

Abstract
Disulfide bridges, which stabilize the native conformation of conotoxins impose a challenge in the synthesis of smaller analogs. In this work, we describe the synthesis of a minimized analog of the analgesic μ-conotoxin KIIIA that blocks two sodium channel subtypes, the neuronal NaV1.2 and skeletal muscle NaV1.4. Three disulfide-deficient analogs of KIIIA were initially synthesized in which the native disulfide bridge formed between either C1-C9, C2-C15 or C4-C16 was removed. Deletion of the first bridge only slightly affected the peptide’s bioactivity. To further minimize this analog, the N-terminal residue was removed and two non-essential Ser residues were replaced by a single 5-amino-3-oxapentanoic acid residue. The resulting “polytide” analog retained the ability to block sodium channels and to produce analgesia. Until now, the peptidomimetic approach applied to conotoxins has progressed only modestly at best; thus, the disulfide-deficient analogs containing backbone spacers provide an alternative advance toward the development of conopeptide-based therapeutics.
Keywords: conopeptide, conotoxin, sodium channels, backbone spacers, disulfide bridges
Introduction
Venoms of Conus snails are a valuable resource of potent and selective ligands for ion channels and receptors. [1-3] Although only a few dozen conotoxins (out of an estimated >50,000 conopeptides) have been characterized in detail thus far, several of them have reached human clinical trials, with one already approved by the FDA to treat neuropathic pain. [4] Indeed, many conotoxins target ion channels in which malfunctioning of these channels has been implicated in diseases of the nervous system. [5-8] While these venom-based peptides provide useful neuropharmacological tools, there is a continuing need for novel strategies to transform them into smaller molecules and thus becoming more amenable for use as therapeutics.
There is an ongoing need for subtype-selective blockers of sodium channels that could be developed into drugs for treatment of neuropathic pain. [9] Small-molecule sodium channel blockers, such as phenytoin, carbamaxepine, lamotrigine, or lidocaine are effective analgesics, but because of their non-selective nature, they also exhibit a range of adverse effects including dizziness, convulsions or anxiety. [10-13] Promising in this regard is the recently reported sodium channel blocker, A-803467, which exhibited high potency and selectivity for human Nav1.8, effectively targeting sensory neurons. [14] Additional examples of subtype selective inhibitors of sodium channels that posses analgesic activities are μO-conotoxin MrVIB that blocks Nav1.8 [15, 16] and ProTx-II that selectively interacts with Nav1.7. [17] Therefore, further development of subtype-selective blockers of sodium channels will not only improve their safety profiles as analgesics, but may also help better understand pain mechanisms at the molecular level.
μ-Conotoxins belong to a family of Conus peptides that are potent and selective blockers of sodium channels. [18-20] All μ-conotoxins share a common disulfide framework with 3 bridges that form 3 intercysteine loops (Fig. 1A). Some of these peptides preferentially block specific subtypes of mammalian sodium channels, including Nav1.4 (e.g., μ-conotoxin GIIIA [21, 22]) or Nav1.2 (e.g., μ-conotoxin KIIIA). [23] Recently, KIIIA and a related peptide, SIIIA, were shown to exhibit very potent analgesic activity in the inflammatory pain assay in mice, following its systemic administration. [23, 24] Thus, this group of conotoxins is an attractive resource for subtype-selective sodium channel ligands. Structure-activity relation (SAR) study of μ-KIIIA identified key non-cysteine amino acid residues that were important for its interactions with the neuronal subtype Nav1.2 and the skeletal muscle subtype Nav1.4. [23] Two residues in the intercysteine loop II (K7 and W8, see Fig. 1A) and three residues in the loop III (R10, H12 and R14) were important for activity. [23] Consistent with the SAR data, the replacement of non-essential Gly-Gly residues in the loop I of μ-SIIIA (Fig. 1A) with non-peptidic backbone spacers did not affect this conotoxin’s ability to block sodium currents in the mouse DRG neurons. [24] We report here further characterization of the structure-function relationships in μ-conotoxins relevant for developing conopeptide-based therapeutics.
Figure 1. Structures of μ-conotoxins.
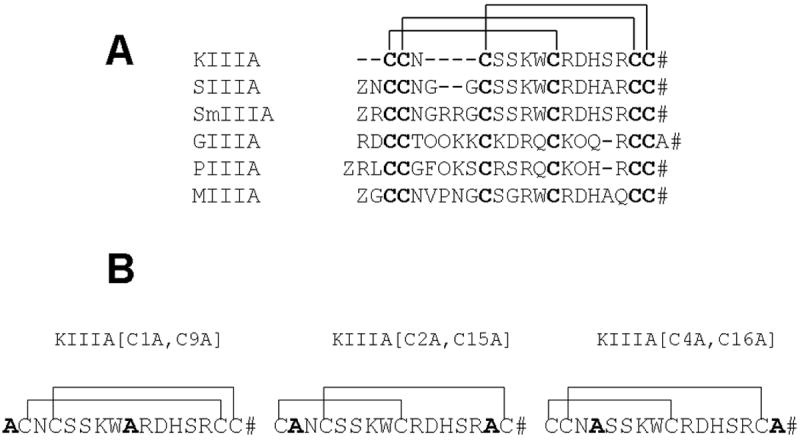
(A) – Alignment of sequences of several representative μ-conotoxins. The Roman numerals label the three intercysteine “loops.” (B) - Structures of the disulfide-deficient analogs of μ-conotoxin KIIIA studied in this report. #, amidated C-terminus; Z, pyroglutamate; O, hydroxyproline.
The remarkable molecular diversity of conotoxins provides a challenge in developing “cono-mimetics”: non-peptidic analogs of conopeptides. Conotoxins possess various disulfide-rich scaffolds, and this, combined with hypervariable amino acid sequences and a variety of posttranslational modifications, makes it difficult to identify a general drug-design strategy applicable to all conopeptides. Recently, we proposed a novel approach, termed “backbone prosthesis”, to produce μ-conotoxin analogs in which non-essential peptidic parts were replaced by non-peptidic spacers. [24] These analogs, called “polytides”, exhibited improved pharmacological properties, despite having simplified and minimized structures. However, a limiting factor in the application of the backbone prosthesis approach is the presence of the disulfide bridges that stabilize the bioactive conformation.
In this work, we examined the role of the disulfide bridges in the structure/function of the analgesic μ-conotoxin KIIIA. Three disulfide-deficient KIIIA analogs were synthesized and characterized by assaying the ability of the analogs to block sodium channels Nav1.2 and Nav 1.4. Removal of the first, C1-C9, native disulfide bridge of KIIIA minimally compromised bioactivity, so we further simplified the structure of the disulfide-deficient KIIIA analog by using a backbone spacer, 5-amino-3-oxapentanoic acid (Aopn), to replace two non-essential Ser residues. The resulting minimized analog, [desC1]KIIIA[S3/4Aopn, C9A], was analgesic and retained the inhibitory activity against NaV1.2 and Nav1.4. These encouraging results are discussed in the context of using disulfide-deficient polytides as templates for designing conopeptide-based mimetics, or “cono-mimetics”.
Results
Chemical synthesis and folding
To investigate the role of each of the native disulfide bridges in stabilizing the bioactive conformation of KIIIA, we designed three analogs in which one or the other of the three bridges was dissected out as shown in Fig. 1B. Linear forms of the analogs were chemically synthesized with an automated peptide synthesizer using Fmoc-based protocols as described in Methods. The peptides were purified by reversed-phase HPLC, and the identity of each analog was confirmed by mass spectrometry. To ensure the formation of the native disulfide bridges, a strategy combining orthogonal Cys-protection with two-step oxidation was applied, as illustrated in Figure 2A. Formation of the first disulfide bridge was accomplished by glutathione-mediated oxidation, whereas the second bridge was closed using iodine. After each folding step, the main oxidized product was purified by HPLC. Figure 2B shows the HPLC elution profile of each purified folding species of the three analogs. Mass spectrometric analysis confirmed that the final oxidation product contained two disulfide bridges: the calculated mass was 1821.9, and the experimental masses were 1821.7 for KIIIA[C1A, C9A], 1821.6 for KIIIA[C2A, C15A], and 1822.0 for KIIIA[C4A, C16A].
Figure 2. Chemical synthesis and folding/unfolding of disulfide-deficient analogs.
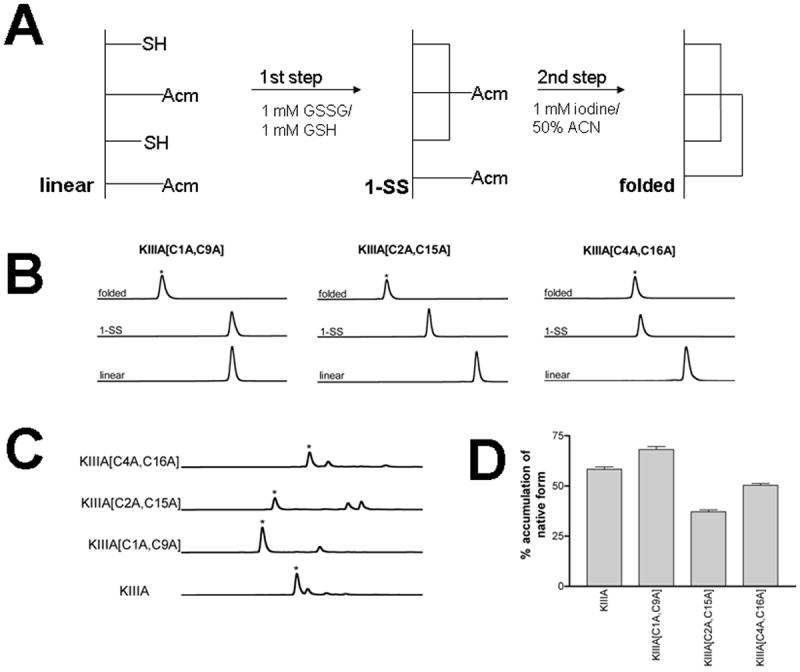
(A) - Two step oxidative folding reactions of KIIIA analogs were carried out in 0.1M Tris-HCl, pH 7.5. The first step was in the presence of 1mM GSSG and 1mM GSH. This was followed by a second oxidation step in 1mM iodine for 10-15 min. Each reaction was quenched with 8% formic acid. (B) - Oxidative folding of KIIIA analogs monitored by reversed-phased HPLC. Elution profiles of fully folded (native), first step folded, and linear forms of each analog are shown. The asterisks signify the peaks of the native forms deduced by mass spectroscopy; (C) - Reductive unfolding of the disulfide-deficient analogs of μ-KIIIA. The folded species were incubated at 25 °C in the presence of 1mM reduced and 1mM oxidized gluthathione at pH 7.5. Shown are HPLC profiles for each analog of the steady-state unfolding reaction (quenched after 30 min). The asterisks signify the native form of the analogs. (D) - Accumulation of the native form in the reductive unfolding reaction at steady-state. Average and s.e. of three independent experiments are represented.
To evaluate the consequences of the removal of the disulfide bridge on the stability of the remaining two native bridges, we conducted reductive unfolding experiments in which each analog was dissolved in a redox solution containing 1 mM reduced and 1 mM oxidized glutathione at pH 7.5 and 25 °C. The equilibrium was achieved within 30 min, since there was no difference between the HPLC elution profiles of the reaction mixtures quenched after 30 or 60 min. Figure 2C shows the HPLC separation of the unfolding reactions at steady-state. For each peptide, the native-like form was the predominant species, suggesting that removal of a single disulfide bridge did not significantly destabilize the remaining two native disulfide bridges. Interestingly, the KIIIA analog missing the Cys1-Cys9 disulfide bridge was more stable than the other two analogs, and the steady-state level of accumulation of the folded species was comparable to that of KIIIA containing all three disulfide bridges (Figure 2D).
Biological activity
In our previous work, KIIIA was found to be a nearly irreversible blocker of neuronal sodium channel subtype NaV1.2, but a readily reversible blocker the skeletal muscle subtype NaV1.4 with an IC50 of 90 nM [23]. Therefore, each of the three analogs of KIIIA was assayed using these two highly susceptible target ion channels. Both subtypes of sodium channels were expressed in oocytes, and their sodium currents were recorded in the absence and presence of 1 μM KIIIA or one of its three, disulfide-deficient, analogs. Representative sodium current traces and the time course of inhibition of Nav1.2 and Nav1.4 by each of the four peptides are shown in Figure 3. Table 1 summarizes on- and off-rates of the block of Nav1.2 and Nav1.4 by the peptides. The % block of the sodium currents by 1 μM peptide is summarized in Figure 4.
Figure 3. Activities of KIIIA and its analogs in blocking NaV1.2 and NaV1.4 expressed in Xenopus oocytes.
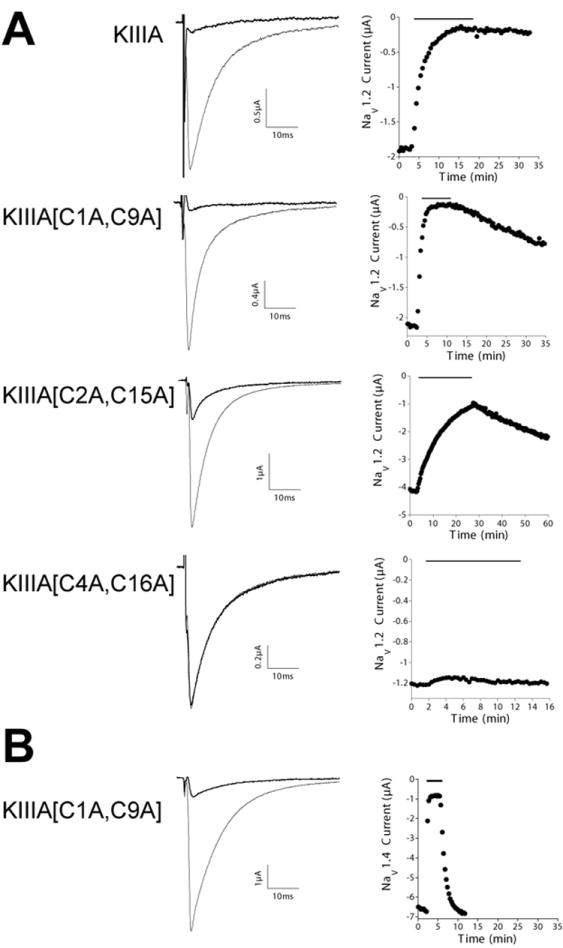
Sodium currents were recorded from oocytes that were two-electrode voltage clamped as described under Experimental Procedures. KIIIA and its analogs were each tested at a concentration of 1 μM on oocytes expressing NaV1.2 (A) or NaV1.4 (B). In each panel, the left column shows representative recordings, where each trace represents the average of 5 responses before (control, gray trace) and during (black trace) exposure to indicated peptide. The right column shows corresponding time course of block and recovery, where the black bar above each plot indicates when peptide was present. Note, block by KIIIA[C1A, C9A] of NaV1.4 (B) is much more rapidly reversible than that of NaV1.4 (2nd row in A), both plots have the same time base to allow direct comparison. Values of kinetic constants of block and recovery in replicate experiments are summarized in Table 1.
Table 1.
Comparison of the kinetics parameters of KIIIA and it disulfide-deficient analogs in blocking NaV1.2 and NaV1.4
| Analog | NaV1.2 | NaV1.4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| %[a] Block | kobs (min-1) |
koff (min-1) |
kon
[b] (μM•min)-1 |
Kd
[c] (μM) |
% Block | kobs (min-1) |
koff (min-1) |
kon
[b] (μM•min)-1 |
Kd
[c] (μM) |
|
| KIIIA | 90±3 | 0.36±0.06 | 0.0016±0.0016 | 0.36±0.06 | 0.004±0.004 | 86±6 | 0.91±0.17 | 0.05±0.017 | 0.86±0.17 | 0.06±0.02 |
| KIIIA[C1A, C9A] | 93±2 | 1.8±0.39 | 0.014±0.002 | 1.8±0.39 | 0.008±0.002 | 85±2 | 4.0±0.5 | 0.78±0.1 | 3.22±0.51 | 0.24±0.05 |
| KIIIA[C2A, C15A] | 66±4 | 0.07±0.02 | 0.01±0.0014 | 0.06±0.02 | 0.17±0.06 | 71±4 | 0.56±0.08 | 0.15±0.03 | 0.41±0.09 | 0.37±0.11 |
| KIIIA[C4A, C16A] | 8±3 | n.a. [d] | n.a. | -- | -- | 1±1 | n.a. | n.a. | -- | -- |
Percent block was determined at steady-state or plateau value of single exponential fit of time course of block during 20 min. of exposure to 1 μM peptide;
kon was estimated from {kobs – koff};
Kd was estimated from {koff/kon};
n.a., unable to be determined because level of block was too small. Values represent mean s.d. from at least three different oocytes.
Figure 4. Relative bioactivities of the disulfide deficient analogs as sodium channel blockers.
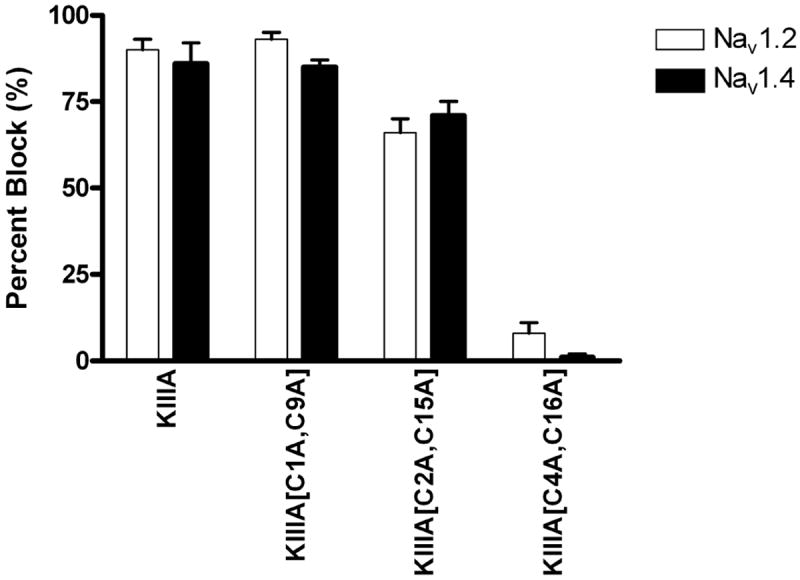
The abilities of the peptides in blocking sodium currents were measured as described in Fig. 3. The graph depicts the percentages of sodium currents in oocytes expressing NaV1.2 (white bars) and NaV1.4 (black bars) blocked by 1 μM of the indicated peptide (mean ± s.e., N ≥ 3 oocytes for each peptide).
Two analogs, KIIIA[C1A, C9A] and KIIIA[C2A, C15A], were able to block both subtypes of sodium channels. The analog missing the Cys1-Cys9 disulfide bridge was almost as active as the native KIIIA in blocking Nav1.2 but less so in blocking Nav1.4; for both channels however, both on- and off-rates were significantly increased (Figure 4 and Table 1). Interestingly, KIIIA[C2A, C15A] was almost equipotent against Nav1.2 and Nav1.4. Its on-rate for blocking NaV1.2 was substantially lower than that of both KIIIA and KIIIA[C1A, C9A], by 6-fold and 30-fold, respectively. The KIIIA[C4A, C16A] analog produced little if any block of either sodium channel subtype (Figure 4), even at a concentration 10 μM (not illustrated).
The KIIIA[C1A, C9A] analog containing a backbone spacer
To investigate whether the KIIIA[C1A, C9A] analog could further accommodate modifications that minimized the bioactive conformation of KIIIA, we replaced two adjacent Ser residues (S5-S6) with the backbone spacer, 5-amino-3-oxapentanoic acid (Aopn), also known as a PEG-spacer, or “backbone spacer”. The backbone spacers were previously used in μ-conotoxin SIIIA (Figure 1) to replace two nonessential G6-G7 residues located in the first intercysteine loop. [24] Such “backbone prosthesis” modifications in SIIIA not only allowed to structurally minimize the conotoxin (by replacing two adjacent amino acid residues with a single backbone spacer), but they also improved the ability of the resulting “polytides” to block sodium currents and to produce analgesic effects. For introduction of the backbone spacer, 5-amino-3-oxapentanoic acid, to KIIIA[C1A, C9A] analog, the two adjacent S5-S6 were selected since individual replacements of these two Ser residues with Ala in KIIIA had minimal effects on the ability of the peptide to block Nav1.2 and Nav1.4 [23]. Furthermore, since the Ala1 residue in the KIIIA[C1A, C9A] analog seemed unlikely to play a major role in the interactions with the sodium channels, we produced an analog of KIIIA[C1A, C9A] with the Ala1 residue deleted, resulting in the peptide [desC1]KIIIA[S3/4Aopn, C9A] whose structure is shown in Fig. 5A.
Figure 5. Disulfide-deficient analog of KIIIA containing the backbone spacer.
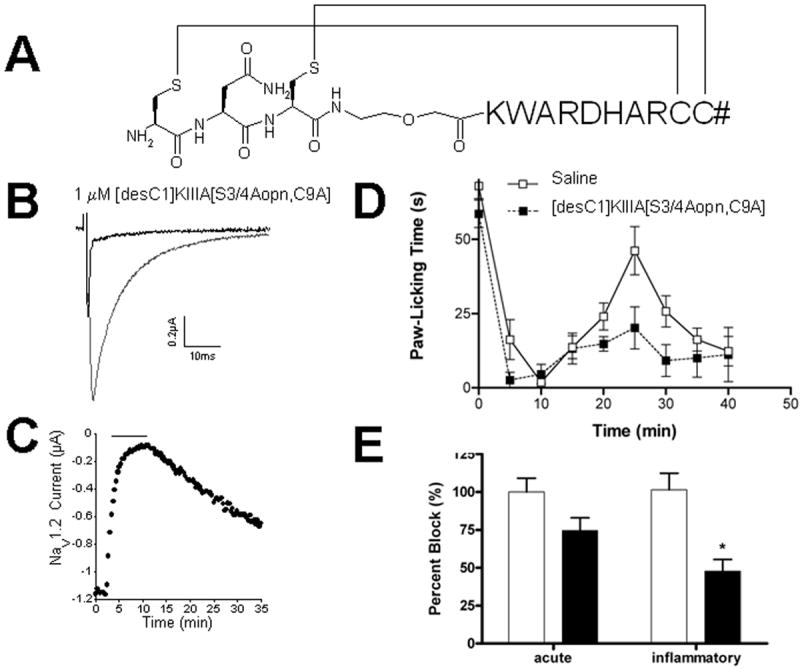
(A) - Amino acid sequence of [desC1, S3, S4]KIIIA[C9A, Aopn4] with disulfide bridge connectivity; (B) - Activity of this analog on neuronal sodium channel subtype Nav1.2; (C) - The on and off rates of this analog on Nav1.2; (D) - Analgesic activity: the cumulative licking time from 0-5 minutes is Phase I (acute), and that from 15-30 minutes is Phase II (inflammatory). Each dose was tested in at least 5 mice. Response to the peptide (dotted line) is compared to the response to saline solution alone (solid line), using Prism Software. (E) - Bar graph of values from panel D. White and black bars represent control and test conditions as do white and black squares in D, respectively. The asterisk indicates that the effect of [desC1, S3, S4]KIIIA[C9A, Aopn4] in the inflammatory phase (phase II) is significantly different from control (p < 0.05).
Chemical synthesis and formation of the two disulfide bridges in this peptide was carried out using protocols identical to those employed in the synthesis of the other disulfide-deficient analogs. The final oxidized product was purified by HPLC, and mass spectrometric analysis confirmed the chemical identity of the analog (MW 1677.7, experimental; 1677.8, calculated). When tested at a concentration of 1 μM, the [desC1]KIIIA[S3/4Aopn, C9A] analog blocked 93 ± 1 % of sodium current of Nav1.2 (Fig. 5B) with kon = 0.64 ± 0.06 μM•min)-1 and koff = 0.03 ± 0.0002 min-1 (Fig. 5C), yielding Kd value of 46 nM. Likewise, 1 μM of this analog blocked 70 ± 2% of Nav1.4-mediated current with kon = 0.64 ± 0.06 μM•min)-1 and koff = 0.03 ± 0.0002 min-1. When tested in the analgesic assay (10 nmoles per mouse, administered intraperitonally), the analog produced a 54% reduction of the phase II nociceptive response (Fig 5D and 5E), indicating that it retained much of the pharmacological properties of the native conotoxin μ-KIIIA.
Structural studies
Recent studies suggested that α-helical conformation is important for the bioactivity of μ-conotoxins. [25, 26] Since all studied KIIIA analogs, except KIIIA[C4A, C16A], retained the ability to block sodium channels to a various degree, we compared their structural properties using circular dichroism (CD) spectroscopy and molecular dynamics (MD) simulations. CD spectra were recorded in phosphate buffer, pH 7.4, and at 25 °C. As shown in Figure 6, all the KIIIA analogs showed some α-helical structure by circular dichroism measurments. Historically, helicity is calculated from the [Θ222] wavelength [27]. Of the analogs, KIIIA[C2A, C15A] exhibited the greatest helical content at 27%, followed by KIIIA[C1A, C9A] with 20% helicity. Native KIIIA was found to have 14% helicity. Analogs KIIIA[C4A, C15A] and [desC1]KIIIA[S3/4Aopn, C9A] both were calculated to have helicities of ~13%; however, proteins and peptides that have degrees of α-helical structure should also have a characteristic absorbance centered at 210 nm. Both of these analogs show a hypsochromatic shift, suggesting that they are less structured overall.
Figure 6. Structural characterization of the disulfide-deficient analogs by circular dichroism.
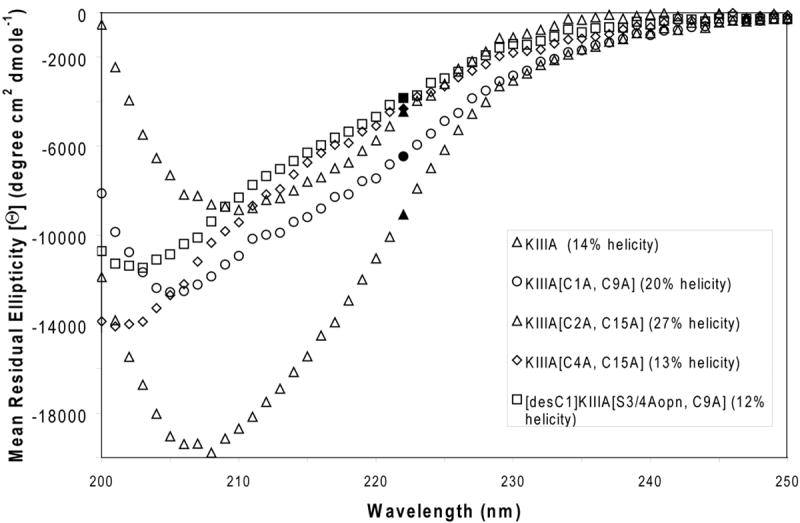
The CD spectra of the KIIIA analogs, recorded at 25°C, in 150 mM phosphate buffered NaF. The % helicity is determined at λ = 222 nm.
To further investigate structural consequences of removing the individual disulfide bridges in the KIIIA, a series of MD simulations were performed on the wild type and three disulfide deficient analogs using AMBER, [28] an implicit solvent (generalized Born, igb=2) representation, and the ff99SB force field. [29] The initial model structure of KIIIA was based on our previous work, [23] and the disulfide-deficient analogs were built by substituting the cross linked cysteine residues with alanine residues. Structures over the course of 50 ns of MD simulation for each alanine-substituted model are shown in Fig. 7A. The top left model shows the canonical KIIIA ribbon structure. During the simulations in implicit solvent, the expected structure is only metastable as alternative geometries with less of the expected secondary structure are sampled. Some level of conformational heterogeneity in the wild-type μ-conotoxins was previously observed, [30-32] although not in μ-SmIIIA. [33] This contrasts with the behavior seen with KIIIA[C1A, C9A], where the MD simulations suggested that a single folded geometry was sampled. Such apparent stabilization of the partially disulfide linked polypeptide is not without precedent, as reduced flexibility has been previously seen in work by Rizzuti et al. where MD simulations on the azurin protein showed similar effects. [34] Independent structural studies of KIIIA and the KIIIA[C1A, C9A] analog using NMR spectroscopy and additional MD simulation confirmed our observations (Khoo KK, et al, manuscript submitted). Removal of the third disulfide linkage (C4-C15) appeared to lead to the most pronounced conformational instabilities (Fig. 7A). The analysis of the secondary structure for KIIIA and the disulfide-deficient analogs using ptraj program followed by DSSP method yielded α-helical fragments in all peptides. Consistent with CD results, the highest amount of α-helical structure was observed in KIIIA[C1A, C9A] and KIIIA[C2A, C15A], 32% and 28%, respectively, whereas KIIIA and KIIIA[C4A, C15A] contained only 5 % and 6 % α-helical content, respectively.
Figure 7. Structural properties of the disulfide-deficient analogs studied by molecular dynamics.
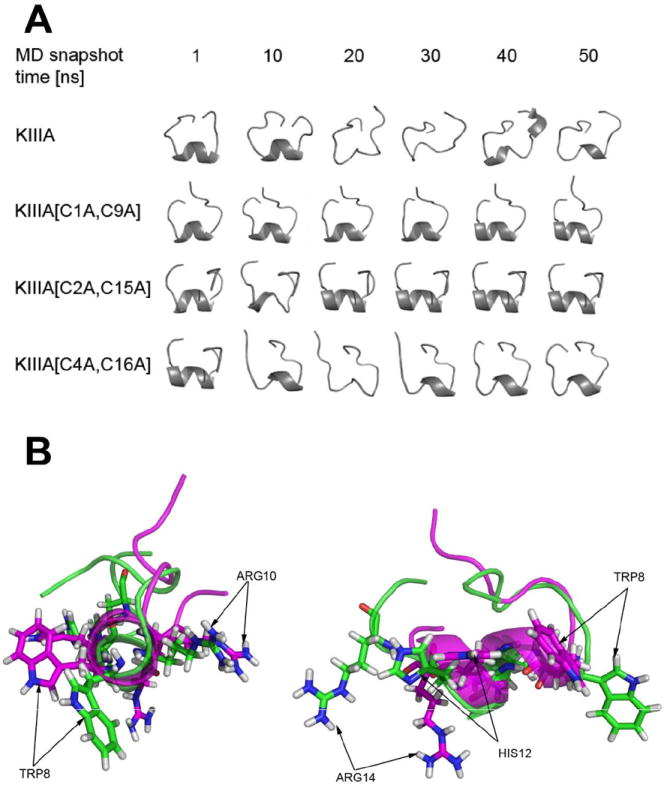
(A) - The total of 6 snapshot trajectories of 1, 10, 20, 30, 40 and 50 ns MD simulations of KIIIA and its analogs missing specific disulfide bridges, specifically KIIIA[C1A, C9A], [C2A, C15A] and [C4A, C16A]; (B) - Conformation of the KIIIA (green) and KIIIA[C1A, C9A] (magenta) backbone, shown as cartoon representation, carrying four pharmacophore residues (W8, R10, H12, R14) indicated by arrows. End-on view (left) and side view (right) of the helical secondary structure are presented.
Discussion
In this work we examined the consequences of removing, one at a time, the native disulfide bridges in μ-conotoxin KIIIA. Our results show that an analog missing the native disulfide bridge C1-C9 retained much of the activity in blocking sodium channels, whereas the analog missing the C4-C16 lost essentially all biological activity. Interestingly, even for the KIIIA[C1A, C9A] analog that retained much of the blocking activity, its off-rates were significantly larger than those of KIIIA, suggesting the important contribution of the disulfide bridges to the interactions between μ-conotoxins and sodium channels. This conclusion was further supported by the substantially reduced activity of KIIIA[C2A, C15A], with a slower on-rate and faster off-rate. Unexpectedly, removal of the C1-C9 disulfide bridge increased the on-rate of block of both Nav 1.2 and 1.4 by about five-fold. In contrast, the removal of the C2-C15 disulfide decreased the on-rate, with a more pronounced effect on the on-rate for blocking Nav1.2 channel (6-fold decrease), as compared to that for blocking Nav1.4 (2-fold decrease). Since the removal of any single one of the native disulfide bridges did not significantly destabilize the remaining two native disulfide bridges in KIIIA (Fig. 2D), the disulfide-deficient analogs may be further exploited in the production of useful probes to study the mechanism of the interactions between conotoxins and ion channels.
Role of the disulfide bridges in the stability and activity of conotoxins was studied previously with four model peptides: ω-GVIA, ω-MVIIA, α -ImI and α -GI. Our findings confirm the critical role of the disulfide bridges in the structure/function of the conotoxins. For ω-MVIIA, removal of the first disulfide bridge (C1-C16) produced a 100-fold loss in binding affinity for calcium channels, whereas the removal of the C15-C25 bridge caused a 104-fold decrease in Ki, comparable to that of the fully unfolded and reduced peptide. [35, 36] In ω -conotoxin GVIA, removal of a single disulfide bridge (between C15-C26) resulted in misfolding and loss of ability to block calcium channels. [37] For the two disulfide bridged α-conotoxin GI, a removal of a single disulfide bridge affected the overall conformation and the folding properties of the peptide, but the NMR studies indicated the presence of the residual secondary structure, in particular for the analog missing the first disulfide bridge. [38] Furthermore, the analog of α -ImI missing the C3-C12 disulfide bridge fully retained the ability to block α7 nAChRs. [39] It thus appears that the structural and functional consequences of removing individual disulfides are context dependant, consistent with our current findings.
A successful approach of minimizing a bioactive conformation of a three-disulfide bridged, 35-residue neurotoxin was reported by the Norton’s group. [40-42] Two out of three disulfide-deficient analogs of the ShK toxin were able to block Kv1.1 potassium channels with nanomolar affinity. [42] Furthermore, a highly minimized analog of ShK containing eight amino acid residues and crosslinked by two disulfide bridges blocked Kv1.3 channels with a Kd of 92 μM. [41] The disulfide-deficient analog of the 34-residue scorpion toxin, maurotoxin, also retained the overall α / β scaffold and the ability to block potassium channels, [43] but no further efforts in minimizing the bioactive conformation of these neurotoxins have been reported to-date. Recently, “molecular pruning” was succesfully applied to α-conotoxin analog [A10L]PnIA: central truncation of three amino acid residues in the second intercysteine loop did not affect the peptide’s ability to block nicotinic acetylcholine receptors. [44] Based on the above examples and our current work, minimization of disulfide-rich toxins will continue to provide novel analogs with a therapeutic potential.
Another important finding of our work is that the disulfide-deficient analog of KIIIA containing the backbone spacer, [desC1]KIIIA[S3/4Aopn, C9A] was able to block sodium channels and retain analgesic activity in the inflammatory pain assay in mice, comparable to that of the wild-type KIIIA conotoxin. When minimizing KIIIA, the Kd values for blocking Nav1.2 were decreasing from 4 nM (for KIIIA) to 8 nM (for KIIIA[C1A, C9A]) to 46 nM (for [desC1]KIIIA[S3/4Aopn, C9A]). The subtypes of sodium channels blocking of which produced this analgesic effect remain unknown, but presumably are similar to those blocked by the wild-type KIIIA. [23] More electrophysiological and pharmacological studies are underway in our laboratory to dissect the mechansim of analgesia mediated by μ-conotoxins and their analogs.
The apparent drug-design implications of our results are that it is possible to reduce the molecular size of a three disulfide-bridged conotoxin (14 AA, 2SS, MW = 1,678 instead of 16 AA, 3SS, MW = 1,884) and still retain the bioactivity. Since Trp8, Arg10, His12 and Arg14 were previously shown to be critical for blocking NaV1.2, our results suggest that the pharmacophore can be maintained with merely two disulfide bridges and the presence of a backbone adjacent to the Lys7-Trp8 within the same intercysteine loop. As illustrated in Fig. 6B, the overall conformation of the backbone carrying four pharmacophore residues (W8, R10, H12, R14) that determine the interactions with NaV1.2 maintained the helical conformation. Our functional studies, which demonstrate that the stepwise size reduction from μ-KIIIA to KIIIA[C1A, C9A] to [desC1]KIIIA[S3/4Aopn, C9A] did not compromise the bioactivity against NaV1.2, encourages further efforts in designing non-peptidic scaffolds for μ-conotoxins. Potential advantages of the backbone prosthesis for conotoxins were previously discussed [24], and involve improving their pharmacological properties. Although the mechanism by which the backbone spacers enhanced the analgesic activity of SIIIA remains unknown, it may include changes in the in vivo stability and/or penetration of the polytides across the biological membranes. Additional structure-function studies on polytides will likely lead to a more rational design of toxin analogs with improved pharmacological properties.
The disulfide-deficient analogs containing backbone spacers provide an advance toward the development of conopeptide-based therapeutics. Previous efforts to apply peptidomimetic approaches to conotoxins achieved limited success: mimetics of ω -conotoxin GVIA had substantially diminished binding affinities, [45, 46] suggesting that more innovative mimetic templates must be developed to transform conotoxins into conomimetics. [47] We would like to speculate that helical peptidomimetic scaffolds for grafting the μ-KIIIA pharmacophore, WXRXHXR, may include peptoids, β - or γ - or β/ γ peptides, lactam-stabilized helical or other synthetic non-peptide mimetics. [48, 49] Furthermore, these scaffolds can be employed to design synthetic conformationally homogenous combinatorial libraries for screening against sodium channels. Taken together, our work raises the possibility that minimization of the bioactive conformation of μ-conotoxins can be accomplished using disulfide-deficient polytides and suggests further directions in the development of conotoxin-based mimetics and small molecules that target sodium channels.
Experimental Section
Chemical synthesis
Peptides were synthesized using standard N-(9-fluorenyl)methoxycarbonyl (Fmoc) chemistry. The peptides were cleaved from the resin by 3-4 h treatment with reagent K (TFA/water/ethanedithiol/phenol/thioanisole; 82.5/5/2.5/5/5 by volume). The cleaved peptides were filtered, precipitated with cold methyl tert-butyl ether (MTBE) and washed several times with cold MTBE. The reduced peptides were purified by reversed-phase HPLC using a semipreparative C18 Vydac column (218TP510, 10 mm × 250 mm) eluted with a linear gradient from 5 to 35% solvent B in 35 min, where solvent A was 0.1% (v/v) TFA in water, and B was 0.1% (v/v) TFA in 90% aqueous acetonitrile (ACN). The flow rate was 5 ml/min, and absorbance was monitored at 220 nm.
Oxidative folding
To prepare KIIIA analogs with the native cysteine connectivity, the first of cysteine pair was protected by S-trityl groups and the second pair was protected by acetamidomethyl groups. To oxidize the first disulfide bridge, the reduced peptides (at 20 μM final concentration) were dissolved in 0.01% TFA and added to the folding mixture containing 0.1M Tris-HCl, 1mM EDTA, 1 mM oxidized glutathione (GSSG) and 1 mM reduced glutathione (GSH). The reactions were carried out at room temperature. After 1 h, the reactions were quenched with formic acid (8% final concentration) and purified by semi-preparative HPLC as described above. To remove the acetamidomethyl groups from the second pair of cysteines and close the remaining disulfide bridge, the peptides were treated with 2 mM iodine in 50% aqueous (acetonitrile) ACN for 10 min (for [desC1]KIIIA[S3/4Aopn, C9A] 1 mM iodine for 10 min) and quenched with ascorbic acid. The analytical HPLC gradient was linear and changed from 5 to 35% solvent B in 35 min, where solvent A was 0.1% (v/v) TFA in water, and B was 0.1% (v/v) TFA in 90% aqueous ACN. The flow rate was 1 ml/min. The identities of final products were confirmed by MALDI-TOF analysis.
Electrophysiology assay
Electrophysiological experiments were carried out essentially as described elsewhere. [23] Briefly, oocytes expressing cloned sodium channels were prepared and placed in a 30 μl chamber containing ND96 and two-electrode voltage clamped with a holding potential of −80 mV. To activate Na channels, the membrane potential was stepped to a value between -20 and 0 mV (depending on NaV subtype) for 50 ms every 20 sec. To apply toxin, the perfusion was halted, 3 μL of toxin solution (at ten times of the final concentration) was applied to the 30 μL bath, and the bath manually stirred for about 5 seconds by gently aspirating and expelling ~ 5 μL of the bath fluid several times with a micropipette. Toxin-exposures were in static baths to conserve material. Off-rate constants were determined from single-exponential fits of the time course of recovery from block during peptide-washout. All recordings were done at room temperature (~21 °C). Unless indicated otherwise, data are expressed as average ± S.E. (N ≥ 3 oocytes for each concentration tested).
Analgesic assay
The formalin test was carried out on 25-30g Swiss Webster mice by intraperitoneal injection of 10 nanomoles of the KIIIA analog or a saline solution, followed (after 30 min) by intraplanar injection of 20 μL of 5% formalin. [23, 24] Mice were placed in open glass cylinders and paw-licking was monitored for 50 minutes. Increased licking-frequency between 0-5 minutes reflected the phase I response, whereas that between 15-30 min was defined as the phase II response. The area under the curve of each phase was calculated using Prism software. All animal procedures followed protocols approved by the University of Utah IACUC.
Circular dichroism
The peptides were reconstituted in a 150 mM NaF buffer solution (50 mg KH2PO4, 54 mg Na2HPO4, 1.55 g NaF, 250 mL H2O, pH 7.4 with Na2HPO4) to a final concentration of 0.1 mg/mL. The solutions placed in a 0.1 cm quartz cuvette (Starna Cells, Inc.) and were then analyzed by an Aviv 400 Circular Dichroism Spectrometer, scanning 200 nm to 250 nm, at 1 nm intervals with a 1 sec dwell time. The percent helicity was calculated as previously reported [27].
Molecular modeling
The initial model structure of KIIIA was based on our previous work. [23] Using LEaP, with the ff99SB AMBER force field, [29] models were built of KIIIA and the models missing individual disulfide bridges, specifically KIIIA[C1A, C9A], KIIIA[C2A, C15A] and KIIIA[C4A, C16A]. The structures were built and minimized in vacuo, followed by MD with an implicit (generalized Born) solvation method (igb=2). [50] A cutoff of 12 Å was applied and temperature was controlled through a Langevin thermostat [51] at 300K with 1 ps factor. A time step of 1.0 fs was employed during the full 50 ns MD simulations. Analysis of the secondary structure for the models was carried out using ptraj program following DSSP method by Kabsch and Sander.
Supplementary Material
Acknowledgments
This work was supported by NIH Program Project Grant GM 48677 (B.M.O.) and R21 NS055845(G.B.). T.S.H. acknowledges grants from the undergraduate research UROP and LEAP programs. We thank Drs. Robert Schackmann and Scott Endicott from the DNA/Peptide Synthesis Core Facility at the University of Utah for the synthesis of peptides, and Philip Catlin for his help with synthetic peptides. T.S.H. would like to express her gratitude to her Family and P.I. for their support and encouragement.
References
- 1.Olivera BM. The Journal of biological chemistry. 2006;281(42):31173–31177. doi: 10.1074/jbc.R600020200. [DOI] [PubMed] [Google Scholar]
- 2.Livett BG, Gayler KR, Khalil Z. Current medicinal chemistry. 2004;11(13):1715–1723. doi: 10.2174/0929867043364928. [DOI] [PubMed] [Google Scholar]
- 3.Norton RS, Olivera BM. Toxicon. 2006;48(7):780–798. doi: 10.1016/j.toxicon.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 4.Miljanich GP. Current medicinal chemistry. 2004;11(23):3029–3040. doi: 10.2174/0929867043363884. [DOI] [PubMed] [Google Scholar]
- 5.Jones RM, Bulaj G. Current pharmaceutical design. 2000;6(12):1249–1285. doi: 10.2174/1381612003399653. [DOI] [PubMed] [Google Scholar]
- 6.Livett BG, Sandall DW, Keays D, Down J, Gayler KR, Satkunanathan N, Khalil Z. Toxicon. 2006;48(7):810–829. doi: 10.1016/j.toxicon.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 7.Armishaw CJ, Alewood PF. Current protein & peptide science. 2005;6(3):221–240. doi: 10.2174/1389203054065437. [DOI] [PubMed] [Google Scholar]
- 8.Han TS, Teichert RW, Olivera BM, Bulaj G. Current pharmaceutical design. 2008;14:2462–2479. doi: 10.2174/138161208785777469. [DOI] [PubMed] [Google Scholar]
- 9.Amir R, Argoff CE, Bennett GJ, Cummins TR, Durieux ME, Gerner P, Gold MS, Porreca F, Strichartz GR. J Pain. 2006;7(5 Suppl 3):S1–29. doi: 10.1016/j.jpain.2006.01.444. [DOI] [PubMed] [Google Scholar]
- 10.Priest BT, Kaczorowski GJ. Expert opinion on therapeutic targets. 2007;11(3):291–306. doi: 10.1517/14728222.11.3.291. [DOI] [PubMed] [Google Scholar]
- 11.Cummins TR, Sheets PL, Waxman SG. Pain. 2007;131(3):243–257. doi: 10.1016/j.pain.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallace MS. The Clinical journal of pain. 2000;16(2 Suppl):S80–85. doi: 10.1097/00002508-200006001-00014. [DOI] [PubMed] [Google Scholar]
- 13.Cummins TR, Rush AM. Expert review of neurotherapeutics. 2007;7(11):1597–1612. doi: 10.1586/14737175.7.11.1597. [DOI] [PubMed] [Google Scholar]
- 14.Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, Kort M, Carroll W, Marron B, Atkinson R, Thomas J, Liu D, Krambis M, Liu Y, McGaraughty S, Chu K, Roeloffs R, Zhong C, Mikusa JP, Hernandez G, Gauvin D, Wade C, Zhu C, Pai M, Scanio M, Shi L, Drizin I, Gregg R, Matulenko M, Hakeem A, Gross M, Johnson M, Marsh K, Wagoner PK, Sullivan JP, Faltynek CR, Krafte DS. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(20):8520–8525. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ekberg J, Jayamanne A, Vaughan CW, Aslan S, Thomas L, Mould J, Drinkwater R, Baker MD, Abrahamsen B, Wood JN, Adams DJ, Christie MJ, Lewis RJ. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(45):17030–17035. doi: 10.1073/pnas.0601819103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bulaj G, Zhang MM, Green BR, Fiedler B, Layer RT, Wei S, Nielsen JS, Low SJ, Klein BD, Wagstaff JD, Chicoine L, Harty TP, Terlau H, Yoshikami D, Olivera BM. Biochemistry. 2006;45(23):7404–7414. doi: 10.1021/bi060159+. [DOI] [PubMed] [Google Scholar]
- 17.Schmalhofer WA, Calhoun J, Burrows R, Bailey T, Kohler MG, Weinglass AB, Kaczorowski GJ, Garcia ML, Koltzenburg M, Priest BT. Molecular pharmacology. 2008;74(5):1476–1484. doi: 10.1124/mol.108.047670. [DOI] [PubMed] [Google Scholar]
- 18.Lewis RJ, Schroeder CI, Ekberg J, Nielsen KJ, Loughnan M, Thomas L, Adams DA, Drinkwater R, Adams DJ, Alewood PF. Molecular pharmacology. 2007;71(3):676–685. doi: 10.1124/mol.106.028225. [DOI] [PubMed] [Google Scholar]
- 19.Zhang MM, Fiedler B, Green BR, Catlin P, Watkins M, Garrett JE, Smith BJ, Yoshikami D, Olivera BM, Bulaj G. Biochemistry. 2006;45(11):3723–3732. doi: 10.1021/bi052162j. [DOI] [PubMed] [Google Scholar]
- 20.French RJ, Terlau H. Current medicinal chemistry. 2004;11(23):3053–3064. doi: 10.2174/0929867043363866. [DOI] [PubMed] [Google Scholar]
- 21.Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D, Moczydlowski E. The Journal of biological chemistry. 1985;260(16):9280–9288. [PubMed] [Google Scholar]
- 22.Cruz LJ, Kupryszewski G, LeCheminant GW, Gray WR, Olivera BM, Rivier J. Biochemistry. 1989;28(8):3437–3442. doi: 10.1021/bi00434a043. [DOI] [PubMed] [Google Scholar]
- 23.Zhang MM, Green BR, Catlin P, Fiedler B, Azam L, Chadwick A, Terlau H, McArthur JR, French RJ, Gulyas J, Rivier JE, Smith BJ, Norton RS, Olivera BM, Yoshikami D, Bulaj G. The Journal of biological chemistry. 2007;282(42):30699–30706. doi: 10.1074/jbc.M704616200. [DOI] [PubMed] [Google Scholar]
- 24.Green BR, Catlin P, Zhang MM, Fiedler B, Bayudan W, Morrison A, Norton RS, Smith BJ, Yoshikami D, Olivera BM, Bulaj G. Chemistry & biology. 2007;14(4):399–407. doi: 10.1016/j.chembiol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Yao S, Zhang MM, Yoshikami D, Azam L, Olivera BM, Bulaj G, Norton RS. Biochemistry. 2008;47(41):10940–10949. doi: 10.1021/bi801010u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schroeder CI, Ekberg J, Nielsen KJ, Adams D, Loughnan ML, Thomas L, Adams DJ, Alewood PF, Lewis RJ. The Journal of biological chemistry. 2008;283(31):21621–21628. doi: 10.1074/jbc.M802852200. [DOI] [PubMed] [Google Scholar]
- 27.Chen YH, Yang JT, Chau KH. Biochemistry. 1974;13(16):3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- 28.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. Journal of computational chemistry. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins. 2006;65(3):712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lancelin JM, Kohda D, Tate S, Yanagawa Y, Abe T, Satake M, Inagaki F. Biochemistry. 1991;30(28):6908–6916. doi: 10.1021/bi00242a014. [DOI] [PubMed] [Google Scholar]
- 31.Wakamatsu K, Kohda D, Hatanaka H, Lancelin JM, Ishida Y, Oya M, Nakamura H, Inagaki F, Sato K. Biochemistry. 1992;31(50):12577–12584. doi: 10.1021/bi00165a006. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen KJ, Watson M, Adams DJ, Hammarstrom AK, Gage PW, Hill JM, Craik DJ, Thomas L, Adams D, Alewood PF, Lewis RJ. The Journal of biological chemistry. 2002;277(30):27247–27255. doi: 10.1074/jbc.M201611200. [DOI] [PubMed] [Google Scholar]
- 33.Keizer DW, West PJ, Lee EF, Yoshikami D, Olivera BM, Bulaj G, Norton RS. The Journal of biological chemistry. 2003;278(47):46805–46813. doi: 10.1074/jbc.M309222200. [DOI] [PubMed] [Google Scholar]
- 34.Rizzuti B, Sportelli L, Guzzi R. Biophysical chemistry. 2001;94(1-2):107–120. doi: 10.1016/s0301-4622(01)00228-9. [DOI] [PubMed] [Google Scholar]
- 35.Price-Carter M, Bulaj G, Goldenberg DP. Biochemistry. 2002;41(10):3507–3519. doi: 10.1021/bi012033c. [DOI] [PubMed] [Google Scholar]
- 36.Price-Carter M, Hull MS, Goldenberg DP. Biochemistry. 1998;37(27):9851–9861. doi: 10.1021/bi9803978. [DOI] [PubMed] [Google Scholar]
- 37.Flinn JP, Pallaghy PK, Lew MJ, Murphy R, Angus JA, Norton RS. Biochimica et biophysica acta. 1999;1434(1):177–190. doi: 10.1016/s0167-4838(99)00165-x. [DOI] [PubMed] [Google Scholar]
- 38.Kaerner A, Rabenstein DL. Biochemistry. 1999;38(17):5459–5470. doi: 10.1021/bi9826658. [DOI] [PubMed] [Google Scholar]
- 39.Lamthanh H, Jegou-Matheron C, Servent D, Menez A, Lancelin JM. FEBS letters. 1999;454(3):293–298. doi: 10.1016/s0014-5793(99)00831-5. [DOI] [PubMed] [Google Scholar]
- 40.Baell JB, Harvey AJ, Norton RS. Journal of computer-aided molecular design. 2002;16(4):245–262. doi: 10.1023/a:1020214720560. [DOI] [PubMed] [Google Scholar]
- 41.Lanigan MD, Pennington MW, Lefievre Y, Rauer H, Norton RS. Biochemistry. 2001;40(51):15528–15537. doi: 10.1021/bi011300b. [DOI] [PubMed] [Google Scholar]
- 42.Pennington MW, Lanigan MD, Kalman K, Mahnir VM, Rauer H, McVaugh CT, Behm D, Donaldson D, Chandy KG, Kem WR, Norton RS. Biochemistry. 1999;38(44):14549–14558. doi: 10.1021/bi991282m. [DOI] [PubMed] [Google Scholar]
- 43.Fajloun Z, Ferrat G, Carlier E, Fathallah M, Lecomte C, Sandoz G, di Luccio E, Mabrouk K, Legros C, Darbon H, Rochat H, Sabatier JM, De Waard M. The Journal of biological chemistry. 2000;275(18):13605–13612. doi: 10.1074/jbc.275.18.13605. [DOI] [PubMed] [Google Scholar]
- 44.Jin AH, Daly NL, Nevin ST, Wang CI, Dutertre S, Lewis RJ, Adams DJ, Craik DJ, Alewood PF. Journal of medicinal chemistry. 2008;51(18):5575–5584. doi: 10.1021/jm800278k. [DOI] [PubMed] [Google Scholar]
- 45.Baell JB, Duggan PJ, Forsyth SA, Lewis RJ, Lok YP, Schroeder CI. Bioorganic & medicinal chemistry. 2004;12(15):4025–4037. doi: 10.1016/j.bmc.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 46.Baell JB, Forsyth SA, Gable RW, Norton RS, Mulder RJ. Journal of computer-aided molecular design. 2001;15(12):1119–1136. doi: 10.1023/a:1015930031890. [DOI] [PubMed] [Google Scholar]
- 47.Pallaghy PK, Norton RS. Biopolymers. 2000;54(3):173–179. doi: 10.1002/1097-0282(200009)54:3<173::AID-BIP30>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 48.Davis JM, Tsou LK, Hamilton AD. Chemical Society reviews. 2007;36(2):326–334. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]
- 49.Mills NL, Daugherty MD, Frankel AD, Guy RK. Journal of the American Chemical Society. 2006;128(11):3496–3497. doi: 10.1021/ja0582051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Onufriev A, Bashford D, Case DA. Proteins. 2004;55(2):383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 51.Wu XW, Brooks BR. Chemical Physics Letters. 2003;381(3-4):512–518. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.