.28, 23.32.
The intermediate obtained above (0.27 g, 0.49 mmol) was dissolved in a solution of KOH in MeOH (10 mL, 0.1 M) and stirred at room temperature for 1 h. The mixture was neutralized using Dowex (H+) resin and the resulting suspension was filtrated and concentrated. Silica gel column purification (EtOAc:MeOH:H2O = 10: 1:0.2, by volume) afforded product 2a (75 mg, 65%). 1H NMR (600 MHz, D2O) δ 5.08 (s, 0.7H), 4.98 (s, 0.3H), 4.30–4.29 (m, 1H), 4.11–4.09 (m, 1H), 3.82 (s, 3H), 3.54 (s, 3H), 3.42–3.35 (m, 2H), 2.04 (s, 3H); 13C NMR (150 MHz, D2O) δ 174.91, 93.11, 77.02, 72.06, 71.18, 68.94, 60.34, 53.53, 22.08.
4.2.3. 2-Acetamido-2,4-dideoxy-d-mannopyranoside (4-Deoxy-ManNAc, 3a)
Compound 16 (0.97 g, 1.81 mmol) and 1,1-thiocarbonyldiimidazole (0.64 g, 3.62 mmol) were dissolved in 1,2-dichloroethane (50 mL). The mixture was refluxed at 90 °C for 24 h. The solvent was removed and the concentrated residue was co-evaporated with toluene and dried under vacuum. To the dry compound was added azobisisobutyronitrile (AIBN) (59 mg, 0.36 mmol), Bu3SnH (2.92 mL, 10.86 mmol), and toluene (80 mL). The resulting mixture was refluxed at 110 °C for 5 h. After removing the solvent under reduced pressure, column chromatography on silica gel (Hexanes:EtOAc = 3:2, by volume) afforded deoxygenated intermediate (0.84 g, 90%). 1H NMR (600 MHz, CDCl3) δ 8.03–7.99 (m, 7H), 7.58–7.42 (m, 11H), 7.18–7.14 (m, 1H), 6.38 (d, J = 1.8 Hz, 0.3H), 6.13 (d, J = 1.8 Hz, 0.7H), 6.05 (d, J = 9.6 Hz, 0.7H), 6.01 (d, J = 9.6 Hz, 0.3H), 5.74–5.71 (m, 0.3H), 5.43–5.40 (m, 0.7H), 4.94 (dd, J = 9.0 and 2.4 Hz, 0.7H), 4.83 (dd, J = 9.6 and 2.4 Hz, 0.3H), 4.54–4.43 (m, 3H), 4.26–4.22 (m, 1H), 2.30–2.27 (m, 1H), 2.15 (s, 2.3H), 2.10 (s, 0.7H), 1.93–1.85 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 170.56, 170.26, 166.37, 165.88, 164.42, 164.28, 133.99, 133.52, 130.24, 130.18, 130.01, 129.86, 129.77, 129.26, 129.08, 128.93, 128.75, 128.68, 128.45, 93.92, 92.22, 71.17, 69.44, 68.30, 67.30, 65.87, 49.04, 48.04, 28.65, 28.44, 23.63, 23.50.
The intermediate obtained above (0.84 g, 1.62 mmol) was dissolved in dry MeOH (15 mL) containing a catalytic amount of NaOMe. The mixture was stirred at room temperature for overnight and neutralized using Dowex (H+) resin. The resulting suspension was filtrated and concentrated. Silica gel column purification (EtOAc:MeOH:H2O = 8:1:0.3, by volume) produced the desired compound 3a (0.3 g, 90%). 1H NMR (600 MHz, D2O) δ 5.21 (d, J = 3.6 Hz, 0.5H), 4.60 (d, J = 8.4 Hz, 0.5H), 4.11–4.08 (m, 0.5H), 3.99–3.95 (m, 0.5H), 3.77–3.48 (m, 4H), 2.03 (s, 1.5H), 2.02 (s, 1.5H), 1.51–1.41 (m, 2H); 13C NMR (150 MHz, D2O) δ 174.99, 174.83, 95.36, 91.58, 72.69, 69.01, 68.59, 65.26, 63.83, 58.41, 55.61, 34.83, 3451, 22.28, 22.01.
4.2.4. 2-Acetamido-4-azido-2,4-dideoxy-d-mannopyranoside (4-N3-ManNAc, 4a)
To a stirring solution of pre-dried ManNAc (8.2 g, 37.07 mmol) in anhydrous pyridine (50 mL) at 0 °C was added a solution of p-toluenesulfonyl chloride (11.31g, 59.31 mmol) in pyridine (20 mL) and the mixture was stirred for overnight at room temperature. The reaction was stopped by adding MeOH (10 mL) and the mixture was concentrated. Purification of the residue by column chromatography (EtOAc:MeOH = 9:1, by volume) provided the tosylated product (9.60 g, 69%). The resulted 6-O-Ts-ManNAc (9.60 g, 25.57 mmol) was dissolved in anhydrous ethanol (100 mL), and 1,8-diazabicycloundec-7-ene (DBU) (7.6 mL, 51.14 mmol) was added drop-wisely. The reaction mixture was stirred at room temperature for overnight. The crude product was dried under reduced pressure and silica gel flash column purification (EtOAc:MeOH = 9:1, by volume) provided 1,6-anhydro compound 17 (4.31 g, 83%).
To a solution of 17 (4.31 g, 21.21 mmol) in dry THF (50 mL) was added PPh3 (6.67 g, 25.45 mmol). Diisopropylazodicarboxylate (DIAD) (4.0 mL, 25.45 mmol) was added to the solution drop-wisely at 0 °C. The reaction was stirred for 1 h at 0 °C followed by 1 h at room temperature. The mixture was concentrated and silica gel column purification (EtOAc) afforded compound 18 (2.90 g, 74%). 1H NMR (600 MHz, CDCl3) δ 5.91 (s, 1H), 5.24 (d, J = 3.6 Hz, 1H), 4.84 (t, J = 4.8 Hz, 1H), 4.36–4.34 (m, 1H), 3.93 (d, J = 6.0 Hz, 1H), 3.73 (t, J = 4.8 Hz, 1H), 3.53 (dd, J = 6.6 and 4.8 Hz, 1H), 3.30 (dd, J = 3.6 and 1.2 Hz, 1H), 2.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 170.09, 97.04, 71.82, 63.99, 55.88, 49.21, 47.10, 23.22.
Compound 18 (2.90 g, 15.66 mmol) was dissolved in anhydrous DMF (25 mL). Dowex (H+) resin (3 g) and NaN3 (4.07 g, 62.64 mmol) were added to the solution. The suspension was heated at 95 °C for overnight. The reaction mixture was cooled down to room temperature, filtered through a plug of Celite, and washed with ethyl acetate and methanol. Silica gel column purification (EtOAc:MeOH = 9.5:0.5, by volume) provided compound 19 (2.43 g, 68%). 1H NMR (600 MHz, CDCl3) δ 5.34 (s, 1H), 4.56 (d, J = 5.4 Hz, 1H), 4.26 (d, J = 7.8 Hz, 1H), 4.14 (t, J = 9.0 Hz, 1H), 3.97 (d, J = 3.4 Hz, 1H), 3.76 (dd, J = 7.2 and 6.0 Hz, 1H), 3.55 (s, 1H), 1.98 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 170.04, 101.00, 74.01, 68.11, 65.63, 63.10, 48.59, 23.20.
To a solution of compound 19 (2.43 g, 10.65 mmol) in pyridine (20 mL) at 0 °C was added acetic anhydride (4 mL). The mixture was stirred at room temperature for 3 h. MeOH (4 mL) was added and the solvents were removed under reduced pressure. Product 20 (2.73 g, 95%) was obtained after silica gel column purification (Hexanes:EtOAc = 3:7, by volume). A mixture of compound 20 (2.73 g, 10.10 mmol) in acetic anhydride (15 mL) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) (1.84 mL, 10.10 mmol) was stirred under nitrogen at 0 °C for 2 h. The reaction was stopped by the addition of a saturated solution of NaHCO3 (50 mL) over 30 min. The mixture was extracted with ethyl acetate and washed with a saturated solution of NaHCO3 and brine. The concentrated residue was subjected to silica gel column chromatography (Hexanes:EtOAc = 1:9, by volume) to yield 21 (2.8 g, 77%). Compound 21 (2.8 g, 7.52 mmol) was dissolved in dry MeOH (30 mL) containing a catalytic amount of NaOMe. The mixture was stirred at room temperature for 2 h. The reaction was neutralized using Dowex (H+) resin. The resulted suspension was filtrated and concentrated. Silica gel column chromatography (EtOAc:MeOH = 9:1) of the crude product afforded final product 4a (1.53 g, 83%). 1H NMR (600 MHz, D2O) δ 5.13 (s, 0.6H), 4.97 (s, 0.4H), 4.45 (d, J = 6.4 Hz, 0.4H), 4.31 (d, J = 6.4 Hz, 0.6H), 4.18–4.12 (m, 1H), 3.89–3.78 (m, 3H), 3.59–3.48 (m, 2H), 2.06 (s, 1.5H), 2.04 (s, 1.5H); 13C NMR (150 MHz, D2O) δ 174.70, 174.48, 93.12, 92.87, 74.63, 71.56, 70.25, 70.00, 62.26, 60.73, 60.67, 53.94, 53.71, 52.65, 21.83, 21.79.
4.2.5. 4-Deoxy-4-fluoro-d-mannopyranoside (4-F-Man, 5a)
To a solution of pre-dried mannose (6.0 g, 33.30 mmol) in anhydrous pyridine (120 mL) was added benzoyl chloride (16.7 mL, 133.2 mmol) at −40 °C drop-wisely for 20 min. The reaction solution was stirred at −40 °C for 2 h. The reaction was stopped by adding MeOH and the reaction mixture was concentrated. The residue was dissolved in CH2Cl2 and washed sequentially with HCl (100 mL, 2M), saturated NaHCO3 (60 mL), and water (100 mL). The product was purified by a silica gel column (Hexanes:EtOAc = 8:2) to give 22 (13.9 g, 70%). 1H NMR (600 MHz, CDCl3) δ 8.15–8.14 (m, 4H), 7.96–7.91 (m, 4H), 7.65–7.63 (m, 1H), 7.55–7.45 (m, 6H), 7.34 (t, J = 7.2 Hz, 2H), 7.28–7.25 (m, 3H), 6.55 (s, 1H), 5.80 (s, 2H), 5.01 (dd, J = 12.0 and 3.0 Hz, 1H), 4.52 (dd, J = 12.0 and 1.8 Hz, 1H), 4.37 (t, J = 9.6 Hz, 1H), 4.24 (d, J = 9.6 Hz, 1H), 3.22 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 167.39, 166.64, 165.26, 164.23, 134.23, 133.77, 133.64, 133.60, 130.32, 130.16, 130.07, 130.05, 129.83, 129.45, 129.38, 128.99, 128.77, 128.62, 91.75, 73.94, 72.30, 69.68, 65.61, 63.10.
To a solution of compound 22 (0.6 g, 1.00 mmol) in CH2Cl2 (20 mL) and pyridine (0.8 mL, 10.00 mmol), trifluoromethanesulfonic acid anhydride (Tf2O) (0.33 mL, 2 mmol) was added drop-wisely at −20 °C. The reaction mixture was stirred at −20 °C for 1 h and then 0 °C to room temperature for 1 h. The solvent was removed and the residue was dissolved in CH2Cl2 and washed sequentially with HCl (5%), NaHCO3, and water. The organic phase was concentrated by rotavap, co-evaporated with toluene, and dried under vacuum for overnight. The product was dissolved in CH3CN (20 mL) and Bu4NNO2 (0.57 g, 2.00 mmol) was added. The reaction was stirred at room temperature for 1 h and at 55 °C for 4 h. The solvent was removed. The residue was dissolved in CH2Cl2 and washed with a saturated brine solution. The crude product was dried and purified using silica gel flash column chromatography (Hexanes:EtOAc = 4:1, by volume) to yield 23 (0.36 g, 60%). 1H NMR (600 MHz, CDCl3) δ 8.11–8.05 (m, 4H), 7.99–7.93 (m, 4H), 7.67–7.60 (m, 2H), 7.56–7.45 (m, 6H), 7.41–7.34 (m, 4H), 6.64 (d, J = 6.0 Hz, 1H), 5.77–5.73 (m, 2H), 4.75–4.71 (m, 1H), 4.65–4.61 (m, 1H), 4.56 (t, J = 6.0 Hz, 1H), 4.45–4.41 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 166.22, 165.85, 165.44, 165.42, 165.15, 165.12, 164.15, 163.80, 134.02, 133.97, 133.95, 133.90, 133.65, 133.60, 133.59, 133.22, 133. 18, 130.17, 130.01, 129.97, 129.89, 129.84, 129.72, 129.71, 129.60, 129.25, 129.06, 128.92, 128.89, 128.85, 128.76, 128.65, 128.50, 128.46, 128.39, 128.38, 94.40, 92.00, 71.33, 69.50, 68.87, 68.20, 67.74, 67.65, 67.01, 62.99, 62.50.
To a solution of 23 (0.36 g, 0.60 mmol) in anhydrous CH2Cl2 (15 mL) in a Teflon flask, DAST (0.59 mL, 4.83 mmol) was slowly added at −40 °C. The reaction was stirred for 3 days at room temperature. After cooling down the reaction mixture to −20 °C, MeOH (1 mL) was added and the solvent was removed under reduced pressure. The residue was diluted with CH2Cl2, washed with water for 3 times, dried over MgSO4, and concentrated. Silica gel flash column purification (Hexanes:EtOAc = 8:2, by volume) yielded fluorinated product (0.23 g, 65%). The fluorinated product (0.27 g, 0.49 mmol) was dissolved in a solution of KOH in MeOH (10 mL, 0.1 M) and stirred at room temperature for 1 h. The mixture was neutralized using Dowex (H+) resin. The resulted suspension was filtrated and concentrated. Silica gel column chromatography (EtOAc:MeOH:H2O = 10:1:0.2) of the crude product afforded product 5a (70 mg, 85%). 1H NMR (600 MHz, D2O) δ 5.16 (s, 1H), 4.59 (t, J = 9.6 Hz, 0.4H), 4.51 (t, J = 10.2 Hz, 0.6H), 4.12–3.58 (m, 5H); 13C NMR (150 MHz, D2O) δ 93.89, 88.80, 87.63, 71.20 (d, J = 9.06 Hz), 69.71 (d, J = 24.16 Hz), 68.55 (d, J = 17.97 Hz), 60.25.
4.2.6. 4-O-Methyl-d-mannopyranoside (4-O-Me-Man, 6a)
Compound 22 (0.40 g, 0.67 mmol) and Ag2O (1.55 g, 6.7 mmol) were dissolved in anhydrous CH2Cl2 (15 mL) containing 4 Å molecular sieves (0.7 g). CH3I (0.42 mL, 6.7 mmol) was added drop-wisely at 0 °C and the suspension was stirred for 48 h at room temperature. The mixture was filtered over Celite and concentrated. Silica gel column purification (Hexanes:EtOAc = 7:3, by volume) yielded the methylated product (0.37 g, 90%). 1H NMR (600 MHz, CDCl3) δ 8.15–7.34 (m, 20H), 6.59 (d, J = 1.8 Hz, 1H), 6.00 (t, J = 9.6 Hz, 1H), 5.86–5.85 (m, 1H), 4.70 (dd, J = 12.0 and 1.8 Hz, 1H), 4.43 (dd, J = 12.0, 4.2 Hz, 2H), 4.14 (dd, J = 9.6, 3.0 Hz, 1H), 3.47 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 166.36, 165.63, 165.54, 164.06, 134.22, 133.73, 133.66, 133.23, 130.28, 130.25, 130.21, 130.19, 130.09, 130.03, 130.01, 129.00, 128.77, 128.75, 128.73, 128.53, 92.02, 77.60, 71.40, 68.20, 67.90, 63.20, 58.50.
The product obtained above (0.37 g, 0.60 mmol) was dissolved in dry MeOH (10 mL) containing a catalytic amount of NaOMe. The mixture was stirred at room temperature for 6 h and neutralized using Dowex (H+) resin. The resulting suspension was filtrated and concentrated. Silica gel column purification (EtOAc:MeOH:H2O = 10:1:0.5, by volume) afforded product 6a (82 mg, 70%). 1H NMR (600 MHz, D2O) δ 5.20 (d, J = 1.8 Hz and 0.6H), 5.14 (d, J = 1.2 Hz, 0.4H), 4.17–3.66 (m, 5H), 3.53–3.40 (m, 4H); 13C NMR (150 MHz, D2O) δ 94.17, 79.87, 77.12, 72.53, 71.57, 71.06, 70.23, 66.48, 65.91, 61.08, 56.36.
4.2.7. 4-Deoxy-d-mannopyranoside (4-Deoxy-Man, 7a)
Compound 22 (0.25 g, 0.42 mmol) and 1,1-thiocarbonyldiimidazole (0.15 g, 0.84 mmol) were dissolved in 1,2-dichloroethane (30 mL). The mixture was refluxed at 90 °C for 24 h. The solvent was removed. The concentrated residue was co-evaporated with toluene and dried under vacuum. To the dry compound was added azobisisobutyronitrile (AIBN) (10 mg, 0.08 mmol), Bu3SnH (0.68 mL, 2.52 mmol), and toluene (60 mL). The resulting mixture was refluxed at 110 °C for 5 h. After removing the solvent under reduced pressure, silica gel column purification (Hexanes:EtOAc = 8:2, by volume) provided the deoxygenated product (0.23 g, 98%). 1H NMR (600 MHz, CDCl3) δ 8.15–8.08 (m, 6H), 7.92 (dd, J = 8.4 and 1.2 Hz, 2H), 7.66–7.35 (m, 12H), 6.63 (d, J = 1.8 Hz, 1H), 5.88–5.85 (m, 1H), 5.70 (s, 1H), 4.54 (s, 3H), 2.46 (dd, J = 24.0, 12.0 Hz, 1H), 2.32–2.26 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 166.36, 165.74, 165.38, 164.12, 133.99, 133.67, 133.47, 133.33, 130.15, 130.01, 129.84, 129.52, 128.83, 128.74, 128.55, 92.40, 68.93, 67.42, 67.35, 65.94, 28.34.
The compound obtained above (0.23 g, 0.39 mmol) was dissolved in dry MeOH (15 mL) containing a catalytic amount of NaOMe. The mixture was stirred at room temperature for overnight and neutralized using Dowex (H+) resin. The resulting suspension was filtrated and concentrated. Silica gel column purification (EtOAc:MeOH:H2O = 8:1:0.3, by volume) afforded product 7a (58 mg, 90%). 1H NMR (600 MHz, D2O) δ 5.19 (s, 0.5H), 4.73 (s, 0.5H), 4.09–3.59 (m, 5H), 1.69–1.46 (m, 2H); 13C NMR (150 MHz, D2O) δ 94.77, 94.12, 72.79, 70.02, 69.02, 68.82, 68.16, 64.85, 64.31, 64.01, 29.20, 28.61.
4.2.8. 4-Azido-4-deoxy-d-mannopyranoside (4-N3-Man, 8a)
To a solution of compound 23 (0.42 g, 0.70 mmol) in CH2Cl2 (15 mL) and pyridine (0.56 mL, 7.0 mmol), trifluoromethanesulfonic acid anhydride (Tf2O) (0.23 mL, 1.4 mmol) was added drop-wisely at −20 °C. The reaction mixture was stirred at −20 °C for 1 h and at 0 °C to room temperature for 1 h. The solvent was removed. The residue was dissolved in CH2Cl2 and washed sequentially with HCl (5%), NaHCO3, and water. The organic phase was concentrated by rotavap, co-evaporated with toluene, and dried under vacuum for overnight. A mixture containing the crude product and Bu4NN3 (0.24 g, 1.05 mmol) in anhydrous toluene (15 mL) was heated at 70 °C for 1 h and at 100 °C for 1.5 h. The mixture was cooled down to room temperature, concentrated, and purified by silica gel flash column chromatography (EtOAc:Hexanes = 2:8, by volume) to afford azido-modified product (0.33 g, 76%). 1H NMR (600 MHz, CDCl3) δ 8.15–8.13 (m, 4H), 8.01–7.97 (m, 4H), 7.67–7.53 (m, 6H), 7.47 (t, J = 7.8 Hz, 2H), 7.41–7.36 (m, 4H), 6.57 (s, 1H), 5.86–5.83 (m, 2H), 4.77 (dd, J = 12.6 and 6.6 Hz, 1H), 4.68 (dd, J = 12.0 and 1.8 Hz, 1H), 4.43 (t, J = 10.2 Hz, 1H), 4.15 (dd, J = 10.2 and 2.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 166.05, 165.44, 164.89, 163.76, 134.12, 133.71, 133.66, 133.31, 130.10, 129.83, 129.80, 128.95, 128.83, 128.65, 128.57, 128.55, 91.24, 71.52, 71.36, 68.34, 62.83, 57.19.
The compound obtained above (0.28 g, 0.45 mmol) was dissolved in a solution of KOH in dry MeOH (0.1 M, 15 mL). The mixture was stirred at room temperature for 2 h and neutralized using Dowex (H+) resin. The resulting suspension was filtrated and concentrated. Silica gel column purification (EtOAc:MeOH:H2O = 10:1:0.2, by volume) afforded product 8a (78 mg, 85%). 1H NMR (600 MHz, D2O) δ 5.18 (s, 1H), 3.95–3.72 (m, 5H), 3.63 (t, J = 10.2 Hz, 0.65H), 3.54 (t, J = 10.2 Hz, 0.35H); 13C NMR (150 MHz, D2O) δ 94.02, 74.43, 72.41, 70.62, 69.93, 69.44, 61.11, 59.03.
4.3. Enzymatic synthesis of sialosides 1b–8b and 1c–8c
4.3.1. General procedure for one-pot three-enzyme preparative-scale synthesis of α2–3- and α2–6-linked sialosides
GalβpNP (1.0 eq, 10 mM, 0.1 mmol), a sialic acid precursor (1.5 eq, 15 mM, 0.15 mmol) chosen from 1a–8a, sodium pyruvate (10.0 eq, 100 mM, 1 mmol), and CTP (2.0 eq, 20 mM, 0.2 mmol) were dissolved in water in a 50 mL centrifuge tube containing Tris-HCl buffer (100 mM, pH 8.5) and MgCl2 (20 mM). After adding appropriate amounts of Pasteurella multocida sialic acid aldolase30 (PmNanA, 0.7–1.1 mg), N. meningitides CMP-sialic acid synthetase31 (NmCSS, 1.0–1.3 mg), and a sialyltransferase (PmST16, 0.1–0.2 mg or Pd2,6ST7, 0.3–0.5 mg), water was added to bring the volume of the reaction mixture to 10 mL. The reaction was incubated at 37 °C in an isotherm incubator with agitating at 120 rpm for 3–16 h using PmST1 or 16–48 h using Pd2,6ST. The product formation was monitored by TLC using EtOAc:MeOH:H2O:AcOH = 7:2:0.5:0.1 (by volume) as the developing solvent and stained with p-anisaldehyde sugar stain solution. The reaction was terminated by adding the same volume (10 mL) of ice-cold EtOH followed by incubation at 4 °C for 30 min. The mixture was centrifuged to remove precipitates. The supernatant was concentrated and passed through a Bio-gel P-2 gel filtration column with water as the eluant to obtain the desired product. Silica gel column purification (EtOAc:MeOH:H2O = 7:2:0.6, by volume) was used for further purification.
4.3.1. 4-Nitrophenyl O-(7-fluoro-5-acetamido-3,5,7-trideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-β-d-galactopyranoside (Neu5Ac7Fα2–3GalβpNP, 1b)
Yield, 98%; white solid. 1H NMR (600 MHz, D2O) δ 8.26 (dd, J = 7.8 and 2.4 Hz, 2H), 7.24 (dd, J = 7.2 and 2.4 Hz, 2H), 5.30 (d, J = 7.8 Hz, 1H), 4.49 (dd, J = 47.4 and 9.0 Hz, 2H), 4.22 (dd, J = 9.6 and 3.0 Hz, 1H), 4.09–4.05 (m, 2H), 3.94–3.64 (m, 9H), 2.77 (dd, J = 12.6 and 4.8 Hz, 1H), 2.03 (s, 3H), 1.86 (t, J = 12.6 Hz, 1H); 13C NMR (150 MHz, D2O) δ 174.98, 173.65, 161.93, 142.69, 126.31, 116.65, 100.50, 99.89, 88.75 (d, J = 179.08 Hz), 75.99, 75.65, 71.79, 69.27, 68.99, 68.32, 67.70, 62.16, 60.81, 51.38, 39.59, 22.39; HRMS (ESI) calculated for C23H30FN2O15− (M-H) 593.1636, found 593.1659.
4.3.2. 4-Nitrophenyl O-(5-acetamido-3,5-dideoxy-7-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-β-d-galactopyranoside (Neu5Ac7OMeα2–3GalβpNP, 2b)
Yield, 95%; white solid. 1H NMR (600 MHz, D2O) δ 8.14 (d, J = 9.6 Hz, 2H), 7.12 (d, J = 9.6 Hz, 2H), 5.15 (d, J = 7.8 Hz, 1H), 4.06 (dd, J = 9.6 and 3.6 Hz, 1H), 3.89 (d, J = 3.0 Hz, 1H), 3.79–3.45 (m, 10H), 3.30 (dd, J = 10.2 and 1.8 Hz, 1H), 3.25 (s, 3H), 2.61 (dd, J = 12.0 and 4.8 Hz, 1H), 1.90 (s, 3H), 1.69 (t, J = 12.0 Hz, 1H); 13C NMR (150 MHz, D2O) δ 174.52, 173.91, 161.98, 142.75, 126.35, 116.72, 100.34, 99.93, 78.29, 75.88, 75.69, 72.95, 71.51, 69.43, 69.06, 67.58, 62.43, 60.94, 60.35, 51.78, 39.77, 22.52; HRMS (ESI) calculated for C24H33N2O16− (M-H) 605.1836, found 605.1867.
4.3.3. 4-Nitrophenyl O-(5-acetamido-3,5,7-trideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-β-d-galactopyranoside (Neu5Ac7Deoxyα2–3GalβpNP, 3b)
Yield, 80%; white solid. 1H NMR (600 MHz, D2O) δ 8.26 (dd, J = 12.6 and 3.6 Hz, 2H), 7.25 (dd, J = 10.2 and 2.4 Hz, 2H), 5.30 (d, J = 7.8 Hz, 1H), 4.23 (dd, J = 10.2 and 3.6 Hz, 1H), 4.05–3.86 (m, 4H), 3.75 (d, J = 6.0 Hz, 4H), 3.71 (d, J = 6.0 Hz, 2H), 3.47 (dd, J = 12.0 and 6.6 Hz, 1H), 2.75 (dd, J = 12.6 and 4.2 Hz, 1H), 2.01 (s, 3H), 1.82 (t, J = 11.4 Hz, 1H), 1.68–1.59 (m, 2H); 13C NMR (150 MHz, D2O) δ 175.02, 174.61, 161.94, 142.73, 126.28, 116.62, 100.22, 99.84, 75.79, 75.63, 71.18, 68.95, 68.37, 68.26, 67.66, 65.44, 60.71, 55.80, 39.76, 34.72, 22.30; HRMS (ESI) calculated for C23H31N2O15− (M-H) 575.1730, found 575.1761.
4.3.4. 4-Nitrophenyl O-(7-azido-5-acetamido-3,5,7-trideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-β-d-galactopyranoside (Neu5Ac7N3α2–3GalβpNP, 4b)
Yield, 91%; white solid. 1H NMR (600 MHz, D2O) δ 8.12 (dd, J = 9.0, 1.8 Hz, 2H), 7.10 (dd, J = 9.0 and 1.8 Hz, 2H), 5.15 (d, J = 7.8 Hz, 1H), 4.04 (dd, J = 10.2 and 3.6 Hz, 1H), 3.88 (d, J = 3.6 Hz, 1H), 3.86–3.83 (m, 1H), 3.80–3.72 (m, 4H), 3.69 (dd, J = 10.2 and 1.8 Hz, 1H), 3.60 (d, J = 6.6 Hz, 2H), 3.56 (s, 2H), 3.34 (dd, J = 9.0 and 1.8 Hz, 1H), 2.61 (dd, J = 12.6 and 4.2 Hz, 1H), 1.89 (s, 3H), 1.73 (t, J = 12.6 Hz, 1H); 13C NMR (150 MHz, D2O) δ 174.42, 173.49, 161.66, 142.44, 126.03, 116.39, 100.39, 99.58, 75.55, 75.35, 71.98, 70.78, 68.71, 68.23, 67.57, 62.40, 60.74, 60.57, 52.36, 39.14, 22.09; HRMS (ESI) calculated for C23H30N5O15− (M-H) 616.1744, found 616.1780.
4.3.5. 4-Nitrophenyl O-(3,7-dideoxy-7-fluoro-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-β-d-galactopyranoside (Kdn7Fα2–3GalβpNP, 5b)
Yield, 98%; white solid. 1H NMR (600 MHz, D2O) δ 8.25 (d, J = 9.0 Hz, 2H), 7.24 (d, J = 9.0 Hz, 2H), 5.29 (d, J = 7.8 Hz, 1H), 4.20 (dd, J = 9.6 and 3.0 Hz, 1H), 4.12–4.08 (m, 1H), 4.04 (d, J = 3.0 Hz, 1H), 3.93–3.84 (m, 3H), 3.76 (d, J = 6.6 Hz, 3H), 3.73–3.62 (m, 4H), 3.58 (t, J = 9.0 Hz, 1H), 2.74 (dd, J = 12.6 and 4.8 Hz, 1H), 1.82 (t, J = 11.4 Hz, 1H); 13C NMR (150 MHz, D2O) δ 173.60, 161.69, 142.46, 126.09, 116.44, 100.29, 99.71, 88.43 (d, J = 179.69 Hz), 75.74, 75.39, 73.05 (d, J = 17.96 Hz), 69.85, 69.34, 69.09 (d, J = 27.18 Hz), 68.77, 67.46, 61.94, 60.59, 39.05; (ESI) calculated for C21H27NO15F− (M-H) 552.1370, found 552.1391.
4.3.6. 4-Nitrophenyl O-(3-deoxy-7-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-β-d-galactopyranoside (Kdn7OMeα2–3GalβpNP, 6b)
Yield, 89%; white solid. 1H NMR (600 MHz, D2O) δ 8.26 (dd, J = 7.2 and 2.4 Hz, 2H), 7.24 (dd, J = 7.2 and 1.8 Hz, 2H), 5.28 (d, J = 7.8 Hz, 1H), 4.18 (dd, J = 9.6 and 3.0 Hz, 1H), 4.01 (d, J = 3.0 Hz, 1H), 3.92–3.84 (m, 4H), 3.75 (d, J = 6.0 Hz, 2H), 3.72 (s, 2H), 3.67 (dd, J = 12.0, 6.0 Hz, 1H), 3.63 (dd, J = 10.2 and 1.8 Hz, 1H), 3.60–3.57 (m, 1H), 3.52 (s, 3H), 2.72 (dd, J = 12.0 and 4.8 Hz, 1H), 1.78 (t, J = 12.0 Hz, 1H); 13C NMR (150 MHz, D2O) δ 173.75, 161.62, 142.46, 126.02, 116.37, 99.95, 99.56, 77.78, 75.50, 75.38, 74.01, 71.17, 70.42, 69.94, 68.72, 67.21, 62.13, 60.60, 60.21, 39.13; HRMS (ESI) calculated for C22H30NO16− (M-H) 564.1570, found 564.1594.
4.3.7. 4-Nitrophenyl O-(3,7-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-β-d-galactopyranoside (Kdn7Deoxyα2–3GalβpNP, 7b)
Yield, 90%; white solid. 1H NMR (600 MHz, D2O) δ 8.26 (dd, J = 8.4, 1.2 Hz, 2H), 7.25 (dd, J = 8.4 and 1.2 Hz, 2H), 5.29 (d, J = 7.8 Hz, 1H), 4.20 (dd, J = 7.2 and 2.4 Hz, 1H), 4.03 (s, 2H), 3.92–3.52 (m, 8H), 3.23 (t, J = 10.2 Hz, 1H), 2.71 (dd, J = 12.6 and 4.8 Hz, 1H), 1.92 (t, J = 12.0 Hz, 1H), 1.78 (t, J = 12.6 Hz, 1H), 1.66 (t, J = 11.4 Hz, 1H); 13C NMR (150 MHz, D2O) δ 174.14, 161.92, 142.73, 126.28, 116.61, 100.21, 99.88, 75.66 (2C), 74.45, 72.26, 69.24, 68.99, 68.51, 67.61, 65.48, 60.77, 39.58, 34.71; HRMS (ESI) calculated for C21H28NO15− (M-H) 534.1464, found 534.1476.
4.3.8. 4-Nitrophenyl O-(7-azido-3,7-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-β-d-galactopyranoside (Kdn7N3α2–3GalβpNP, 8b)
Yield, 98%; white solid. 1H NMR (600 MHz, D2O) δ 8.27 (dd, J = 9.6 and 2.4 Hz, 2H), 7.25 (dd, J = 9.0 and 1.8 Hz, 2H), 5.29 (d, J = 7.8 Hz, 1H), 4.17 (dd, J = 9.6 and 3.0 Hz, 1H), 4.05–4.02 (m, 2H), 3.93–3.87 (m, 3H), 3.76–3.73 (m, 4H), 3.67 (dd, J = 9.6 and 2.4 Hz, 1H), 3.65–3.59 (m, 2H), 2.72 (dd, J = 12.6 and 4.8 Hz, 1H), 1.87 (t, J = 12.6 Hz, 1H); 13C NMR (150 MHz, D2O) δ 173.85, 161.76, 142.50, 126.17, 116.54, 100.73, 99.73, 75.52, 75.39, 73.77, 70.74, 70.59, 70.09, 68.79, 67.83, 62.56, 60.67, 59.80, 38.66; HRMS (ESI) calculated for C21H27N4O15− (M-H) 575.1478, found 575.1505.
4.3.9. 4-Nitrophenyl O-(7-fluoro-5-acetamido-3,5,7-trideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→6)-O-β-d-galactopyranoside (Neu5Ac7Fα2–6GalβpNP, 1c)
Yield, 92%; white solid. 1H NMR (600 MHz, D2O) δ 8.15 (dd, J = 6.6 and 1.8 Hz, 2H), 7.14 (dd, J = 6.6 and 3.0 Hz, 2H), 5.06 (d, J = 7.8 Hz, 1H), 4.34 (dd, J = 47.4 and 8.4 Hz, 1H), 3.92–3.48 (m, 12H), 2.62 (dd, J = 12.6 and 4.8 Hz, 1H), 1.89 (s, 3H), 1.54 (t, J = 12.0 Hz, 1H); 13C NMR (150 MHz, D2O) δ 174.88, 173.28, 162.06, 142.76, 126.35, 116.75, 101.02, 100.09, 88.36 (d, J = 180.89 Hz), 74.19, 72.57, 71.73 (d, J = 16.31 Hz), 70.55, 69.22 (d, J = 28.69 Hz), 68.63, 68.39, 63.57, 62.13, 51.49, 40.57, 22.42; HRMS (ESI) calculated for C23H30FN2O15− (M-H) 593.1636, found 593.1665.
4.3.10. 4-Nitrophenyl O-(5-acetamido-3,5-dideoxy-7-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→6)-O-β-d-galactopyranoside (Neu5Ac7OMeα2–6GalβpNP, 2c)
Yield, 57%; white solid. 1H NMR (600 MHz, D2O) δ 8.29 (d, J = 7.2 Hz, 2H), 7.26 (d, J = 7.2 Hz, 2H), 5.18 (d, J = 7.2 Hz, 1H), 4.01 (s, 2H), 3.93–3.58 (m, 10H), 3.39 (d, J = 7.8 Hz, 1H), 3.33 (s, 3H), 2.71 (dd, J = 11.0 and 1.8 Hz, 1H), 2.02 (s, 3H), 1.66 (t, J = 12.0 Hz, 1H); 13C NMR (150 MHz, D2O) δ 174.48, 173.37, 162.06, 142.77, 126.35, 116.61, 100.41, 100.04, 78.26, 74.19, 72.53, 72.25, 71.58, 70.47, 69.17, 68.58, 63.23, 62.67 (2C), 51.88, 40.12, 22.43; HRMS (ESI) calculated for C24H33N2O16−(M-H) 605.1836, found 605.1869.
4.3.11. 4-Nitrophenyl O-(5-acetamido-3,5,7-trideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→6)-O-β-d-galactopyranoside (Neu5Ac7Deoxyα2–6GalβpNP, 3c)
Yield, 88%; white solid. 1H NMR (600 MHz, D2O) δ 8.29 (d, J = 9.0 Hz, 2H), 7.27 (d, J = 9.0 Hz, 2H), 5.18 (d, J = 7.8 Hz, 1H), 4.02 (d, J = 3.6 Hz, 1H), 3.99–3.89 (m, 2H), 3.84 (t, J = 7.8 Hz, 1H), 3.78 (dd, J = 9.6 and 3.0 Hz, 1H), 3.71–3.68 (m, 3H), 3.60–3.56 (m, 1H), 3.52 (dd, J = 12.0 and 3.0 Hz, 1H), 3.47 (t, J = 9.6 Hz, 1H), 3.40 (dd, J = 11.4 and 7.2 Hz, 1H), 2.73 (dd, J = 12.6 and 4.8 Hz, 1H), 1.98 (s, 3H), 1.64 (t, J = 12.0 Hz, 1H), 1.59–1.54 (m, 1H), 1.36–1.30 (m, 1H); 13C NMR (150 MHz, D2O) δ 174.59, 173.48, 161.72, 142.42, 125.97, 116.48, 100.27, 99.58, 73.99, 72.27, 70.65, 70.24, 68.37, 68.08, 67.84, 65.13, 55.75, 40.46, 34.33, 21.99; HRMS (ESI) calculated for C23H31N2O15−(M-H) 575.1730, found 575.1759
4.3.12. 4-Nitrophenyl O-(7-azido-5-acetamido-3,5,7-trideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→6)-O-β-d-galactopyranoside (Neu5Ac7N3α2–6GalβpNP, 4c)
Yield, 98%; white solid. 1H NMR (600 MHz, D2O) δ 8.29 (d, J = 9.0 Hz, 2H), 7.26 (d, J = 9.6 Hz, 2H), 5.18 (d, J = 7.8 Hz, 1H), 4.02 (d, J = 3.0 Hz, 1H), 3.99 (dd, J = 7.2 and 4.2 Hz, 1H), 3.96–3.92 (m, 1H), 3.89–3.83 (m, 4H), 3.77 (dd, J = 9.6 and 3.6 Hz, 1H), 3.72 (s, 1H), 3.68–3.60 (m, 3H), 3.50 (dd, J = 9.0 and 1.8 Hz, 1H), 2.73 (dd, J = 12.0 and 4.2 Hz, 1H), 2.02 (s, 3H), 1.68 (t, J = 12.0 Hz, 1H); 13C NMR (150 MHz, D2O) δ 174.33, 173.18, 161.78, 142.43, 126.05, 116.44, 100.56, 99.78, 73.93, 72.27, 71.70, 70.97, 70.23, 68.30, 68.16, 63.09, 62.38, 61.03, 52.50, 40.04, 22.08; HRMS (ESI) calculated for C23H30N5O15− (M-H) 616.1744, found 616.1775.
4.3.13. 4-Nitrophenyl O-(3,9-dideoxy-7-fluoro-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→6)-O-β-d-galactopyranoside (Kdn7Fα2–6GalβpNP, 5c)
Yield, 95%; white solid. 1H NMR (600 MHz, D2O) δ 8.26 (dd, J = 7.2 and 1.8 Hz, 2H), 7.25 (dd, J = 7.2 and 2.4 Hz, 2H), 5.18 (d, J = 7.8 Hz, 1H), 4.06–3.58 (m, 12H), 3.48 (t, J = 10.2 Hz, 1H), 2.71 (dd, J = 12.0 and 4.8 Hz, 1H), 1.63 (t, J = 12.6 Hz, 1H); 13C NMR (150 MHz, D2O) δ 173.10, 161.78, 142.50, 126.09, 116.49, 100.77, 99.82, 88.50 (d, J = 179.2 Hz), 73.96, 72.87 (d, J = 17.9 Hz), 72.32, 70.29, 69.84, 69.49 (d, J = 4.9 Hz), 69.11 (d, J = 27.6 Hz), 68.39, 63.36, 61.90, 40.01; HRMS (ESI) calculated for C21H27NO15F− (M-H) 552.1370, found 552.1386.
4.3.14. 4-Nitrophenyl O-(3-deoxy-7-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→6)-O-β-d-galactopyranoside (Kdn7OMeα2–6GalβpNP, 6c)
Yield, 53%; white solid. 1H NMR (600 MHz, D2O) δ 8.28 (dd, J = 7.2 and 2.4 Hz, 2H), 7.25 (dd, J = 7.2 and 2.4 Hz, 2H), 5.17 (d, J = 7.2 and 2.4 Hz, 1H), 3.99–3.98 (m, 1H), 3.89 (t, J = 10.2 Hz, 1H), 3.86–3.82 (m, 2H), 3.77 (dd, J = 10.2 and 4.8 Hz, 1H), 3.71 (s, 3H), 3.68–3.53 (m, 5H), 3.44 (s, 2H), 3.40 (t, J = 10.2 Hz, 1H), 2.69 (dd, J = 12.6 and 4.8 Hz, 1H), 1.59 (t, J = 12.6 Hz, 1H); 13C NMR (150 MHz, D2O) δ 173.48, 161.77, 142.48, 126.06, 116.32, 100.21, 99.75, 77.77, 73.94, 73.67, 72.25, 71.33, 70.35, 70.20, 70.08, 68.32, 62.95, 61.34, 60.20, 39.77, HRMS (ESI) calculated for C22H30NO16− (M-H) 564.1570, found 564.1585.
4.3.15. 4-Nitrophenyl O-(3,7-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→6)-O-β-d-galactopyranoside (Kdn7Deoxyα2–6GalβpNP, 7c)
Yield, 58%; white solid. 1H NMR (600 MHz, D2O) δ 8.29 (d, J = 9.6 Hz, 2H), 7.27 (d, J = 9.0 Hz, 2H), 5.18 (d, J = 7.8 Hz, 1H), 4.01 (d, J = 3.6 Hz, 1H), 3.99–3.97 (m, 2H), 3.88 (t, J = 10.8 Hz, 1H), 3.84 (t, J = 10.2, 1H), 3.77 (dd, J = 10.2 and 3.6 Hz, 1H), 3.70–3.65 (m, 1H), 3.60–3.52 (m, 2H), 3.46 (dd, J = 11.4 and 6.6 Hz, 1H), 3.31 (t, J = 18.0 Hz, 1H), 3.16 (d, J = 18.0 Hz, 1H), 3.11 (t, J = 9.6 Hz, 1H), 2.69 (dd, J = 12.6 and 4.8 Hz, 1H), 1.87–1.83 (m, 1H), 1.61 (t, 1H), 1.45–1.42 (m, 1H); 13C NMR (150 MHz, D2O)δ 173.53, 161.73, 142.42, 125.99, 116.47, 100.34, 99.65, 74.54, 73.95, 72.24, 71.65, 70.24, 69.56, 68.35, 68.21, 65.21, 63.17, 40.11, 34.40; HRMS (ESI) calculated for C21H28NO15− (M-H) 534.1464, found 534.1471.
4.3.16. 4-Nitrophenyl O-(7-azido-3,7-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→6)-O-β-d-galactopyranoside (Kdn7N3α2–6GalβpNP, 8c)
Yield, 89%; white solid. 1H NMR (600 MHz, D2O) δ 8.28 (d, J = 9.0 Hz, 2H), 7.25 (d, J = 9.0 Hz, 2H), 5.18 (d, J = 7.2 Hz, 1H), 4.01 (d, J = 3.0 Hz, 1H), 3.99–3.93 (m, 2H), 3.91–3.77 (m, 5H), 3.74–3.66 (m, 3H), 3.62–3.57 (m, 1H), 3.49 (t, J = 9.0 Hz, 1H), 2.71 (dd, J = 12.6 and 4.2 Hz, 1H), 1.66 (t, J = 12.0 Hz, 1H); 13C NMR (150 MHz, D2O) δ 173.32, 161.75, 142.41, 126.01, 116.42, 100.56, 99.72, 73.93, 73.33, 72.24, 70.86, 70.72, 70.21, 69.84, 68.29, 63.08, 62.41, 60.14, 39.68; HRMS (ESI) calculated for C21H27N4O15− (M-H) 575.1478, found 575.1498.
4.4. Sialidase substrate specificity assays
4.4.1. Materials
Clostridium perfringens sialidase (type VI), Vibrio cholerae sialidase (type III), and β-galactosidase from Aspergillus oryzae were purchased from Sigma. Salmonella typhimurium sialidase and Streptococcus pneumoniae sialidase were bought from Prozyme. All of these enzymes were used without further purification. PmSTl6 and NEU214 were expressed in Escherichia coli and purified as described previously. Sodium pyruvate (Fisher Biotech), N-acetylmannosamine (ManNAc, Sigma), mannose (Acros Organics), para-nitrophenyl β-D-galactopyranoside (GalβpNP, Sigma), and cytidine 5’-triphosphate disodium salt (CTP, Sigma) were from commercially available sources. 384-Well plates for sialidase assays were from Fisher Biotech.
4.4.2. Assays
All sialidase assays were carried out at 37 °C in duplicate in 384-well plates in a final volume of 20 µL containing a sialoside substrate (0.3 mM) and the β-galactosidase (12 µg, 126 mU). The amount of the β-galactosidase required to completely hydrolyze GalβpNP (0.3 mM) within the time frame of the assay was predetermined. The assay conditions for various sialidases were as follows: Salmonella typhimurium sialidase (6 mU) in sodium acetate buffer (100 mM, pH 5.5) containing NaCl (100 mM); Clostridium perfringens sialidase (4 mU) in MES buffer (100 mM, pH 5.0); Vibrio cholerae sialidase (2.8 mU) in sodium acetate buffer (100 mM, pH 5.5) containing NaCl (150 mM) and CaCl2 (10 mM); Streptococcus pneumoniae sialidase (0.8 mU) in sodium acetate buffer (100 mM, pH 6.0); PmST1 (25 µg) in sodium acetate buffer (100 mM, pH 5.5); NEU2 (18 µg) in MES buffer (100 mM, pH 5.0); NanH2 (10 ng) in sodium acetate buffer (100 mM, pH 5.0). The reactions were stopped at 60 min by adding 40 µL of N-cyclohexyl-3-aminopropane sulfonic acid (CAPS) buffer (0.5 M, pH 10.5). The amount of para-nitrophenolate formed was determined by measuring the A405nm of the reaction mixtures using a microplate reader. Reactions of GalβpNP and an excess amount of β-galactosidase were used as controls.
Supplementary Material
Highlights.
Convenient and efficient one-pot three-enzyme chemoenzymatic approaches for synthesizing sialosides containing C7-modified sialic acids which have not been synthesized previously.
Novel probes for detecting the substrate specificity of sialidases.
Sialosides containing C7-deoxy Neu5Ac have been found to be selective substrates for all bacterial sialidases tested but not for human NEU2.
Information obtained from sialidase substrate specificity studies can be used for guide the design of inhibitors that are selectively against bacterial sialidases.
Acknowledgements
This work was supported byNIH grant R01HD065122, NSF grant CHE-1012511, and Beckman Young Investigator Award. X.C. is a Camille Dreyfus Teacher-Scholar and a UC-Davis Chancellor’s Fellow. The authors thank the organizers of the EuroCarb17 especially Professor Timor Baasov.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data: 1H and 13C spectra of sialosides 1b–8b and 1c–8c.
References
- 1.Yu H, Chen X. Org. Biomol. Chem. 2007;5:865–872. doi: 10.1039/b700034k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angata T, Varki A. Chem. Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 3.Schauer R. Glycoconj. J. 2000;17:485–499. doi: 10.1023/A:1011062223612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen X, Varki A. ACS Chem. Biol. 2010;5:163–176. doi: 10.1021/cb900266r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu H, Chokhawala HA, Huang S, Chen X. Nat. Protoc. 2006;1:2485–2492. doi: 10.1038/nprot.2006.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X. J. Am. Chem. Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 7.Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X. Angew. Chem. Int. Ed. 2006;45:3938–3944. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu H, Cheng J, Ding L, Khedri Z, Chen Y, Chin S, Lau K, Tiwari VK, Chen X. J. Am. Chem. Soc. 2009;131:18467–18477. doi: 10.1021/ja907750r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chokhawala HA, Yu H, Chen X. Chembiochem. 2007;8:194–201. doi: 10.1002/cbic.200600410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao H, Li Y, Lau K, Muthana S, Yu H, Cheng J, Chokhawala HA, Sugiarto G, Zhang L, Chen X. Org. Biomol. Chem. 2009;7:5137–5145. doi: 10.1039/b916305k. [DOI] [PubMed] [Google Scholar]
- 11.Khedri Z, Muthana MM, Li Y, Muthana SM, Yu H, Cao H, Chen X. Chem. Commun. 2012;48:3357–3359. doi: 10.1039/c2cc17393j. [DOI] [PubMed] [Google Scholar]
- 12.Yu H, Cao H, Tiwari VK, Li Y, Chen X. Bioorg. Med. Chem. Lett. 2011;21:5037–5040. doi: 10.1016/j.bmcl.2011.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Cao H, Dao N, Luo Z, Yu H, Chen Y, Xing Z, Baumgarth N, Cardona C, Chen X. Virology. 2011;415:12–19. doi: 10.1016/j.virol.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Cao H, Yu H, Chen Y, Lau K, Qu J, Thon V, Sugiarto G, Chen X. Mol. BioSyst. 2011;7:1060–1072. doi: 10.1039/c0mb00244e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khedri Z, Li Y, Cao H, Qu J, Yu H, Muthana MM, Chen X. Org. Biomol. Chem. 2012;10:6112–6120. doi: 10.1039/c2ob25335f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartmann M, Christian R, Zbiral E. Liegigs Ann. Chem. 1990;1990:83–91. [Google Scholar]
- 17.Schreiner E, Zbiral CE. Liegigs Ann. Chem. 1990;1990:93–97. [Google Scholar]
- 18.Thomson R, von Itzstein M. Carbohydr. Res. 1995;274:29–44. [Google Scholar]
- 19.Honda T, Masuda T, Yoshida S, Arai M, Kobayashi Y, Yamashita M. Bioorg. Med. Chem. Lett. 2002;12:1921–1924. doi: 10.1016/s0960-894x(02)00328-1. [DOI] [PubMed] [Google Scholar]
- 20.Halcomb RL, Fitz W, Wong CH. Tetrahedron: Asymm. 1994;5:2437–2442. [Google Scholar]
- 21.Calveras J, Nagai Y, Sultana I, Ueda Y, Higashi T, Shoji M, Sugai T. Tedrahedron. 2010;66:4284–4291. [Google Scholar]
- 22.Kong DCM, von Itzstein M. Tetrahedron Lett. 1995;36:957–960. [Google Scholar]
- 23.Honda T, Kubo S, Masuda T, Arai M, Kobayashi Y, Yamashita M. Bioorg. Med. Chem. Lett. 2009;19:2938–2940. doi: 10.1016/j.bmcl.2009.04.067. [DOI] [PubMed] [Google Scholar]
- 24.Hartlieb S, Gunzel A, Gerardy-Schahn R, Munster-Kuhnel AK, Kirschning A, Drager G. Carbohydr. Res. 2008;343:2075–2082. doi: 10.1016/j.carres.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Moller H, Bohrsch V, Bentrop J, Bender J, Hinderlich S, Hackenberger CP. Angew. Chem. Int. Ed. 2012;51:5986–5990. doi: 10.1002/anie.201108809. [DOI] [PubMed] [Google Scholar]
- 26.Hasegawa A, Adachi K, Yoshida M, Kiso M. Carbohydr. Res. 1992;230:273–288. doi: 10.1016/0008-6215(92)84038-t. [DOI] [PubMed] [Google Scholar]
- 27.Sawada N, Ishida H, Collins BE, Schnaar RL, Kiso M. Carbohydr. Res. 1999;316:1–5. doi: 10.1016/s0008-6215(99)00081-6. [DOI] [PubMed] [Google Scholar]
- 28.Lau K, Thon V, Yu H, Ding L, Chen Y, Muthana MM, Wong D, Huang R, Chen X. Chem. Commun. 2010;46:6066–6068. doi: 10.1039/c0cc01381a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watt GM, Flitsch SL, Fey S, Elling L, Kragl U. Tetrahedron: Asymm. 2000;11:621–628. [Google Scholar]
- 30.Li Y, Yu H, Cao H, Lau K, Muthana S, Tiwari VK, Son B, Chen X. Appl. Microbiol. Biotechnol. 2008;79:963–970. doi: 10.1007/s00253-008-1506-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu H, Yu H, Karpel R, Chen X. Bioorg. Med. Chem. 2004;12:6427–6435. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.