

Abstract
Metformin is a biguanide drug that is widely prescribed for type 2 diabetes. Metformin suppresses hepatic gluconeogenesis and increases fatty acid oxidation. Although studies have suggested that metformin acts, at least in part, via activation of the liver kinase B1 (LKB1)/AMP-activated protein kinase (AMPK) pathway, the specific molecular mechanisms underlying metformin's regulation of glucose and lipid metabolism have not been well delineated. Recently, we have shown that inositol polyphosphate multikinase (IPMK) plays an important role in cellular energy metabolism and glucose-mediated AMPK regulation. Here we investigated the role of IPMK in metformin-induced AMPK activation. We observed that metformin-mediated activation of AMPK was impaired in the absence of IPMK. Overexpression of wild-type IPMK was sufficient to restore LKB1-AMPK activation by either metformin or AICAR in IPMK−/− murine embryonic fibroblast cells, suggesting that IPMK may act as an upstream regulator of LKB1-AMPK signaling in response to metformin. Moreover, this regulation was mediated by protein-protein interaction between IPMK and LKB1 as a dominant-negative peptide, which abrogates this interaction, attenuated metformin's ability to activate AMPK. Our data demonstrate that IPMK plays an important role in LKB1/AMPK signaling and may be targeted for treatment of metabolic diseases.
AMP-activated protein kinase (AMPK) is a serine/threonine kinase that is evolutionarily conserved from yeast to humans. AMPK senses the energy status of cells and regulates fuel availability (1–3). AMPK forms a heterotrimeric complex comprising a catalytic α- and 2 regulatory β- and γ-subunits, and liver kinase B1 (LKB1) enhances AMPK activity through the phosphorylation of the α-subunit at T172 (4, 5). Another upstream kinase, calmodulin-dependent protein kinase kinase β (CaMKKβ) activates AMPK in the presence of high cytosolic calcium (6, 7). AMPK is allosterically activated by the accumulation of AMP molecules, due to metabolic stresses that inhibit ATP production (eg, hypoxia and glucose deprivation) or stimulate ATP consumption (1, 3, 5) and is also activated by drugs used to treat type 2 diabetes, including metformin (8).
LKB1 is a 50-kDa serine/threonine kinase that was originally identified as the product of the gene mutated in the autosomal dominantly inherited Peutz-Jeghers cancer syndrome (9, 10). Like AMPK, LKB1 forms a heterotrimeric complex with regulatory proteins termed STE20-like pseudokinase (STRAD) and the armadillo repeat-containing mouse protein (MO25), which are required for its activation and cytosolic localization (11–13). The LKB1 complex itself is not stimulated by AMP and appears to be constitutively active in cells (14, 15). LKB1 regulates cellular energy metabolism through its capacity to phosphorylate and activate AMPK as well as other members of the AMPK subfamily (16, 17). Activated AMPK restores ATP levels by promoting ATP-producing catabolic processes (eg, glycolysis) and blocking ATP-consuming biosynthetic processes (including mammalian target of rapamycin [mTOR]-directed protein synthesis). LKB1-mediated regulation of AMPK has tissue-specific effects on glucose metabolism (15, 18, 19).
Metformin is a biguanide compound that is widely used as an insulin sensitizer in type 2 diabetes. Evidence from clinical and animal studies demonstrates that metformin suppresses gluconeogenesis and reduces hepatic glucose output (20, 21). However, the molecular mechanisms by which metformin regulates glucose metabolism have not been well delineated. It has been shown that the biguanides inhibit mitochondrial respiratory chain complex I, which leads to a reduction in cellular energy status and activation of AMPK/LKB1 (8, 22). It has been demonstrated that metformin activated AMPK in rat hepatocytes, and subsequent studies also showed that AMPK-mediated phosphorylation of key metabolic enzymes contributed to the therapeutic action of metformin (8). LKB1 deficiency in the liver induces hyperglycemia due to increased gluconeogenesis and abolishes the antidiabetic action of metformin (23). Importantly, LKB1 single-nucleotide polymorphism has been linked to diabetes (24) and nonresponse to metformin (25). However, other studies have described AMPK-independent actions of metformin (21, 26–28).
Inositol polyphosphate multikinase (IPMK, also called Ipk2 and Arg82) accounts for phosphorylation of InsP3 to InsP as well as the production of several other lipid-inositol molecules (29–32). Diverse cellular stimuli result in phospholipase C activation and subsequent formation of inositol phosphate (InsP) second messengers (30), which are involved in multiple cellular processes such as intracellular calcium mobilization (33) and apoptosis (34–36). Mammalian IPMK is a multifunctional protein in cellular signal transduction regulating the Akt pathway by its phosphatidylinositol 3-kinase (PI3K) activity or mTOR complex1 signaling as a cofactor (37–40). Moreover, IPMK is a novel AMPK binding protein, and its binding to AMPK is dynamically regulated by glucose availability (41).
Although numerous studies have established a critical role of LKB1 in AMPK activation, the upstream signaling that controls LKB1-dependent AMPK activation remains still elusive. Previously, we have shown that IPMK plays a critical role in glucose-mediated AMPK regulation (41). These interfaces of IPMK and the LKB1/AMPK pathway prompted us to further explore the role of IPMK in metformin action. In the present study, we have found that metformin-mediated activation of AMPK is IPMK-dependent, in particular, via protein interaction between IPMK and its upstream kinase LKB1.
Materials and Methods
Cell culture and transfection
GT1–7 cells (a generous gift from Dr Pamela Mellon, University of California, San Diego, CA), HeLa (American Type Culture Collection; CCL-2), 293T cells, and MEFs (IPMK MEFs [fl/fl], IPMK−/− MEF) (41, 42) were maintained in DMEM with 10% fetal bovine serum and 2mM l-glutamine at 37°C with a 5% CO2 atmosphere in a humidified incubator. For transient transfection of cells with expression constructs, we used PolyFect reagent (QIAGEN) according to the manufacturer's protocols. Thirty hours after transfection, cells were treated with metformin (1mM–8mM) or AICAR (0.1mM–4mM) for 2 hours (Sigma). For Cre recombinase experiments, IPMK MEFs (flox/flox) cells were transfected with either Myc or Myc-Cre targeting IPMK with PolyFect reagent, and 30 hours after transfection, cells were treated with metformin or AICAR.
Electroporation was performed using the neon electroporator (Invitrogen) according to the manufacturer's recommendation. Briefly, 1 × 107 trypsinized IPMK−/− MEFs were centrifuged at 1200 rpm for 5 minutes at room temperature and resuspended in 1 mL Invitrogen R buffer (Invitrogen), and then 100 μL of cells were gently transferred in buffer R with 15 to 30 μg Myc or Myc-IPMK plasmid and gently mixed. The cell/plasmid mixture was sucked into a neon pipette tube filled with electrolyte buffer. The mixture received 2 pulses (pulse voltage, 1100 V; pulse width, 20 milliseconds each), and the electroporated cells were transferred into 6-cm tissue culture plates containing 5 mL DMEM with 10% fetal bovine serum (no antibiotics) and incubated at 37°C for 24 hours, unless otherwise indicated. For rescue experiments, IPMK−/− cells were transfected with Myc, Myc-tagged wild-type (WT), or atIpk2β (Arabidopsis, plant) IPMK plasmid using electroporation method.
Reagents and antibodies
Metformin and AICAR were obtained from Sigma-Aldrich. Anti-Myc was obtained from Roche. The following were from Cell Signaling Technologies: AMPK, phospho-AMPK, acetyl coenzyme A carboxylase (ACC), phospho-ACC, Raptor, phospho-Raptor, LKB1, and glyceraldehyde-3phosphate dehydrogenase. Anti-IPMK was a custom rabbit polyclonal antibody from Covance raised against a synthetic peptide starting with Cys followed by mouse IPMK amino acids 295 to 311 (SKAYSRHRKLYAKKHQS).
Immunoprecipitation
Cells were lysed in 50mM Tris (pH 7.4), 150mM NaCl, 1% Triton X-100, 15% glycerol, phosphatase inhibitor mixture 2 and 3 (Sigma), and protease inhibitor mixture 1 and 2 (Roche). Total protein (500 μg) was incubated with 2 μg IPMK antibodies as indicated for 16 hours at 4°C and precipitated with 50 μL TrueBlot antirabbit Ig immunoprecipitation beads (eBioscience) for an additional 3 hours. The samples were washed 5 times with lysis buffer, and SDS-loading sample buffer was added. Samples were separated by SDS-PAGE and analyzed by immunoblotting as described before (41). For coimmunoprecipitates of Myc-LKB1 blots, Mouse TrueBlot ULTRA antimouse Ig horseradish peroxidase-conjugated secondary antibody (eBioscience) was used for detection with SuperSignal West Pico chemiluminescence reagent (Thermo Scientific).
Glutathione S-transferase pull-down assay
Glutathione S-transferase (GST)-tagged IPMK was coexpressed in 293T cells with either Myc-control of Myc-tagged LKB1 fragment plasmids. For GST pull-down assays, cells were lysed in lysis buffer (100mM Tris [pH 7.4], 150mM NaCl, 1% Triton X-100, 15% glycerol, and 1mM phenylmethylsulfonyl fluoride) supplemented with a complete protease inhibitor tablet (Roche) and phosphatase inhibitors (Sigma). Total protein (0.3 mg) was incubated with glutathione-Sepharose beads for 3 hours at 4°C and washed 3 times in wash buffer (100mM Tris [pH 7.4], 500mM NaCl, 1% Triton X-100, 15% glycerol). Beads were quenched in sample buffer (100mM Tris [pH 6.8], 10% glycerol, 250mM β-mercaptoethanol, 2% sodium dodecyl sulfate, and bromophenol blue). The proteins were resolved in SDS-PAGE, and tagged LKB1 was detected by anti-myc horseradish peroxidase antibody.
[3H]Inositol labeling of cells
Cells were seeded at a density of 2 × 106 cells per 10-cm dish and then labeled with 200 μCi (1 Ci = 37 GBq) [3H]inositol (PerkinElmer Life Sciences) for 4 days. Soluble inositol phosphates were extracted from labeled cells as described previously. Inositol incorporated into lipids were measured by extracting the remaining cell pellet with 0.1M NaOH and 0.1% Triton X-100 overnight at room temperature with shaking and counting a fraction of the solubilized material in a liquid scintillation counter. The 3H-labeled inositol phosphates were resolved by HPLC as described above. Soluble inositol phosphate levels were normalized against total lipid inositol content for each cell line.
Glucose uptake assay
Cell (3–4 × 106) were seeded in 6-well plates. One day later, glucose uptake was measured with an Abcam glucose uptake assay kit according to the manufacturer's instructions.
Results
Deletion of IPMK impairs metformin-mediated activation of LKB1/AMPK
It has been shown that deletion of LKB1 abolished the therapeutic effects of metformin (23). We have previously demonstrated that IPMK plays a pivotal role in glucose-mediated regulation of AMPK activation (41). Hence we first investigated whether IPMK has any role in glucose uptake. First, we deleted IPMK in MEF cells isolated from IPMK flox/flox mice by treating them with retrovirus containing Cre recombinase and selected a colony of cells that constitutively lost expression of IPMK. We confirmed a complete loss of IPMK expression in cells treated with retrovirus containing Cre recombinase (Figure 1A). We measured the glucose uptake in MEF cells described above (fl/fl vs knockout [KO]). We found that deletion of Ipmk in MEFs significantly reduced basal glucose uptake (Figure 1B).
Figure 1.
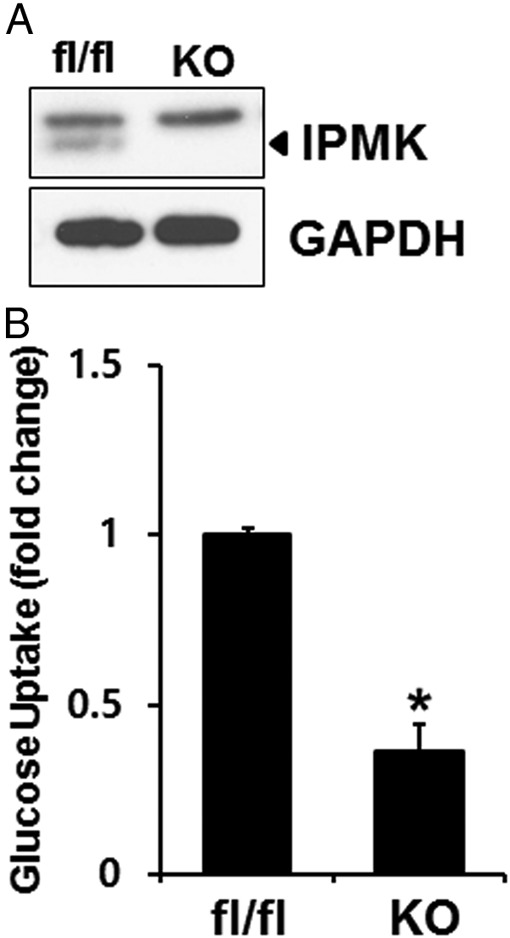
IPMK deletion reduces glucose uptake. A, IPMK in MEFs isolated from Ipmkloxp/loxp mice is deleted with retrovirus containing Cre recombinase. Blots are representative of at least 3 separate experiments. B, Glucose uptake was measured in WT (fl/fl) and Ipmk−/− (KO) mice as described in Materials and Methods. The graph represent the mean ± SE of 3 independent uptake analysis performed in at least triplicate. *, P < .005, Student's t test.
Metformin is the most widely used insulin sensitizer for type 2 diabetes and regulates glucose metabolism. Hence, we examined whether IPMK has any effects on metformin response. As we reported before (41), a deletion of IPMK led to a slight increase in basal level of phospho-AMPK and its downstream target ACC, but these changes were not statistically significant. We also incubated these cells with either 4mM metformin or 2mM AICAR for 2 hours and examined the levels of phospho-AMPK as well as its downstream target phospho-ACC. In IPMK flox/flox cells, the levels of phospho-AMPK were increased by treatment with metformin or AICAR, compared with IPMK−/− (KO MEF) cells (Figure 2). Because AMPK activation requires the phosphorylation of Thr172 in the activation loop of α1 and α2 subunits (4), AMPK activity was determined by Western blots detecting the expression levels of phospho-AMPK. Consistent with this observation, we detected an increase in phosphorylation of AMPK substrates such as ACC at Ser79 in WT MEF cells but not in KO MEF cells (Figure 2). We also confirmed that total AMPK levels were similar in both cell lines. Importantly, the failure to increase phosphorylation of AMPK in IPMK KO cells was not due to a loss of LKB1, because the expression levels of LKB1 were not affected by deletion of IPMK in MEF cells. However, we noticed that a deletion of IPMK led to a reduction in metformin-mediated phosphorylation of LKB1 at Ser428 (Supplemental Figure 1). We also transiently depleted IPMK by transfecting Cre recombinase plasmid in IPMK flox/flox cells. As shown in Figure 1C, a transient overexpression of Cre recombinase in these MEFs abolished IPMK expression and did not significantly activate AMPK after metformin or AICAR treatment (Supplemental Figure 2). AMPK is phosphorylated by not only LKB1 but also CaMKKβ. We treated either WT or KO MEFs with calcium ionophore A23187 and found that deletion of IPMK does not affect phosphorylation of AMPK by CaMKKβ (Supplemental Figure 3). Therefore, our data suggest that IPMK is required for metformin or AICAR-induced LKB1-AMPK activation.
Figure 2.
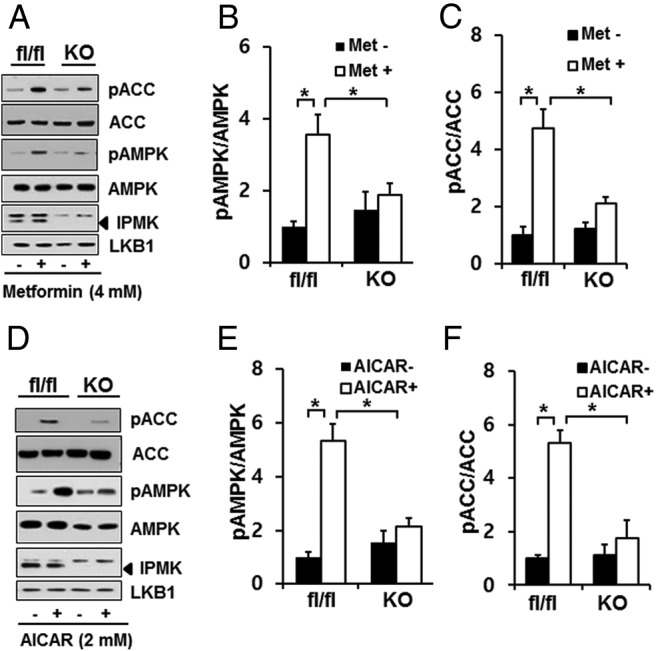
IPMK depletion attenuates LKB1-AMPK signaling in response to AMPK agonists. Cells were treated with 4mM metformin (A–C) or 2mM AICAR (D–F) for 2 hours. Cell lysates were subjected to SDS-PAGE and immunoblotting for phospho-AMPK (pAMPK) and its substrates phospho-ACC (pACC) and phospho-Raptor. Blots are representative of at least 3 separate experiments. Relative quantifications of pAMPKα2 or pACC levels are shown. Values are normalized by total levels of AMPKα2 or ACC, respectively. Values are expressed as means ± SE of 3 determinations. *, P < .005, Student's t test.
Ectopic overexpression of IPMK restored LKB1/AMPK activation by AMPK activator in IPMK−/−MEF cells
To further understand the role of IPMK in metformin action, we overexpressed WT-IPMK plasmid in IPMK−/− (KO MEF) cells and examined metformin responsiveness. We have reported previously that IPMK functions as a cofactor for mTOR complex 1 and enzyme activity of IPMK is not required because an inactive enzyme can also rescue a loss of mTOR signaling in IPMK−/− cells (38). We have overexpressed WT-IPMK as well as kinase-dead (KD)-IPMK in IPMK−/− MEF cells. First we have confirmed that overexpression of WT-IPMK restored the inositol levels, whereas KD-IPMK did not (Figure 3C). These cells were treated with metformin (2mM and 4mM), and we observed a dose-dependent activation of AMPK and its downstream target only in cells overexpressing WT-IPMK (Figure 3, A and B). As a multikinase, IPMK possesses both inositol phosphate kinase and PI3K activity, which regulates AKT signaling (39). To further examine which enzyme activities mediate the activation of LKB1-AMPK in the treatment with metformin, we overexpressed WT or plant (atIpkβ, Arabidopsis thaliana) orthologs of IPMK, which lack PI3K activity (39), in IPMK−/− MEF cells. Both WT and atIpk2 restored InsP5 and InsP6 levels even though the molecular weight of atIpk2 is lower than that of mammalian IPMK (Supplemental Figure 4). Moreover, both overexpressed WT and atIpk2β IPMK rescue AMPK activation as well as phospho-ACC in response to metformin treatment in IPMK−/− (KO MEF) cells (Supplemental Figure 4, A and B). Thus, the PI3K activity of IPMK is not required for mediating IPMK's regulation of LKB1-AMPK signal in the treatment with AMPK activators. To further understand the effect of IPMK in metformin responsiveness, we overexpressed different levels of WT-IPMK in IPMK−/− (KO MEF) cells and treated with a fixed concentration of metformin (4mM) or AICAR (2mM). Interestingly, we found that expression levels of IPMK determine the levels of phospho-AMPK activation upon metformin as well as AICAR treatment (Figure 3, D–I), suggesting that IPMK may be a limiting factor for metformin-mediated AMPK activation.
Figure 3.

Overexpression of catalytically active IPMK restores LKB1-AMPK signaling in IPMK-depleted (KO) MEFs cells. A and B, Cells (1 × 106) were transfected (electroporation) with 30 μg Myc-tagged WT IPMK or KD IPMK. Thirty hours after electroporation, cells were treated with 4mM metformin for 2 hours. Cell lysates were analyzed for phospho-AMPK (pAMPK) and phospho-ACC (pACC). Blots are representative of at least 3 separate experiments. C, Soluble InsPs (labeled IP4, IP5, IP6, and IP7) were separated by HPLC. D–I, Cells (1 × 106) were transfected (electroporation) with 15 or 30 μg of Myc-tagged WT IPMK for 30 hours. Cells were then treated with 4mM metformin (D–F) or 2mM AICAR (G–I) for 2 hours. Blots are representative of at least 3 separate experiments. Relative quantifications of pAMPKα2 or pACC levels are shown. Values are normalized by total levels of AMPKα2 or ACC, respectively, and are expressed as means ± SE of 3 determinations. *, P < .005, Student's t test.
The regulation of AMPK phosphorylation by metformin is dependent on the interaction between IPMK and LKB1
To examine the potential mechanism by which IPMK regulates metformin-mediated activation of the LKB1/AMPK pathway, we tested whether IPMK physically interacts with LKB1. In an overexpression system, we confirmed an interaction between GST-IPMK and Myc-LKB1 (Figure 4A). We have also generated truncated human LKB1 fragments (1–433, 97–433, and 197–433 amino acids) to determine the binding site for IPMK. As shown in Figure 4B, deletion of exon 1 of the LKB1 (remaining residues, 97–433 amino acids) abrogated an interaction between IPMK and LKB1, indicating IPMK binds to the N-terminal region (1–90) of LKB1 (Figure 4B).
Figure 4.
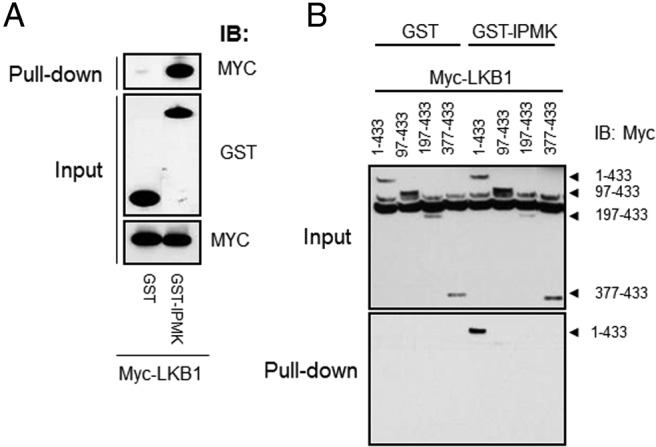
An interaction between LKB1 and IPMK is necessary for metformin-mediated AMPK activation. A, 293T cells were cotransfected with GST or GST-IPMK and Myc-LKB1 for 30 hours, followed by GST pull-down and immunoblotting for Myc-LKB1. B, Cells were cotransfected with plasmids encoding various N-terminal truncated fragments (1–433, 97–433, 197–433, or 377–433 amino acids) and either GST or a GST-IPMK in HEK293T cells for 48 hours, followed by pull-down and immunoblotting for Myc-LKB1 fragments.
We also examined the interaction region in IPMK using GST-fused fragments of IPMK described previously (38, 43). We overexpressed GST-IPMK fragments and Myc-LKB1 plasmids in HEK293T cells and performed a GST pull-down assay. We found that exon 3 and exon 6 areas bind to LKB1 (Supplemental Figure 5). However, we observed that IPMK does not affect the formation of LKB1 ternary complex with MO25 and STRAD (data not shown), nor does an interaction between LKB1 and AMPKα (Supplemental Figure 6). Furthermore, we found that IPMK enzyme activity does not affect its interaction with LKB1 (Supplemental Figure 7). To further investigate the functional consequences of IPMK-LKB1 interaction, we overexpressed the N-terminal fragment of LKB1 (encoding residues1–90) as a dominant-negative peptide in MEFs (Figure 5, A and B). Overexpression of LKB1 (1–90) disrupts endogenous IPMK-LKB1 interaction in dose-dependent manner and shows a reduction in basal phospho-ACC and phospho-Raptor levels (Figure 5A). We then examined whether endogenous IPMK-LKB1 interaction is necessary for the phospho-AMPK activation by treatment with metformin. As shown in Figure 5B, overexpression of the LKB1 (1–90) fragment abolished the metformin-mediated activation of phospho-AMPK and phospho-ACC in MEF cells. Moreover, we confirmed that overexpression of LKB1 N-terminal fragment did not influence IPMK activity (Supplemental Figure 8). Thus, IPMK and LKB1 can interact physically in the basal state, and this interaction is necessary for metformin-mediated activation of AMPK and potentially its therapeutic actions.
Figure 5.
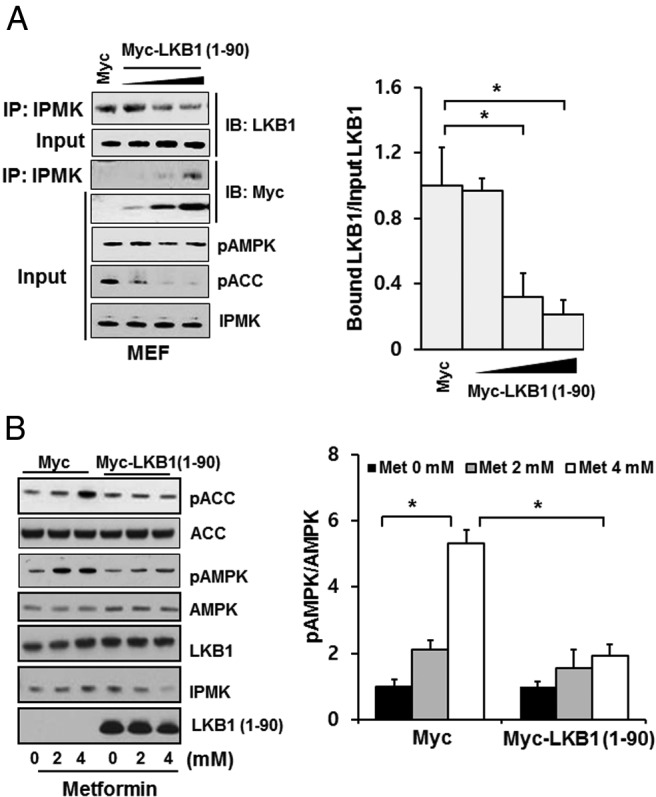
An interaction between LKB1 and IPMK is necessary for metformin-mediated AMPK activation. A, Cells were transfected with Myc or 1 to 4 μg Myc-LKB1 (1–90) for 30 hours, immunoprecipitation was performed with endogenous IPMK antibody, and bound LKB1 was detected. AMPK phosphorylation and its substrates ACC and Raptor protein expression were analyzed by Western blotting. Blots are representative of at least 3 separate experiments. Relative quantifications of bound LKB1 to IPMK are shown. Values are normalized by total levels of input LKB1 and expressed as means ± SE of 3 determinations. *, P < .005, Student's t test. B, Overexpression of LKB1 (1–90) interferes with AMPK activation in response to AICAR. MEF cells were transfected with Myc or 4 μg Myc-LKB1 (1–90) for 30 hours, followed by treatment with various concentrations of metformin for 2 hours. Cell lysates were prepared and analyzed for phospho-AMPK (pAMPK) and phospho-ACC (pACC). Blots are representative of at least 3 separate experiments. Values are normalized by total levels of AMPK and are expressed as means ± SE of 3 determinations. *, P < .005, Student's t test.
Discussion
Abnormal glucose and lipid metabolism is a hallmark of type 2 diabetes. Metformin, the most widely prescribed drug for patients with type 2 diabetes, acts at least in part by enhancing AMPK activity (8, 20, 22, 44). As the rate-limiting enzyme for the generation of higher inositol phosphate species, IPMK is involved in regulation of insulin secretion from pancreatic β-cells (45) and the development of familial type2 diabetes (46). Recently, our group reported that IPMK physically binds and regulates AMPK activity in response to glucose availability (41).
In the present study, we provide evidence that IPMK functions as an upstream regulator of LKB1-AMPK signaling in response to AMPK agonist (metformin or AICAR) treatment. We demonstrated that the metformin-induced activation of AMPK was abolished in cells lacking IPMK, and it was comparable to the effects observed in LKB1-deficient HeLa cells as reported before (23, 40). Furthermore, we found that IPMK activity was necessary to regulate metformin-mediated activation of AMPK. This regulation was independent of PI3K activities because ectopic reintroduction of A. thaliana IPMK, which lacks PI3K activity unlike the mammalian ortholog, was sufficient to rescue metformin responsiveness in IPMK−/− (KO MEF) cells (39, 40).
LKB1 contains a central serine-threonine kinase domain, an N-terminal region, and a C-terminal regulatory region (4). Our mapping studies suggest that the N-terminal area of LKB1 (1–90) or exon 3 region (amino acids 93–124) of IPMK are the minimal binding regions for its interaction between 2 proteins. In this report, we revealed that overexpressed LKB1 fragment (amino acids 1–90) not only blocks endogenous interaction between IPMK and LKB1 but also abrogates an increase in phospho-AMPK elicited by metformin treatment. It is prudent to mention that IPMK also binds to various other related signaling molecules. We have previously reported that the N-terminal fragment of IPMK (1–60) binds to mTOR/Raptor and functions as a cofactor to stabilize the mTOR complex 1 (42). More recently, we demonstrated in another study that the exon 4 area of IPMK is responsible for its interaction with AMPK, and this interaction is dynamically regulated under glucose availability (41). However, these interactions and modulation of target molecules do not require enzymatic activities of IPMK. On the other hand, our current studies identified that another region of IPMK was responsible for its interaction with LKB1 and, importantly, its catalytic activity to produce water-soluble inositol is crucial to mediate metformin responsiveness.
LKB1 is activated by interaction with STRAD and MO25, which are required for its activation and cytosolic localization (12, 13, 47). Recent studies have shown that IPMK is a nucleocytoplasmic shuttling protein (48). Our current data showed that IPMK did not directly affect LKB1/MO25/STRAD ternary complex formation or an interaction between AMPK and LKB1. Moreover, we found that IPMK did not appear to modulate LKB1 localization in the basal condition (data not shown), and yet it is possible this may occur under certain stress conditions. In fact, recent studies have shown that metformin-enhanced AMPK activity is likely via nuclear export of LKB1 (49, 50). Interestingly, we have observed that deletion of IPMK reduces metformin-mediated phosphorylation of LKB1 at Ser428, which plays a role in an export of LKB1 from nucleus to cytosol. Hence it is tempting to speculate that IPMK or its product, inositol, may play a role in metformin-mediated localization of LKB1 to modulate AMPK activation. However, further studies are needed to delineate an exact mechanism by which soluble inositols produced by IPMK regulates the LKB1/AMPK pathway.
In summary, we have identified that an interaction of IPMK and LKB1 interaction is essential for metformin-mediated activation of the LKB1/AMPK pathway. The consequence of this interaction results in subsequent AMPK signaling cascades. Our findings enhance the understanding of the role of the IPMK in regulating LKB1 functions and cellular energy balance and suggest a possible mechanism for metformin-induced AMPK activation. IPMK acts as an upstream regulator of LKB1-AMPK signaling in response to AMPK agonist (metformin or AICAR), and hence compounds that regulate IPMK activity, in particular, a production of soluble inositols may provide novel drugs for metabolic diseases such as obesity and diabetes.
Additional material
Supplementary data supplied by authors.
Acknowledgments
This work was supported by grants from the American Diabetes Association (7-13-BS-004), National Institutes of Health (DK084336), Abramson Cancer Center Core Pilot Project, and the National Alliance for Research on Schizophrenia and Depression.
Disclosure Summary: The authors have nothing to declare.
Footnotes
- ACC
- acetyl coenzyme A carboxylase
- AMPK
- AMP-activated protein kinase
- CaMKKβ
- calmodulin-dependent protein kinase kinase β
- IPMK
- inositol polyphosphate multikinase
- InsP
- inositol phosphate
- KD
- kinase-dead
- KO
- knockout
- MO
- armadillo repeat-containing mouse protein
- mTOR
- mammalian target of rapamycin
- PI3K
- phosphatidylinositol 3-kinase
- STRAD
- STE20-like pseudokinase
- WT
- wild-type.
References
- 1. Carling D. The AMP-activated protein kinase cascade–a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. [DOI] [PubMed] [Google Scholar]
- 2. Hardie DG, Carling D. The AMP-activated protein kinase–fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. [DOI] [PubMed] [Google Scholar]
- 3. Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase–development of the energy sensor concept. J Physiol. 2006;574:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. [DOI] [PubMed] [Google Scholar]
- 5. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. [DOI] [PubMed] [Google Scholar]
- 6. Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. [DOI] [PubMed] [Google Scholar]
- 7. Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. [DOI] [PubMed] [Google Scholar]
- 8. Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. [DOI] [PubMed] [Google Scholar]
- 10. Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. [DOI] [PubMed] [Google Scholar]
- 11. Baas AF, Boudeau J, Sapkota GP, et al. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003;22:3062–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boudeau J, Baas AF, Deak M, et al. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22:5102–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dorfman J, Macara IG. STRADalpha regulates LKB1 localization by blocking access to importin-alpha, and by association with Crm1 and exportin-7. Mol Biol Cell. 2008;19:1614–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lizcano JM, Göransson O, Toth R, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Woods A, Johnstone SR, Dickerson K, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. [DOI] [PubMed] [Google Scholar]
- 16. Hezel AF, Bardeesy N. LKB1; linking cell structure and tumor suppression. Oncogene. 2008;27:6908–6919. [DOI] [PubMed] [Google Scholar]
- 17. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hawley SA, Boudeau J, Reid JL, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hundal RS, Krssak M, Dufour S, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Viollet B, Guigas B, Sanz GN, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012;122:253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 23. Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keshavarz P, Inoue H, Nakamura N, Yoshikawa T, Tanahashi T, Itakura M. Single nucleotide polymorphisms in genes encoding LKB1 (STK11), TORC2 (CRTC2) and AMPK alpha2-subunit (PRKAA2) and risk of type 2 diabetes. Mol Genet Metab. 2008;93:200–209. [DOI] [PubMed] [Google Scholar]
- 25. Legro RS, Barnhart HX, Schlaff WD, et al. Ovulatory response to treatment of polycystic ovary syndrome is associated with a polymorphism in the STK11 gene. J Clin Endocrinol Metab. 2008;93:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Foretz M, Hébrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalender A, Selvaraj A, Kim SY, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hatch AJ, York JD. SnapShot: inositol phosphates. Cell. 2010;143:1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Irvine RF, Schell MJ. Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Biol. 2001;2:327–338. [DOI] [PubMed] [Google Scholar]
- 31. Saiardi A, Nagata E, Luo HR, et al. Mammalian inositol polyphosphate multikinase synthesizes inositol 1,4,5-trisphosphate and an inositol pyrophosphate. Proc Natl Acad Sci U S A. 2001;98:2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shears SB. How versatile are inositol phosphate kinases? Biochem J. 2004;377:265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. 1983;306:67–69. [DOI] [PubMed] [Google Scholar]
- 34. Verbsky J, Majerus PW. Increased levels of inositol hexakisphosphate (InsP6) protect HEK293 cells from tumor necrosis factor (alpha)- and Fas-induced apoptosis. J Biol Chem. 2005;280:29263–29268. [DOI] [PubMed] [Google Scholar]
- 35. Xu R, Paul BD, Smith DR, et al. Inositol polyphosphate multikinase is a transcriptional coactivator required for immediate early gene induction. Proc Natl Acad Sci U S A. 2013;110:16181–16186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu R, Sen N, Paul BD, et al. Inositol polyphosphate multikinase is a coactivator of p53-mediated transcription and cell death. Sci Signal. 2013;6:ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. [DOI] [PubMed] [Google Scholar]
- 38. Kim S, Kim SF, Maag D, et al. Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab. 2011;13:215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maag D, Maxwell MJ, Hardesty DA, et al. Inositol polyphosphate multikinase is a physiologic PI3-kinase that activates Akt/PKB. Proc Natl Acad Sci U S A. 2011;108:1391–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blind RD, Suzawa M, Ingraham HA. Direct modification and activation of a nuclear receptor-PIP(2) complex by the inositol lipid kinase IPMK. Sci Signal 2012;5:ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bang S, Kim S, Dailey MJ, et al. AMP-activated protein kinase is physiologically regulated by inositol polyphosphate multikinase. Proc Natl Acad Sci U S A. 2012;109:616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim S, Snyder SH. Nutrient amino acids signal to mTOR via inositol polyphosphate multikinase. Cell Cycle. 2011;10:1708–1710. [DOI] [PubMed] [Google Scholar]
- 43. Bang S, Steenstra C, Kim SF. Striatum specific protein, Rhes regulates AKT pathway. Neurosci Lett. 2012;521:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med. 2000;6:998–1003. [DOI] [PubMed] [Google Scholar]
- 45. Illies C, Gromada J, Fiume R, Leibiger B, Yu J, Juhl K, Yang SN, Barma DK, Falck JR, Saiardi A, Barker CJ, Berggren PO. Requirement of inositol pyrophosphates for full exocytotic capacity in pancreatic beta cells. Science. 2007;318:1299–1302. [DOI] [PubMed] [Google Scholar]
- 46. Kamimura J, Wakui K, Kadowaki H, et al. The IHPK1 gene is disrupted at the 3p21.31 breakpoint of t(3;9) in a family with type 2 diabetes mellitus. J Hum Genet. 2004;49:360–365. [DOI] [PubMed] [Google Scholar]
- 47. Song P, Xie Z, Wu Y, Xu J, Dong Y, Zou MH. Protein kinase Czeta-dependent LKB1 serine 428 phosphorylation increases LKB1 nucleus export and apoptosis in endothelial cells. J Biol Chem. 2008;283:12446–12455. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Meyer R, Nalaskowski MM, Ehm P, et al. Nucleocytoplasmic shuttling of human inositol phosphate multikinase is influenced by CK2 phosphorylation. Biol Chem. 2012;393:149–160. [DOI] [PubMed] [Google Scholar]
- 49. Xie Z, Dong Y, Scholz R, Neumann D, Zou MH. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu H, Moriasi CM, Zhang M, Zhao Y, Zou MH. Phosphorylation of serine 399 in LKB1 protein short form by protein kinase Cζ is required for its nucleocytoplasmic transport and consequent AMP-activated protein kinase (AMPK) activation. J Biol Chem. 2013;288:16495–16505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.