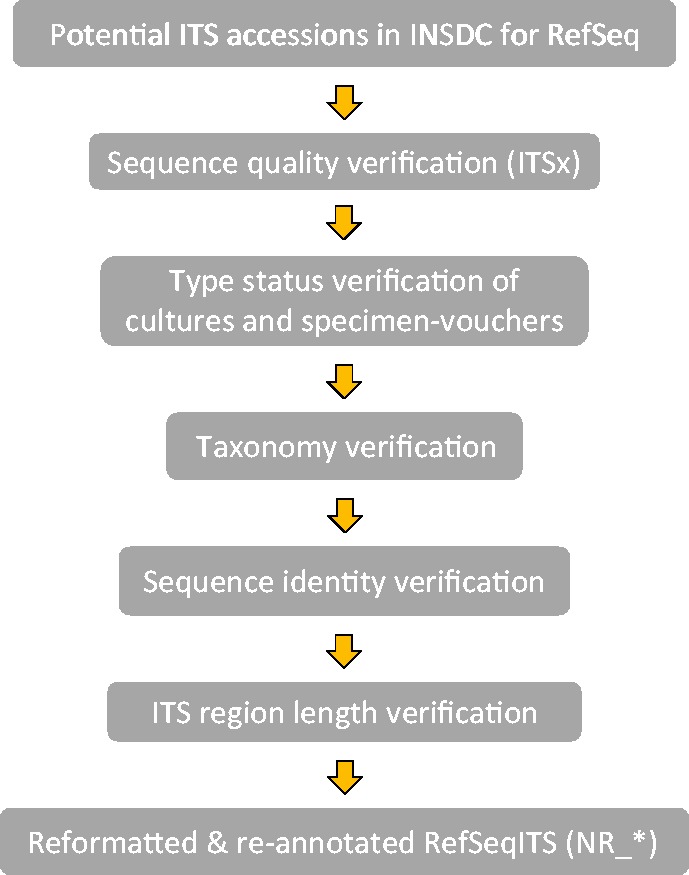
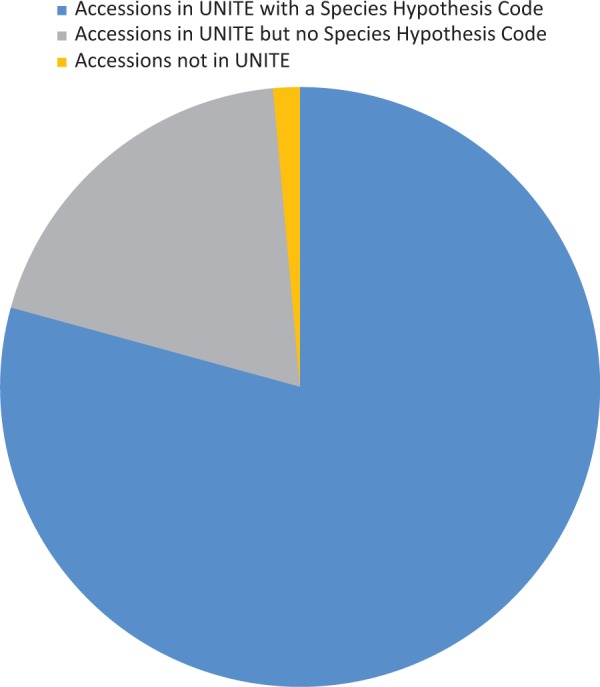
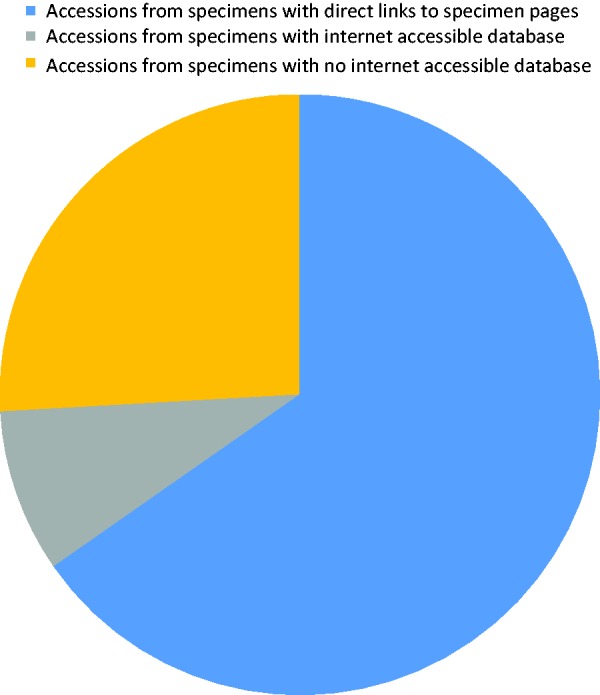
Conrad L Schoch
Conrad L Schoch
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
1,*,†,
Barbara Robbertse
Barbara Robbertse
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
1,
Vincent Robert
Vincent Robert
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
2,
Duong Vu
Duong Vu
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
2,
Gianluigi Cardinali
Gianluigi Cardinali
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
3,
Laszlo Irinyi
Laszlo Irinyi
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
4,
Wieland Meyer
Wieland Meyer
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
4,
R Henrik Nilsson
R Henrik Nilsson
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
5,
Karen Hughes
Karen Hughes
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
6,
Andrew N Miller
Andrew N Miller
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
7,
Paul M Kirk
Paul M Kirk
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
8,
Kessy Abarenkov
Kessy Abarenkov
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
9,
M Catherine Aime
M Catherine Aime
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
10,
Hiran A Ariyawansa
Hiran A Ariyawansa
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
11,
Martin Bidartondo
Martin Bidartondo
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
12,
Teun Boekhout
Teun Boekhout
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
2,
Bart Buyck
Bart Buyck
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
13,
Qing Cai
Qing Cai
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
14,
Jie Chen
Jie Chen
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
11,
Ana Crespo
Ana Crespo
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
15,
Pedro W Crous
Pedro W Crous
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
2,
Ulrike Damm
Ulrike Damm
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
16,
Z Wilhelm De Beer
Z Wilhelm De Beer
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
17,
Bryn T M Dentinger
Bryn T M Dentinger
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
8,
Pradeep K Divakar
Pradeep K Divakar
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
15,
Margarita Dueñas
Margarita Dueñas
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
18,
Nicolas Feau
Nicolas Feau
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
19,
Katerina Fliegerova
Katerina Fliegerova
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
20,
Miguel A García
Miguel A García
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
21,
Zai-Wei Ge
Zai-Wei Ge
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
14,
Gareth W Griffith
Gareth W Griffith
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
22,
Johannes Z Groenewald
Johannes Z Groenewald
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
2,
Marizeth Groenewald
Marizeth Groenewald
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
2,
Martin Grube
Martin Grube
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
23,
Marieka Gryzenhout
Marieka Gryzenhout
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
24,
Cécile Gueidan
Cécile Gueidan
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
25,
Liangdong Guo
Liangdong Guo
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
26,
Sarah Hambleton
Sarah Hambleton
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
27,
Richard Hamelin
Richard Hamelin
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
19,
Karen Hansen
Karen Hansen
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
28,
Valérie Hofstetter
Valérie Hofstetter
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
29,
Seung-Beom Hong
Seung-Beom Hong
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
30,
Jos Houbraken
Jos Houbraken
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
2,
Kevin D Hyde
Kevin D Hyde
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
11,
Patrik Inderbitzin
Patrik Inderbitzin
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
31,
Peter R Johnston
Peter R Johnston
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
32,
Samantha C Karunarathna
Samantha C Karunarathna
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
11,
Urmas Kõljalg
Urmas Kõljalg
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
9,
Gábor M Kovács
Gábor M Kovács
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
33,34,
Ekaphan Kraichak
Ekaphan Kraichak
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
35,
Krisztina Krizsan
Krisztina Krizsan
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
36,
Cletus P Kurtzman
Cletus P Kurtzman
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
37,
Karl-Henrik Larsson
Karl-Henrik Larsson
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
38,
Steven Leavitt
Steven Leavitt
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
35,
Peter M Letcher
Peter M Letcher
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
39,
Kare Liimatainen
Kare Liimatainen
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
40,
Jian-Kui Liu
Jian-Kui Liu
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
11,
D Jean Lodge
D Jean Lodge
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
41,
Janet Jennifer Luangsa-ard
Janet Jennifer Luangsa-ard
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
42,
H Thorsten Lumbsch
H Thorsten Lumbsch
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
35,
Sajeewa SN Maharachchikumbura
Sajeewa SN Maharachchikumbura
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
11,
Dimuthu Manamgoda
Dimuthu Manamgoda
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
11,
María P Martín
María P Martín
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
18,
Andrew M Minnis
Andrew M Minnis
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
43,
Jean-Marc Moncalvo
Jean-Marc Moncalvo
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
44,
Giuseppina Mulè
Giuseppina Mulè
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
45,
Karen K Nakasone
Karen K Nakasone
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
46,
Tuula Niskanen
Tuula Niskanen
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
40,
Ibai Olariaga
Ibai Olariaga
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
28,
Tamás Papp
Tamás Papp
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
36,
Tamás Petkovits
Tamás Petkovits
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
36,
Raquel Pino-Bodas
Raquel Pino-Bodas
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
47,
Martha J Powell
Martha J Powell
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
39,
Huzefa A Raja
Huzefa A Raja
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
48,
Dirk Redecker
Dirk Redecker
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
49,
J M Sarmiento-Ramirez
J M Sarmiento-Ramirez
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
18,
Keith A Seifert
Keith A Seifert
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
27,
Bhushan Shrestha
Bhushan Shrestha
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
50,
Soili Stenroos
Soili Stenroos
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
47,
Benjamin Stielow
Benjamin Stielow
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
2,
Sung-Oui Suh
Sung-Oui Suh
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
51,
Kazuaki Tanaka
Kazuaki Tanaka
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
52,
Leho Tedersoo
Leho Tedersoo
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
9,
M Teresa Telleria
M Teresa Telleria
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
18,
Dhanushka Udayanga
Dhanushka Udayanga
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
11,
Wendy A Untereiner
Wendy A Untereiner
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
53,
Javier Diéguez Uribeondo
Javier Diéguez Uribeondo
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
18,
Krishna V Subbarao
Krishna V Subbarao
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
31,
Csaba Vágvölgyi
Csaba Vágvölgyi
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
36,
Cobus Visagie
Cobus Visagie
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
2,
Kerstin Voigt
Kerstin Voigt
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
54,
Donald M Walker
Donald M Walker
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
55,
Bevan S Weir
Bevan S Weir
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
32,
Michael Weiß
Michael Weiß
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
56,
Nalin N Wijayawardene
Nalin N Wijayawardene
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
11,
Michael J Wingfield
Michael J Wingfield
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
17,
J P Xu
J P Xu
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
57,
Zhu L Yang
Zhu L Yang
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
14,
Ning Zhang
Ning Zhang
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
58,
Wen-Ying Zhuang
Wen-Ying Zhuang
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
26,
Scott Federhen
Scott Federhen
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA, 2CBS-KNAW Fungal Biodiversity Centre, P.O. Box 85167, 3508 AD Utrecht, The Netherlands, 3Department of Pharmaceutical Sciences – Microbiology, Università degli Studi di Perugia, Perugia, Italy, 4Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, Australia, 5Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Göteborg, Sweden, 6Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37920, USA, 7Illinois Natural History Survey, University of Illinois, 1816 South Oak Street, Champaign, IL 61820, USA, 8Mycology Section, Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3DS, UK, 9Natural History Museum, University of Tartu, 46 Vanemuise, 51014 Tartu, Estonia, 10Purdue University, Department of Botany and Plant Pathology, 915 W. State Street, West Lafayette, IN 47907, USA, 11Institute of Excellence in Fungal Research, and School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand, 12Imperial College London, Royal Botanic Gardens, Kew TW9 3DS, England, UK, 13Muséum National d’Histoire Naturelle, Dépt. Systématique et Evolution CP39, UMR7205, 12 Rue Buffon, F-75005 Paris, France, 14Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, P. R. China, 15Departamento de Biología Vegetal II, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040, Spain, 16Senckenberg Museum of Natural History Görlitz, PF 300 154, 02806 Görlitz, Germany, 17Department of Microbiology and Plant Pathology, Forestry Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0001, South Africa, 18Real Jardín Botánico, RJB-CSIC, Plaza de Murillo 2, 28014 Madrid, Spain, 19Department of Forest and Conservation Sciences, Faculty of Forestry, The University of British Columbia, 2424 Main Mall, Vancouver, BC, Canada, 20Institute of Animal Physiology and Genetics, Czech Academy of Sciences, v.v.i., Videnska 1083, Prague, Czech Republic, 21Department of Biology, University of Toronto, 3359 Mississagua Road, Mississagua, Ontario L5L 1C6, Canada, 22Institute of Biological, Environmental and Rural Sciences, Prifysgol Aberystwyth, Aberystwyth, Ceredigion Wales SY23 3DD, UK, 23Institute of Plant Sciences, Karl-Franzens-University, Holteigasse 6, 8010 Graz, Austria, 24Department of Plant Sciences, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa, 25CSIRO-Plant Industry, CANBR, GPO Box 1600, Canberra ACT 2601, Australia, 26State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, 27Biodiversity (Mycology and Microbiology), Agriculture and Agri-Food Canada, 960 Carling Avenue, Ottawa, Ontario, Canada, 28Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05, Stockholm, Sweden, 29Agroscope Changins-Wädenswil Research Station ACW, Département de recherche en Protection des végétaux grandes cultures et vigne/Viticulture et oenologie, CP 1012, CH-1260 Nyon, Switzerland, 30Korean Agricultural Culture Collection, National Academy of Agricultural Science, RDA, Suwon, 441-707, Korea, 31University of California, Davis Department of Plant Pathology Davis, CA 95616, USA, 32Landcare Research, Private Bag 92170, Auckland 1142, New Zealand, 33Eötvös Loránd University, Institute of Biology, Department of Plant Anatomy, Pázmány Péter sétány 1/c, 1117 Budapest, Hungary, 34Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, H-1525, Hungary, 35Science and Education, The Field Museum, 1400 S. Lake Shore Drive, Chicago, IL 60605, USA, 36University of Szeged, Faculty of Science and Informatics, Department of Microbiology, Kozep fasor 52, Szeged, H-6726, Hungary, 37Bacterial Foodborne Pathogens and Mycology Research Unit, U.S. Department of Agriculture, National Center for Agricultural Utilization Research, Agricultural Research Service, 1815 North University Street, Peoria, IL 61604, USA, 38Natural History Museum, P.O. Box 1172 Blindern, 0318 Oslo, Norway, 39Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA, 40Plant Biology, Department of Biosciences, P.O. Box 65, 00014 University of Helsinki, Finland, 41USDA Forest Service, NRS, PO Box 1377, Luquillo, Puerto Rico, 42National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Paholyothin Road, Pathum Thani 12120 Thailand, 43Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, 1550 Linden Drive, Madison, WI 53706, USA, 44Department of Natural History, Royal Ontario Museum, and Department of Ecology Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada, 45Institute of Sciences of Food Production, National Research Council (CNR), Via Amendola 122/O, Bari, Italy, 46Center for Forest Mycology Research, Northern Research Station, U.S. Forest Service, One Gifford Pinchot Drive, Madison, WI 53726-2398, USA, 47Botanical Museum, Finnish Museum of Natural History, FI-00014 University of Helsinki, Finland, 48The University of North Carolina at Greensboro, Department of Chemistry and Biochemistry, 457 Sullivan Science Building, P.O. Box 26170, Greensboro, NC 27402-6170, USA, 49Université de Bourgogne, UMR1347 Agroécologie, BP 86510, F-21000 Dijon, France, 50Institute of Life Science and Biotechnology, Sungkyunkwan University,Suwon 440-746, Korea, 51Mycology and Botany Program, American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110, USA, 52Faculty of Agriculture and Life Science, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan, 53Department of Biology, Brandon University, Brandon, Manitoba, Canada, 54Jena Microbial Resource Collection, Leibniz Institute for Natural Product Research and Infection Biology and University of Jena, Jena, Germany, 55Department of Natural Sciences, The University of Findlay, Findlay, OH 45840, USA, 56Institute of Evolution and Ecology, University of Tübingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 57Department of Biology, McMaster University, Hamilton, Ontario L8S 4K1, Canada and 58Department of Plant Biology and Pathology, Rutgers University, New Brunswick, NJ 08901, USA
1