

ABSTRACT
The process of transgenesis involves the introduction of a foreign gene, the transgene, into the genome of an animal. Gene transfer by pronuclear microinjection (PNI) is the predominant method used to produce transgenic animals. However, this technique does not always result in germline transgenic offspring and has a low success rate for livestock. Alternate approaches, such as somatic cell nuclear transfer using transgenic fibroblasts, do not show an increase in efficiency compared to PNI, while viral-based transgenesis is hampered by issues regarding transgene size and biosafety considerations. We have recently described highly successful transgenesis experiments with mice using a piggyBac transposase-based vector, pmhyGENIE-3. This construct, a single and self-inactivating plasmid, contains all the transpositional elements necessary for successful gene transfer. In this series of experiments, our laboratories have implemented cytoplasmic injection (CTI) of pmGENIE-3 for transgene delivery into in vivo-fertilized pig zygotes. More than 8.00% of the injected embryos developed into transgenic animals containing monogenic and often single transgenes in their genome. However, the CTI technique was unsuccessful during the injection of in vitro-fertilized pig zygotes. In summary, here we have described a method that is not only easy to implement, but also demonstrated the highest efficiency rate for nonviral livestock transgenesis.
Keywords: pig, piggyBac, pmGENIE-3, transgenesis, transposase
The production of transgenic pigs by cytoplasmic injection of piggyBac-based pmGENIE-3 plasmids into in vivo-produced one cell zygotes results in efficient transgenesis rates; at least one transgenic piglet was born every time a sow farrowed.
INTRODUCTION
Mammalian transgenesis is an important tool in biomedicine, as it allows researchers to create transgenic animals (TAs) that can be used as model systems for human disease and drug discovery. The original and often-used protocol for transgenesis was developed by Gordon et al. [1], who demonstrated that the pronucleus microinjection (PNI) of exogenous linear transgene DNA into the male mouse pronucleus of zygotes resulted in germline TAs. PNI was subsequently also applied to rabbits, sheep, and pigs [2]. Two decades later, intracytoplasmic sperm injection transgenesis (ICSI-Tr) was developed and applied to the mouse. Here, exogenous linear DNA is complexed with freeze-thawed spermatozoa and subsequently, after removal of their tail, spermatozoon heads are injected into metaphase II (MII) -arrested oocytes [3]. Both PNI and ICSI-Tr are passive transgenesis techniques, which rely on the DNA double-stranded break (DSB) repair mechanisms of zygote nuclei for transgene integration [4]. Physical and chemical mutagens can cause DSBs within the chromosomes of zygotes and have to be repaired quickly in order for embryo development to proceed. Integration of exogenous transgene DNA into the host genome takes place at these DSB sites by nonhomologous DNA recombination, where the linear transgene is usually inserted as a head-to-tail concatemer [5]. This concatenation occurs prior to chromosomal integration, and is considered an extrachromosomal event through homologous recombination that joins circular and linear DNA molecules [5]. Because DSBs occur randomly in the zygote genome and then induce the cellular DNA repair machinery, no control over the transgene integration site can be exercised. As the yield of TAs obtained by PNI or ICSI-Tr techniques is low, in the range of 1%–4.6% of embryos injected (ei) resulting in transgenic founders, the nontargeted integrations are thought to be the result of rare intranuclear events. While functional in mice, ICSI-Tr has proven more challenging in livestock and pigs in particular [6–8]. Recently, a transgenic pig and one transgenic fetus were produced when injecting stored bore spermatozoa into 702 in vitro-matured (IVM) oocytes during ICSI-Tr. When nuclear donor cells were collected from this ICSI-Tr transgenic piglet and used for somatic cell nuclear transfer, six live transgenic clones were obtained with the same transgene copy number in the cloned piglets as was detected in the original ICSI-Tr-produced founder pig [9]. The same group of researchers was then able to use ICSI-Tr to generate transgenic pigs with large linearized bacterial artificial chromosome constructs [10], and was also able to produce a number of transgenic fetuses by ICSI-Tr with moderately sized EGFP transgenes [11]. Therefore, the ability to use the passive ICSI-Tr procedure in pigs effectively might be subject to the different technical approaches used by each individual experimenter, and may result in alternate outcomes from different laboratories.
The first active transgenesis procedure introduced was based on lentiviral transgene vectors [12], initially developed in rodents and then extended to farm animals [13]. While efficient in germline transgenesis, this method is not commonly used for the production of founder animals, as a large number of embryos do not survive its implementation. However, the ratio of animals born (ab) to TAs is large, usually around 80.0% (TA/ab). Other important issues with lentiviral vector use are well documented elsewhere [14, 15].
Mobile genetic elements known as transposons have emerged as an alternative means of active transgenesis [16, 17]. These plasmid-based DNA elements are capable of self-directed excision and subsequent reintegration within the host genome. However, they lack the ability of viruses to traverse the plasma membrane of the cell, and therefore have to be delivered into the cytosol or nucleus by either chemical or physical means. Several class II transposases catalyzing this cut-and-paste DNA transposition in mammalian cells have been described, among them piggyBac (pB), Sleeping Beauty (SB), and Tol2 [18]. Specifically, these enzymes recognize and bind to the terminal repeat elements (TREs) flanking transposons, cut this DNA segment from the donor and reinsert it into the recipient host's genome. We have evaluated several binary, plasmid-based transposase/transposon systems, where the donor plasmid contains the transgenes within the TREs of the transposon, while the helper plasmid encodes the transposase. In our hands, the piggyBac transposase (pBt) isolated from the moth, Trichoplusia ni, proved the most efficient during the transfection of several mammalian cell lines [18], with similar observations being reported by other investigators [19, 20]. These same investigators additionally compared a hyperactive version of the pBt with the most active SB transposase version (SB100X) with similar conclusions [19, 21]. Both SB100X and the hyperactive pBt have now been applied to mammalian transgenesis, demonstrating germline transmission of the transgene [17, 22]. In the case of SB, donor plasmid is coinjected into zygote pronuclei together with mRNA coding for the SB100X transposase [22]. For pBt-mediated transgenesis, we developed an alternative approach, where the helper and donor plasmid components were incorporated into a single plasmid construct known as pmGENIE-3 [23]. Recently, these pBt-based self-inactivating plasmids with a large cargo capacity were additionally engineered to contain the hyperactive form of pBt and with an antibiotic-free selection in bacteria (pmhyGENIE-3) [17] to conform to regulatory recommendations for the elimination of antibiotic resistance markers during transgenesis [24]. In comparison to SB100X, due to the high transposition efficiency of the pBt, it is not necessary to use its mRNA for effective transgenesis. Therefore, the cumbersome process of producing and handling mRNA can be avoided. Indeed, the efficiency of transposition for these pmhyGENIE-3 plasmids by transposase-enhanced pronuclear microinjection (te-PNI) is high enough to support direct zygote cytoplasmic injection (CTI) of the constructs for the production of transgenic mice [17]. The ei efficiency values for such CTIs are comparable with those described for lentiviral transgenesis [12]. Additionally, concerns regarding the safety of pmhyGENIE-3 plasmids, due to the fact that a CAG promoter remains in the transposon, potentially influencing neighboring genes in the host genome [25], have proven unfounded [26]. The preventive measures undertaken during the construction of the pmhyGENIE-3 plasmids, such as the inclusion of stop codons in the chimeric intron engineered into the pBt, are sufficient to prevent aberrant gene activation from surrounding genes near the site of transposon insertion [26]. During in vivo gene therapy experiments, this transpositional efficiency of the pmhyGENIE-3 plasmids is able to support durable hepatic gene delivery of transgenes by ultrasound-targeted microbubble destruction in mice [27], further demonstrating its ability as an effective transgene integrating vector.
The SB100X transposase has recently been applied to active pig transgenesis with an efficiency rate for ei by CTI of 5.8% for transgenic piglets born, or 6.8% if recovered transgenic fetuses are also included for the rate calculations [28]. Such efficiencies are higher than the currently available nonviral passive methods for producing transgenic pigs, including pigs cloned from transfected transgenic fibroblast [7, 29, 30]. We sought to apply our efficient mouse transgenesis strategy, CTI, to pigs. This approach allowed us to avoid centrifugation of pig embryos deemed necessary to visualize their pronuclei usually masked by the dark lipids found in their cytoplasm [31]. CTI in pigs with pmGENIE-3 proved successful, resulting in monogenic transgene insertions into the host genome with the observed transgenesis percentages (ei) being significantly higher than those reported for alternative pig transgenesis methods [7, 9, 28–30].
MATERIALS AND METHODS
Ethics Statements
This study was carried out in strict accordance with The Instructive Notions with Respect to Caring for Laboratory Animals, issued by the Ministry of Science and Technology of China. The animal experimental protocol was approved by the Institutional Animal Care and Use Committee of South China Agricultural University. All efforts were made to minimize animal suffering.
Plasmids
Plasmid pmGENIE-3-EGFP was constructed as previously described [17, 23]. Plasmid mPB was a kind gift from the Wellcome Trust Sanger Institute (Cambridgeshire, UK), and it was constructed as previously described [32]. Plasmid pEGFP-N1 was purchased from Clontech (Mountain View, CA).
Animals
In vivo-fertilized embryo (IVFZ) donors (Yorkshire and Duroc sows) and embryo recipients (Landrace sows) were raised in a swine farm located in Yunfu City, Guangdong Province, China.
Ovary Collection and Oocyte Maturation
Porcine ovaries were purchased from the Guangzhou Tianhe slaughterhouse located at Tianhe district, Guangzhou city, Guangdong Province, China. We obtained permission from the slaughterhouse to use the porcine ovaries for our current experiments. Cumulus-oocyte complexes (COCs) were aspirated from the ovaries and matured in vitro for 42–44 h following the protocol described by Deng et al. [33]. Matured COCs were freed from cumulus cells by repeated pipetting in 0.10% hyaluronidase. IVM oocytes with the first polar body were selected for parthenogenetic activation (PA) or in vitro-fertilization (IVF).
Preparation of PA Embryos
At 3 h after CTI with different concentrations of plasmid solution, IVM oocytes were activated by a single 80-μs DC pulse at 1.4 kV/cm, using an electrofusion instrument (model CF-150/B; BLS Company, Budapest, Hungary). The PA embryos were then cultured in porcine zygote medium 3 (PZM3) containing Cytochalasin B (5 μg/ml) for 4 h postactivation.
Preparation of IVF Embryos
Preparation of porcine IVF embryos was performed as previously described [34]. Briefly, freshly collected semen was incubated at 39°C for 30 min. The semen was then purified by a two-step Percoll gradient method by first washing twice in Dulbecco PBS supplemented with 0.10% (w/v) bovine serum albumin and centrifugation at 250 × g for 4 min. The pellet was resuspended and layered on top of a 45:90 discontinuous Percoll gradient and centrifuged at 300 × g for 30 min. After discarding the final supernatant, the pellet was resuspended in 5 ml porcine gamete medium (PGM) and washed twice at 150 × g for 5 min. The spermatozoa were diluted with PGM to 2 × 106 cells/ml and capacitated by incubating in a 5.00% CO2 incubator at 39°C for 20 min. IVM oocytes were transferred into PGM medium and incubated with capacitated spermatozoa for 6 h; subsequently, oocytes were washed three times with PZM3 and then cultured with PZM3 in a 5.00% CO2 incubator at 39°C.
Collection of IVFZs at the One-Cell Zygote Stage
Sows showing signs of spontaneous estrus were artificially inseminated three times with Yorkshire or Duroc boar semen (3 billion spermatozoa in 80 ml of semen extender) with an interval of 12 h between each insemination. At 12 h after the last insemination, sows were anesthetized with ketamine and xylazine for induction and 3.00% isofluorane for maintenance. Both of their oviducts were removed surgically and IVFZs were flushed out from the detached oviducts with PBS solution using a syringe with a 12-gauge needle.
CTI of pmGENIE-3-EGFP Plasmids
The pmGENIE-3-EGFP plasmids of high purity and in supercoiled conformation were diluted in 10 mM Tris-HCl (pH 7.6) and 0.25 mM EDTA (pH 8.0), and backfilled into a bevel-tipped injection capillary pipette with a 10-μm internal diameter. IVM oocytes prior to PA, or one-cell zygote-stage embryos 16–18 h after IVFZs, were immobilized by suction to a holding pipette, while the injection capillary pipette was manually pushed though the zona pellucida and plasma membrane. Approximately 10 pl of 20 ng/μl plasmid solution was injected into the embryo cytoplasm using a pressure-controlled Eppendorf transjector 5246 (Eppendorf, Hamburg, Germany). For cleaved (two-cell stage) in vivo-derived embryos, each blastomere was injected with approximately 10 pl of plasmid solution.
In Vitro Culture of Embryos
Injected embryos were transferred into PZM3 medium and cultured at 39°C, 5.00% CO2, 7.00% O2, 88.00% N2, and 100% humidity for 168 h (7 days). The time of fertilization (for IVF embryo) or activation for PA embryos was defined as 0 h. The fusion rate, cleavage rate, and blastocyst formation rate of cultured embryos was assessed at 1, 24, and 168 h, respectively. The embryos expressing EGFP were counted under fluorescence microscopy.
Embryo Transfer
The pmGENIE-3-EGFP plasmid-injected embryos were cultured in PZM3 at 39°C with 5.00% CO2, 7.00% O2, 88.00% N2, and 100% humidity for 20 h, and then loaded into a transparent transfer tube, which was kept in a portable incubator (Minitube, WI) during transportation to the farm where the recipient sows are housed. Estrous-synchronized sows were used as embryo recipients for all transfer experiments. They were anesthetized with ketamine and xylazine for induction and with 3.00% isofluorane for maintenance. One oviduct was exposed by surgery. The plasmid-injected embryos (ranging in number from 14 to 33 for each recipient sow) were delivered directly into the oviduct of the recipient sows using the transparent transfer tube attached to the syringe. The transfer tube was examined subsequently via microscope to ensure the transfer of all the embryos. The pregnancy status of the recipient sows was monitored using an ultrasound machine equipped with a convex transducer at approximately 1 mo after embryo transfer.
Delivery of Piglets
If spontaneous farrowing did not occur until Gestation Day 116, the recipient sows were injected with a prostaglandin analog (200 μg/recipient) and, after 24 h, they were delivered by vaginal birth under supervision or with assistance. The newborn piglets were weighed and clinically examined.
Observation of EGFP Expressed by Epifluorescence in Transgenic Pigs and Their Internal Organs and Tissues
Transgenic pigs and their organs or tissues expressing EGFP were visualized by the Living Organism's Fluorescent Protein Observation System, consisting of a blue light lamp with maximum excitation at 488 nm and goggles with light filters (model FBL; BLS Ltd., Hungary). Photographs of transgenic pigs, wild-type pigs, and their organs and tissues were taken under blue light or bright light by a digital camera with and without light filters.
PCR Analysis
Genomic DNA was isolated from biopsies of fetuses or piglets using a Tissue DNA extraction kit (Omega Bio-Tek, Norcross, GA). A 538-bp fragment of the EGFP gene, a 443-bp fragment of pB 3′-TRE, a 509-bp fragment of pB 5′-TRE, and a 114-bp fragment of the internal control β-actin gene were amplified by PCR using primer sets P1+P2, P3+P4, P5+P6, and P7+P8, respectively (for primer sequences and their location in the pmGENIE-3-EGFP plasmid see Supplemental Table S1 and Supplemental Fig. S1A; all Supplemental Data are available online at www.biolreprod.org). The PCR products were sequenced to confirm their identity.
Southern Blot Analysis
Tail genomic DNA (10 μg) from each of the four firs-born transgenic piglets was restriction digested with BamH I, purified by ethanol precipitation, and then separated by electrophoresis in a 0.80% agarose gel. The DNA was subsequently transferred to a nylon membrane (GE Lifesciences, Shanghai, China) by the capillary transfer method. The membrane was then prehybridized overnight at 42°C and then hybridized for 5 h at 42°C with a probe amplified by primer sets P19+P20 (for primer sequences see Supplemental Table S1). The probe is complementary to mouse codon bias piggyBac gene start region and piggyBac 3′-TRE region (Supplemental Fig. S1B), and labeled with digoxigenin (DIG) by using a PCR DIG Probe Synthesis Kit (Roche Applied Science, China). Hybridization and washing were performed with DIG-High Prime DNA Labeling and Detection Starter KitII (Roche Applied Science, China). After hybridization, the membrane was incubated for 30 min in blocking solution and subsequently incubated for a further 30 min in anti-DIG-AP antibody solution. The membrane was subsequently exposed for 5–20 min to incubation with 1 ml of CSPD ready-to-use solution, and a Southern blot photograph was captured with an EC3 imaging system (UPA, CA).
After delivery of all 10 piglets, a second Southern blot was generated for their genomic DNA and analyzed with a probe to the EGFP transgene in the transposon. The genomic DNA of the stillborn transgenic pig #9 (TG9) was found to be degraded, and was omitted from the Southern blot. Tail genomic DNA (10 μg) from each transgenic piglet was restriction digested with Hind III, purified by ethanol precipitation, and then separated by electrophoresis in a 0.80% agarose gel. The DNA was subsequently transferred to a nylon membrane (GE Life sciences, Shanghai, China) by the capillary transfer method, as described above. The membrane was prehybridized for 1 h and then hybridized overnight at 55°C with a probe amplified by primer sets P17+P18 (for primer sequences see Supplemental Table S1). The probe is complementary to the EGFP gene (Supplemental Fig. S1C), and was labeled with DIG by using a PCR DIG Probe Synthesis Kit (Roche Applied Science, China). Hybridization and washing were performed with DIG-High Prime DNA Labeling and Detection Starter Kit II (Roche Applied Science, China). After hybridization, the membrane was incubated as described above for 30 min in blocking solution and subsequently incubated for a further 30 min in anti-DIG-AP antibody solution. The membrane was subsequently exposed for 5–20 min following incubation with 1 ml of CSPD ready-to-use solution, and a Southern blot photograph was captured with an EC3 imaging system (UPA).
Real-Time Quantitative PCR Analysis
A real-time quantitative PCR (real-time qPCR) method, as described previously [35], was used to determine the copy number of EGFP transgenes integrated into the genome of transgenic pigs. This number was compared to that obtained by counting the number of bands detected in Southern blots performed on the four first-born transgenic piglets. Briefly, 20 ng of genomic DNA was analyzed by real-time qPCR using the SYBR Green-Taq polymerase kit and ABI Prism 7700 PCR instrument. Primer sets P9+P10 and P11+P12 (for primer sequences see Supplemental Table S1) were used to amplify the EGFP transgenes and the reference gene β-actin, respectively. The copy number of the β-actin gene was used as reference to estimate the copy number of EGFP transgenes in the genome. The sequence of the 100-bp fragment of porcine β-actin gene amplified by P11 and P12 primers (Supplemental Table S1) was blasted against the Sus scrofa (pig) genomic DNA sequence database (build Sscrofa10.2) on NCBI BLAST website. A number of two-blast hits was found per haploid genome. Since the DNA sample used for real-time qPCR was isolated from diploid cells of transgenic pigs, we estimated that there are four copies of 100-bp β-actin amplicon in each diploid genome of transgenic pigs. This number was used as a normalization standard for the calculation of copy number for the EGFP transgenes in the genome of transgenic pigs, using the 2−ΔΔCt method based on the threshold cycle (Ct) values, as described by Livak and Schmittgen [36].
Inverse PCR Analysis
Genomic DNA (1 μg) was digested with BamHI. Restriction sites for BamHI are located between the two CAG promoters, one driving the expression of the EGFP gene in the transposon, and the other controlling the expression of the mPB gene. A second BamHI restriction site is situated in the mpB gene just past the chimeric intron, which contains the pB 3′-TRE in the pmGENIE-3-EGFP plasmid (Supplemental Fig. S1A). The digestion product was purified by a DNA purification column (Qiagen) and eluted with 100 μl of double-distilled H2O (ddH2O). After adjustment with ddH2O and T4 ligase buffer to a final required volume prior to ligase enzyme addition, T4 ligase was added to a final concentration of 10 U/μl in a 1000-μl final ligation mixture. The ligation reaction was allowed to proceed overnight by incubation at 16°C, and ligated DNA was purified via a Qiagen DNA purification column after elution from the column with 100 μl of ddH2O. A 2-μl elution aliquot was used as a template for the PCR reaction with primer sets P13+P14 or P15+P16 (for primer sequences and their location in the pmGENIE-3-EGFP plasmid see Supplemental Table S1 and Supplemental Fig. S1A). The resulting PCR products were cloned by ligation into a TA vector (Life Technologies, Carlsbad, CA) and sequenced to prove their accuracy. The obtained sequences were blasted against the sequence of the transgene vector pmGENIE-3-EGFP and the Sus scrofa (pig) genomic DNA sequence database (build Sscrofa10.2) on the NCBI BLAST website to determine the integration sites of 5′-TRE of the pB transposon.
Statistical Analysis of Data in Tables
The positive rate of each group in columns labeled as “No. of cleaved embryos expressing EGFP (% positive rate)” in Supplemental Tables S3, S4, and S5, were statistically compared. Data were analyzed by the GENMOD Procedure in the SAS program (SAS Institute, Cary, NC). The assumed distribution is binomial, and the link function is logit. Values labeled with group name as superscript indicate that it is statistically different (P < 0.05) from the value of labeled groups in the same column. Values without the label of group name as superscript indicate that it is not statistically different (P > 0.05) from the values of other groups in the same column.
RESULTS
Parthenogenetic Production of Porcine Embryos Expressing EGFP after CTI, with Different Concentrations of pmGENIE-3-EGFP Plasmid
To determine the optimal plasmid concentration for the production of transgenic embryos by CTI, different concentrations of pmGENIE-3-EGFP plasmids [23] (Supplemental Fig. S1A) were injected into IVM oocytes prior to PA. Injection with 10 ng/μl and 50 ng/μl of pmGENIE-3-EGFP resulted in cleaved green fluorescent embryos at rates of 57.55% and 52.63%, respectively (Supplemental Table S2). This rate is significantly higher for cleaved green fluorescent embryos than the other plasmid concentrations used (Supplemental Table S2), and also resulted in higher numbers of EGFP+ blastocysts. This observation suggests that the ideal concentration of pmGENIE-3-EGFP during CTI is between 10 and 50 ng/μl of plasmid DNA for PA-IVM oocytes, and we therefore used a pmGENIE-3-EGFP concentration of 20 ng/μl for all consecutive experiments. A microscopic image of CTI and resulting EGFP+ embryos are depicted in Figure 1.
FIG. 1.

Generation of EGFP+ pig embryos by CTI. A) Microscopic image of CTI performed on pig one-cell embryos with pmGENIE-3-EGFP plasmid. B) Microscopic image of EGFP+ porcine cleaved embryos derived from CTI with pmGENIE-3-EGFP plasmids. Bars = 100 μm.
To analyze whether pBt-mediated transgene integration could be confirmed in EGFP+ embryos, 9 EGFP+ blastocysts produced by CTI (using 20 ng/μl pmGENIE-3-EGFP) and after PA were randomly selected. Genomic DNA from each of these nine blastocysts was analyzed by PCR. The results (Fig. 2) indicated that 88.80% (8/9) of the analyzed green fluorescence-expressing blastocysts harbored the EGFP transgene, and that this transgene is inserted into the genome of 87.50% (7/8) of such blastocysts via pB transposition. Using primer set P5+P6, we obtained a 509-bp PCR product from a region of the plasmid that is not transferred to the host genome during transpositional insertion (Fig. 2 and Supplemental Fig. S1A) [23], indicating the presence of either episomal plasmid within this blastocyst embryo or nontranspositional pmGENIE-3-EGFP fragment insertions (Fig. 2, blastocyst #8). As we did not detect this amplicon in any of the remaining DNA samples, we assumed transpositional transgene insertion for these blastocyst embryos.
FIG. 2.

PCR analysis of EGFP-expressing porcine blastocysts produced by CTI with pmGENIE-3-EGFP plasmid. Top panel: a 538-bp PCR amplicon generated from genomic DNA obtained from EGFP+ blastocysts. Middle panel: a 509-bp amplicon spanning the pB 5′-TRE region sequence and an adjacent sequence in the plasmid backbone of pmGENIE-3-EGFP. It is only possible to generate this amplicon if circular pmGENIE-3-EGFP is present in EGFP+ blastocyst, or if the plasmid fragments have been inserted into the host genome by a nontranspositional mechanism. Lower panel: amplification of β-actin was used as internal loading control. MW, molecular weight marker; PC, pmGENIE-3-EGFP plasmid control used as template; WT, use of genomic DNA from a noninjected blastocyst as PCR template wild-type control.
Production of Transgenic Porcine Fetuses by CTI with pmGENIE-3-EGFP Plasmid into Unfertilized IVM Oocytes prior to Parthenogenetical Activation
To explore whether CTI with the pmGENIE-3-EGFP plasmid could be used to produce transgenic porcine fetuses that would implant into the sow's uterus, we injected plasmid constructs at a concentration 20 ng/μl into 180 IVM oocytes followed by PA. Subsequent to this activation, 126 CTI-treated cleaved embryos were transferred to two recipient surrogate sows. We recovered 10 fetuses from one of the pregnant surrogates at Gestation Day 27, three of which expressed EGFP (Fig. 3). This observation indicated that 1.66% (3/180) of CTI- and PA-treated oocytes (ei), 2.38% (3/126) of transferred embryos (et), or 30.00% (3/10) of the recovered fetuses are transgenic.
FIG. 3.

EGFP expression in CTI-treated, PA, transgenic porcine fetuses. TG, transgenic; WT, wild type. Bar = 10 mm.
The PCR analysis depicted in Figure 4 indicated that all three transgenic fetuses (TG1-TG3) carried the EGFP transgene. In two transgenic fetuses (TG2 and TG3), the transgene had been integrated into the genome via pB transposition, while, for one fetus (TG1), we detected pB 3′-TRE and 5′-TRE and regions corresponding to the plasmid backbone that generated PCR amplicons, indicating nontranspositional integration of the transgene. This insert was probably a linearized pmGENIE-3 fragment, integrated into the genome of the fetus by the DNA repair machinery (Fig. 4 and Supplemental Fig. S1A).
FIG. 4.

PCR analysis of genomic DNA from the transgenic fetuses recovered. Top panel: 538-bp EGFP amplicon. Second panel: PCR amplification product of a 443-bp region spanning the pB transposon 3′-TRE sequence and an adjacent sequence of the pBt gene outside of the transposon. Third panel: 509-bp amplicon spanning the pB transposon 5′-TRE sequence and an adjacent sequence on the plasmid backbone, outside the region of the pB transposon. Fourth panel: amplification of β-actin was used as internal loading control. MW, molecular weight marker; PC, pmGENIE-3-EGFP plasmid control used as template; WT, genomic DNA from a wild-type pig served as PCR template.
To assess the transgene copy number in these fetuses, we performed a real-time qPCR analysis. We noted that the genome of fetus TG1 contained about 14 copies of the EGFP transgene. This transgene copy number is indicative of a concatemerized insertion of a broken linearized plasmid (Fig. 5) [37–40]. It is also much higher than that observed in fetuses TG2 and TG3, for which we demonstrated transpositional integration by the lack of a PCR product for pB 3′- and 5′-TRE with backbone regions of the plasmid (Fig. 4 and Supplemental Fig. S1A).
FIG. 5.

Real-time qPCR analysis of three PA, transferred into surrogate mothers and Gestation Day 27 recovered transgenic fetuses, and all 10 transgenic (TG) born piglets. The data are representative of 10 repeat real-time qPCRs (error bars represent SEM). WT1, wild-type fetus; WT2, wild-type pig.
Production of Porcine Embryos Expressing EGFP by CTI Coinjection of pmGENIE-3-EGFP and mPB Plasmids
As the pBt does not demonstrate overexpression inhibition [17, 23, 26, 32, 41], we explored whether the coinjection of pmGENIE-3-EGFP and mPB (a pBt-only-expressing plasmid) could increase the rate of transgenic embryos produced, as compared to embryos resulting from only pmGENIE-3-EGFP injection (Supplemental Table S2). To that effect, we performed the following CTI, for which 20 ng/μl pmGENIE-3-EGFP and different concentrations of mPB were coinjected into IVM oocytes prior to PA. The results indicated (Supplemental Table S3) that coinjection of pmGENIE-3-EGFP with various concentrations of mPB (10.0, 20.0, and 40.0 ng/μl) enhanced the rate of cleaved EGFP+-expressing embryos in a positive way (66.10%, 74.51%, or 79.55%), as compared to injections of pmGENIE-3-EGFP plasmid alone (46.94%). However, the rate of blastocysts expressing EGFP was reduced in all cases when compared to cleaved embryo rates (12.82%, 15.79%, and 20.00%).
Comparison of Transgene Integration Efficiencies Between the pmGENIE-3-EGFP Plasmid and a Noninsertional Plasmid During CTI
To investigate if the transgenesis efficiency mediated by the pBt-based plasmid, pmGENIE-3-EGFP, is higher than that associated with a noninsertional plasmid, the previously tested pB plasmid combination (20 ng/μl of pmGENIE-3-EGFP + 40 ng/μl of mPB) (Supplemental Table S4) and a smaller noninsertional EGFP-expressing plasmid (8.4 ng/μl of linearized pEGFP-N1) were injected by CTI into IVM oocytes prior to PA. The results (Supplemental Table S4) demonstrated that 51.61% of cleaved embryos injected with the pB plasmids expressed EGFP, while only 15.69% of cleaved embryos receiving the smaller linearized pEGFP-N1 plasmid expressed the chromophore. This suggests that the pB plasmids resulted in a much higher transgene expression efficiency during CTI in comparison with that observed from noninsertional plasmids.
Production of Porcine Embryos Expressing EGFP by CTI Coinjection of pmGENIE-3-EGFP and mPB Plasmids at Different Time Points after IVF
To determine the best time point for CTI treatment of IVF embryos, the previously tested plasmid combination (20 ng/μl of pmGENIE-3-EGFP + 40 ng/μl of mPB) was delivered by CTI into IVF-generated zygotes at 7–8 h, 10–11 h, 13–14 h, or 16–18 h post-IVF. We achieved the best results for CTI performed at 16–18 h after IVF (Supplemental Table S5), resulting in 73.08% EGFP+ embryos. At all other time points, this EGFP+ embryo efficiency was reduced (53.66%, 54.55%, or 62.50%). To reiterate, the rates of all, and particularly of EGFP-expressing blastocysts, were greatly reduced when compared to the cleaved embryo rates (Supplemental Table S5). The rate of blastocyst formation for the noninjected controls was 15.32%, but for the injected embryos, the highest rate obtained was 2.24%, with the rates for EGFP+ blastocyst being even lower (Supplemental Table S5).
Attempts to Produce Transgenic Pigs by CTI Coinjection of pmGENIE-3-EGFP and mPB Plasmids into IVF-Derived Zygotes
To explore whether CTI coinjection of pmGENIE-3-EGFP and mPB plasmids into IVF-derived embryos could be used to produce transgenic pigs, the previously tested pB plasmid combination (20 ng/μl of pmGENIE-3-EGFP + 40 ng/μl of mPB) was delivered by CTI into 641 IVF-derived zygotes at 16–18 h postfertilization. These injected embryos were then transferred to six recipient sows. No live piglets were born from all six surrogates receiving IVF embryos (Supplemental Table S6), suggesting that the resilience of IVF-produced zygotes for transgenic pig attainment is not optimal.
Production of Transgenic Pigs by CTI with the pmGENIE-3-EGFP Plasmid Delivered into IVFZs
Because our previous experiments with mice demonstrated that in vivo-produced embryos are able to reach term during CTI [17], we tested if transgenic pigs could be produced by the same injection method of pmGENIE-3-EGFP plasmids into IVFZ-derived pig zygotes. We collected 177 embryos from the oviducts of 20 artificially inseminated sows, and CTI treated these embryos at 16–18 h after in vivo fertilization with 10 pl of 20 ng/μl pmGENIE-3-EGFP. We transferred 166 injected embryos to seven recipient sows, five of which farrowed and gave birth to live piglets. Of the 25 piglets delivered from these five pregnant recipients, 10 expressed EGFP (Fig. 6 and Table 1). The overall transgenesis efficiency was 5.60% for ei, or 6.00% for et, and 40.00% for piglets born (ab). The efficiency for transgenic pig production during CTI improves to 8.10% for ei, or 8.80% for et, and 40.00% for (ab) when we removed the two nonfarrowed sows from the transgenesis rate calculations. As shown in Table 1, the transgenic efficiencies for individual experiments ranged from 0.00% for all parameters measured to 15.00% for ei, 21.40% for et, or 60.00% for ab. In summary, the overall efficiency of CTI for zygotes derived from in vivo-fertilized oocytes was at 71.42% (5/7) for these experiments, and we demonstrated that every time a sow farrowed, at least one transgenic piglet was born. All of the transgenic piglets expressed EGFP by epifluorescence, with one of them showing variegated EGFP expression (Supplemental Fig. S2A). Dissection of a transgenic piglet demonstrated that all its internal organs expressed EGFP (Supplemental Fig. S2B).
FIG. 6.

Transgenic and wild-type piglets under bright field and fluorescence lighting. Transgenic piglet expresses even EGFP by epifluorescence.
TABLE 1.
Production of transgenic pigs by CTI with pmGENIE-3-EGFP plasmid delivered into IVFZ embryos.
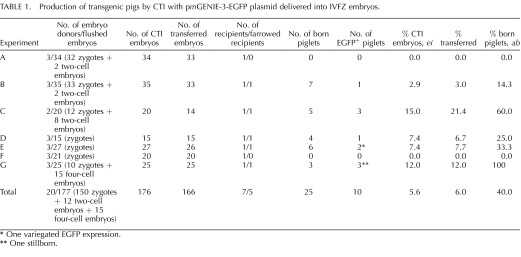
One variegated EGFP expression.
* One stillborn.
A PCR analysis confirmed transpositional EGFP integration (Fig. 7), as PCR reactions with primers spanning the 3′- and 5′-TREs and the plasmid backbone did not generate amplicons (Supplemental Fig. S1A), whereas we obtained EGFP amplicons with the appropriate primers.
FIG. 7.

PCR analysis of genomic DNA from the ten transgenic piglets born. Top panel: 538-bp EGFP amplicon. Second panel: PCR amplification product of a 443-bp region spanning the pB transposon 3′-TRE sequence and an adjacent sequence of the pBt gene outside of the transposon. Third panel: 509-bp amplicon spanning the pB transposon 5′-TRE sequence and an adjacent sequence on the plasmid backbone, outside the region of the pB transposon. Fourth panel: amplification of β-actin was used as internal loading control. MW, molecular weight marker; PC, pmGENIE-3-EGFP plasmid control used as template; WT, genomic DNA from a wild-type pig served as PCR template.
The real-time qPCR result revealed that TG1 and TG4 piglets carried two copies of the EGFP transgene, while TG2 and TG3 piglets harbored only one copy of the EGFP transgene (Fig. 5). The piggyBac gene start and 3′-TRE region of the transposon for the first-born four transgenic piglets was visualized by Southern blot analysis (Fig. 8), confirming that, as detected by real-time qPCR (Fig. 5), two copies of the transgene region were integrated into the genome of TG1 and TG4 transgenic piglets, while only one copy of the same transgene region could be detected in the genomes of TG2 and TG3 transgenic piglets. After all the piglets were born, a Southern blot performed for detection of the EGFP transgene confirmed the transgene copy number detected in the first four piglets done with a probe to the piggyBac gene start and 3′-TRE region of the transposon. For all the other piglets, except for TG9, the genomic DNA of which had degraded, the EGFP transgene copy number mirrored that detected by real-time qPCR (Supplemental Fig. S3; Fig. 5). The genomic location of the EGFP transgene insertion was analyzed by inverse PCR, and one insertion site for the transgene was identified for each of the first-born four transgenic piglets (Supplemental Fig. S4).
FIG. 8.

Southern blot analysis of the first four born transgenic pigs, done with a probe to the pB gene start and 3′-TRE. MW, molecular weights.
DISCUSSION
The most popular transgenesis method, known as PNI, was developed 33 yr ago in mice [1]. It was subsequently introduced into larger animals 5 yr after these original rodent experiments [2], and has remained as a reliable method for the production of TAs. However, its use in agricultural TA production remains limited, because of the inadequate efficiency of the method in inserting recombinant DNA in to host genomes. This inadequacy is further compounded by the lack of two pronuclear stage zygotes availability from livestock, in most cases. To address some of these issues, the Institute for Biogenesis Research at the University of Hawaii developed a procedure in 1999 known as ICSI-Tr [3]. Although this technique allowed the insertion of very large transgenes (>500 kb) into the genomes of mice [42], it still does not achieve such productivity when applied to livestock. However, progress was recently reported for its application in pigs, and future experimenters may have better success with it [9–11]. A more efficient method for the production of TAs is the lentiviral approach developed in 2002 [12]. The advantages of this method are its applicability to many species; however, due to the lentiviral packaging size constraints, larger transgenes cannot be introduced by this approach. The efficiency, however, of lentiviral-based transgenesis is unsurpassed, as 80.0% of ab can be transgenic during its implementation. Recently, a CRISPR/Cas-mediated mouse transgenesis method for small transgenes has been described [43]. The delivery of larger transgenes with this method would result in the most effective in vivo genome engineering procedure to date.
Transposon-based systems are highly efficient in integrating transgenes into a host genome. However they do not possess the ability to traverse the protective membranes of cells as lentiviruses do, but need to be introduced into cells by chemical or physical means. We have engineered unique plasmid vectors that contain the transposon cargo and the pBt gene on a single-helper independent plasmid, pmGENIE-3) [23], and its hyperactive pBt alternative, pmhyGENIE-3 [17]. In the latter's case, this pBt is based on a previously described hyperactive mammalian codon-optimized pBt variant [44]. During CTI in mice, pmhyGENIE-3 worked very effectively when IVFZs were used as transgene recipients. We hypothesized that CTI could be implemented in pigs, where, recently, the SB transposase introduced in its mRNA form during CTI together with a donor plasmid had demonstrated the ability to contribute to the production of transgenic pigs [28].
Our attempts to utilize IVM oocytes and IVF-generated zygotes proved unsuccessful for the production of transgenic pigs. There are two possible reasons why we could not obtain live piglets from IVF-derived embryos treated with CTI. For one, it could be that the coinjection of mPB with pmGENIE-3-EGFP is detrimental to embryo survival. Injections of additional pBt genes may excessively enhance pB transposition activity, which significantly increases the copy numbers of integrated transposons per genome. The resulting overinsertion of transgenes could have been lethal to embryo survival due to insertional mutagenesis. However, an examination of the data in Supplemental Table S2 indicated that, when considering PA embryos in the control sample, which are not injected, of the 198 embryos activated, 116 made it to the blastocyst stage. This represents 58.59% of embryos becoming blastocysts. If we examine the rest of the data for injected PA-cleaved embryos for all plasmid concentrations, none approaches this rate, with most of the rates being 50% of the control or lower (27.00%, 26.70%, 25.00%, 15.15%, 12.50%, 15.60%, 17.71%, and 13.68%). Analysis of the numbers of blastocysts expressing EGFP from such cleaved embryos demonstrates that this rate drops to even lower levels. Similar observations can be made for the injections of PA embryos in Supplemental Table S4 and for IVF-generated embryos in Supplemental Table S5. From this last table, it is apparent that the rate of blastocyst development for control IVF embryos (15.32%) is considerably lower than the PA-activated and noninjected embryos in Supplemental Table S2 (58.59%). The blastocyst development rate for the injected IVF embryos in Supplemental Table S5 decreases to the extremely low rates of 1.49%, 0.74%, 2.24%, and 0.75%. The number of EGFP+ blastocysts under these conditions is very low (Supplemental Table S5). It becomes apparent that IVM oocytes do not tolerate microinjection very well after IVF. Therefore, the most likely reason might be that the use of IVM oocytes and IVF generated one cell zygotes for CTI are contributing to the decreased developmental potential of injected embryos, since several studies have shown that porcine IVF embryos have a very poor full-term developmental ability when certain media conditions are lacking [45–48]. Before improvements to culture media, observations first described by Charles Thibault in 1972 [49] postulated that the quality of IVM oocytes was lower than that of those allowed to mature in ovarian follicles [50]. Thibault theorized that IVM oocytes lack a “male pronucleus growth factor (MPGF).” Improvements made to culture media for most species have eclipsed the mention of MPGF, but this may not be accurate for pig oocytes. We find it improbable that not a single IVF-generated and CTI-treated pig embryo, either EGFP+ or EGFP−, did not reach term (Supplemental Table S6), and that this lack of development was due to overinsertion of pB transposons. We do not observe such lack of development when in vivo-fertilized mouse zygotes are injected with the same concentration of the more active pmhyGENIE-3-EGFP plasmid by CTI. In fact, this plasmid supports the generation of transgenic mice by CTI at the high concentration of 50 ng/μl [17]. Therefore, the most probable reason for the lack of IVF-produced pig embryo development to term is the concentration and energy substrate availability in the culture medium in which the IVF embryos are cultured. Recently, the sequential addition of nonessential amino acids and essential amino acids have produced a modified culture medium based on the original North Carolina State University 23 medium. This new, improved medium was demonstrated to better suit the changing metabolic needs of pig embryos [51]. Perhaps future use of such a modified culture medium could result in some of the IVF-produced pig embryos reaching term during CTI-based transgenesis experiments. As IVM and IVF embryos are more desirable for transgenesis experiments than alternatively produced embryos, because of their ease of production, such culture medium anomalies should be further explored in future porcine transgenesis experiments, in order to improve their outcome. Nonetheless, during the course of these experiments, we determined the optimal conditions for manipulating CTI with the pmGENIE-3-EGFP construct. When we applied these finding to the generation of transgenic pigs with IVFZs, every farrowed sow delivered at least one transgenic piglet. Our approach is also less cumbersome than the SB-based method, as it does not require the use of mRNA as a source for the transposase. We transferred CTI embryos to seven recipient sows, five of which delivered live offspring. A total of 10 of the 25 piglets were transgenic, one of which was stillborn. All of the transgenic piglets were EGFP+, with only one of them showing variegated EGFP expression (Supplemental Fig. S2A). Germline transgenesis requires the transgenic pigs to reach adulthood. Since the dissection of a transgenic piglet demonstrated that not only its skin, but also all its organs, including its testis, expressed EGFP, we assume that sperm produced from such a pig will pass on the transgene to the next generation of piglets (Supplemental Fig. S2B).
We analyzed the genomic DNA of the first four piglets (TG1–TG4) by Southern blot to a region of the transposon near its 3′-TRE and real-time qPCR for EGFP: two piglets had one transgene and the other two had two transgenes incorporated into their genome. This observation was similar to that in mice [17], where we applied the same analysis techniques. A second Southern blot probing for the EGFP transgene and a real-time qPCR data to EGFP for all the transgenic piglets (TG1–TG10), except for TG9, the genomic DNA of which had degraded, demonstrated that all transgenic piglet copy numbers identified for EGFP mirrored the numbers detected by real-time qPCR. Therefore, three different detection procedures to two regions of the same transposon detect the same number of transgene copies and verify that the use of real-time qPCR for predicting transgene copy numbers is a valid and accurate detection method during transgenesis experiments.
The ability to produce TAs in two different species carrying only one transgene per genome on a regular basis is encouraging for the application of CTI to other livestock species, where animals carrying a single transgene copy may be desirable. In the experiments described herein, we did not employ our newly developed and more efficient transgenesis method, te-PNI [17]. This method, implemented with the more effective pmhyGENIE-3-EGFP construct, may result in even greater transgenesis rates in pigs, similar to the improvements that we observed for mouse transgenesis [17]. If that is so, it might prove advantageous to centrifuge the pig IVFZ embryos in order to visualize their nuclear structures, usually impeded during light microscopic techniques by the high lipid content of their cytoplasm. When linked with use of the antibiotic gene free backbone of the pmhyGENIE-3-EGFP construct [17], which conforms to the Food and Drug Administration's requirement for the elimination of antibiotic marker gene releases into the environment, te-PNI or CTI may become powerful tools for TA production.
Supplementary Material
ACKNOWLEDGMENT
We thank the Sanger Institute (Cambridge, England, UK) for the mouse codon-optimized piggyBac plasmid.
Footnotes
Supported by National High Technology Research and Development Program of China (863 Program) grant 2011AA100304, National Science Foundation for Young Scholars of China grant 31101689, Department of Science and Technology of Guangdong grants 2011A020102003 and 2011A020201009, and National Institutes of Health grants 5P20RR024206 and R01 GM083158-01A1 to S.M.
These authors contributed equally to this work.
REFERENCES
- Gordon JW, Scangos GA, Plotkin DJ, Barbarosa JA, Ruddle FH. Genetic transformation of mouse embryos by microinjection of purified DNA. PNAS 1980; 77: 7380–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer RE, Pursel VG, Rexroad CE, Jr., , Wall RJ, Bolt DJ, Ebert KM, Palmiter RD, Brinster RL. Production of transgenic rabbits, sheep and pigs by microinjection. Nature 1985; 315: 680–683. [DOI] [PubMed] [Google Scholar]
- Perry AC, Wakayama T, Kishikawa H, Kasai T, Okabe M, Toyoda Y, Yanagimachi R. Mammalian transgenesis by intracytoplasmic sperm injection. Science 1999; 284: 1180–1183. [DOI] [PubMed] [Google Scholar]
- Perry AC. Hijacking oocyte DNA repair machinery in transgenesis? Mol Reprod Dev 2000; 56: 319–324. [DOI] [PubMed] [Google Scholar]
- Smith K. Theoretical mechanisms in targeted and random integration of transgene DNA. Reprod Nutr Dev 2001; 41: 465–485. [DOI] [PubMed] [Google Scholar]
- Garcia-Vazquez FA, Ruiz S, Grullon LA, de Ondiz A, Gutierrez-Adan A, Gadea J. Factors affecting porcine sperm mediated gene transfer. Res Vet Sci 2011; 91: 446–453. [DOI] [PubMed] [Google Scholar]
- Garcia-Vazquez FA, Ruiz S, Matas C, Izquierdo-Rico MJ, Grullon LA, De Ondiz A, Vieira L, Aviles-Lopez K, Gutierrez-Adan A, Gadea J. Production of transgenic piglets using ICSI-sperm-mediated gene transfer in combination with recombinase RecA. Reproduction 2010; 140: 259–272. [DOI] [PubMed] [Google Scholar]
- Wu Y, Liu CJ, Wan PC, Hao ZD, Zeng SM. Factors affecting the efficiency of producing porcine embryos expressing enhanced green fluorescence protein by ICSI-mediated gene transfer method. Anim Reprod Sci 2009; 113: 156–166. [DOI] [PubMed] [Google Scholar]
- Kurome M, Ueda H, Tomii R, Naruse K, Nagashima H. Production of transgenic-clone pigs by the combination of ICSI-mediated gene transfer with somatic cell nuclear transfer. Transgenic Res 2006; 15: 229–240. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Kurome M, Matsunari H, Nakano K, Umeyema K, Shiota A, Nakauchi H, Nagashima H. The creation of transgenic pigs expressing human proteins using BAC-derived, full-length genes and intracytoplasmic sperm injection-mediated gene transfer. Transgenic Res 2012; 21: 605–618. [DOI] [PubMed] [Google Scholar]
- Umeyama K, Saito H, Kurome M, Matsunari H, Watanabe M, Nakauchi H, Nagashima H. Characterization of the ICSI-mediated gene transfer method in the production of transgenic pigs. Mol Reprod Dev 2012; 79: 218–228. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 2002; 295: 868–872. [DOI] [PubMed] [Google Scholar]
- Hofmann A, Kessler B, Ewerling S, Weppert M, Vogg B, Ludwig H, Stojkovic M, Boelhauve M, Brem G, Wolf E, Pfeifer A. Efficient transgenesis in farm animals by lentiviral vectors. EMBO Rep 2003; 4: 1054–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J, Yao S. Retrovirus silencing and vector design: relevance to normal and cancer stem cells? Curr Gene Ther 2005; 5: 367–373. [DOI] [PubMed] [Google Scholar]
- Park F. Lentiviral vectors: are they the future of animal transgenesis? Physiol Genomics 2007; 31: 159–173. [DOI] [PubMed] [Google Scholar]
- Katter K, Geurts AM, Hoffmann O, Mates L, Landa V, Hiripi L, Moreno C, Lazar J, Bashir S, Zidek V, Popova E, Jerchow B. et al. Transposon-mediated transgenesis, transgenic rescue, and tissue-specific gene expression in rodents and rabbits. FASEB J 2013; 27: 930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marh J, Stoytcheva Z, Urschitz J, Sugawara A, Yamashiro H, Owens JB, Stoytchev I, Pelczar P, Yanagimachi R, Moisyadi S. Hyperactive self-inactivating piggyBac for transposase-enhanced pronuclear microinjection transgenesis. Proc Natl Acad Sci U S A 2012; 109: 19184–19189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SC, Meir YJ, Coates CJ, Handler AM, Pelczar P, Moisyadi S. Kaminski JM. piggyBac is a flexible and highly active transposon as compared to Sleeping Beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci U S A 2006; 103: 15008–15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Kong J, Stalker J, Bradley A. Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis 2009; 47: 404–408. [DOI] [PubMed] [Google Scholar]
- Wilson MH, Coates CJ, George AL., Jr. PiggyBac transposon-mediated gene transfer in human cells. Mol Ther 2007; 15: 139–145. [DOI] [PubMed] [Google Scholar]
- Doherty JE, Huye LE, Yusa K, Zhou L, Craig NL, Wilson MH. Hyperactive piggyBac gene transfer in human cells and in vivo. Hum Gene Ther 2012; 23: 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katter K, Geurts AM, Hoffmann O, Mates L, Landa V, Hiripi L, Moreno C, Lazar J, Bashir S, Zidek V, Popova E, Jerchow B. et al. Transposon-mediated transgenesis, transgenic rescue, and tissue-specific gene expression in rodents and rabbits. FASEB J 2013; 27: 930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urschitz J, Kawasumi M, Owens J, Morozumi K, Yamashiro H, Stoytchev I, Marh J, Dee JA, Kawamoto K, Coates CJ, Kaminski JM, Pelczar P. et al. Helper-independent piggyBac plasmids for gene delivery approaches: strategies for avoiding potential genotoxic effects. Proc Natl Acad Sci U S A 2010; 107: 8117–8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Carnes AE, Hodgson CP. Plasmid DNA vaccine vector design: impact on efficacy, safety and upstream production. Biotechnol Adv 2009; 27: 353–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett PB, Largaespada DA, Switzer KC, Cooper LJ. Evaluating risks of insertional mutagenesis by DNA transposons in gene therapy. Transl Res 2013; 161: 265–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urschitz J, Moisyadi S. Transpositional transgenesis with. Mob Genet Elements 2013; 3: e25167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CD, Urschitz J, Khemmani M, Owens JB, Moisyadi S, Shohet RV, Walton CB. Ultrasound directs a transposase system for durable hepatic gene delivery in mice. Ultrasound Med Biol 2013; 39: 2351–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrels W, Mates L, Holler S, Dalda A, Taylor U, Petersen B, Niemann H, Izsvak Z, Ivics Z, Kues WA. Germline transgenic pigs by Sleeping Beauty transposition in porcine zygotes and targeted integration in the pig genome. PLoS One 2011; 6: e23573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen JE, Li J, Kragh PM, Moldt B, Lin L, Liu Y, Schmidt M, Winther KD, Schyth BD, Holm IE, Vajta G, Bolund L. et al. Pig transgenesis by Sleeping Beauty DNA transposition. Transgenic Res 2011; 20: 533–545. [DOI] [PubMed] [Google Scholar]
- Uchida M, Shimatsu Y, Onoe K, Matsuyama N, Niki R, Ikeda JE, Imai H. Production of transgenic miniature pigs by pronuclear microinjection. Transgenic Res 2001; 10: 577–582. [DOI] [PubMed] [Google Scholar]
- Wall RJ, Pursel VG, Hammer RE, Brinster RL. Development of porcine ova that were centrifuged to permit visualization of pronuclei and nuclei. Biol Reprod 1985; 32: 645–651. [DOI] [PubMed] [Google Scholar]
- Cadinanos J, Bradley A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res 2007; 35: e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Yang D, Zhao B, Ouyang Z, Song J, Fan N, Liu Z, Zhao Y, Wu Q, Nashun B, Tang J, Wu Z. et al. Use of the 2A peptide for generation of multi-transgenic pigs through a single round of nuclear transfer. PLoS One 2011; 6: e19986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Li Z, Yu B, He X, Shi J, Zhou R, Liu D, Wu Z. Effects of DNMT1 and HDAC inhibitors on gene-specific methylation reprogramming during porcine somatic cell nuclear transfer. PLoS One 2013; 8: e64705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Xu Z, Zou X, Zeng F, Shi J, Liu D, Urschitz J, Moisyadi S, Li Z. Pig transgenesis by piggyBac transposition in combination with somatic cell nuclear transfer. Transgenic Res 2013; 22: 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(t)) method. Methods 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Bishop JO, Smith P. Mechanism of chromosomal integration of microinjected DNA. Mol Biol Med 1989; 6: 283–298. [PubMed] [Google Scholar]
- Dellaire G, Yan J, Little KC, Drouin R, Chartrand P. Evidence that extrachromosomal double-strand break repair can be coupled to the repair of chromosomal double-strand breaks in mammalian cells. Chromosoma 2002; 111: 304–312. [DOI] [PubMed] [Google Scholar]
- Folger KR, Wong EA, Wahl G, Capecchi MR. Patterns of integration of DNA microinjected into cultured mammalian cells: evidence for homologous recombination between injected plasmid DNA molecules. Mol Cell Biol 1982; 2: 1372–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth DB, Wilson JH. Relative rates of homologous and nonhomologous recombination in transfected DNA. Proc Natl Acad Sci U S A 1985; 82: 3355–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett PB. Integrating DNA vectors for gene therapy. Mol Ther 2007; 15: 10–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira PN, Pozueta J, Giraldo P, Gutierrez-Adan A, Montoliu L. Generation of yeast artificial chromosome transgenic mice by intracytoplasmic sperm injection. Methods Mol Biol 2006; 349: 151–161. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013; 153: 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, Craig NL. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A 2011; 108: 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ock SA, Lee SL, Kim JG, Kumar BM, Balasubramanian S, Choe SY, Rho GJ. Development and quality of porcine embryos in different culture system and embryo-producing methods. Zygote 2007; 15: 1–8. [DOI] [PubMed] [Google Scholar]
- Kwak SS, Jeung SH, Biswas D, Jeon YB, Hyun SH. Effects of porcine granulocyte-macrophage colony-stimulating factor on porcine in vitro-fertilized embryos. Theriogenology 2012; 77: 1186–1197. [DOI] [PubMed] [Google Scholar]
- Kim S, Lee SH, Kim JH, Jeong YW, Hashem MA, Koo OJ, Park SM, Lee EG, Hossein MS, Kang SK, Lee BC, Hwang WS. Anti-apoptotic effect of insulin-like growth factor (IGF)-I and its receptor in porcine preimplantation embryos derived from in vitro fertilization and somatic cell nuclear transfer. Mol Reprod Dev 2006; 73: 1523–1530. [DOI] [PubMed] [Google Scholar]
- Abeydeera LR, Wang WH, Cantley TC, Rieke A, Prather RS, Day BN. Presence of epidermal growth factor during in vitro maturation of pig oocytes and embryo culture can modulate blastocyst development after in vitro fertilization. Mol Reprod Dev 1998; 51: 395–401. [DOI] [PubMed] [Google Scholar]
- Thibault C. Final stages of oocyte maturation. : Biggers JDSA. (ed.), Oogenesis. Baltimore: University Press; 1972: 397–411. [Google Scholar]
- Kren R, Kikuchi K, Nakai M, Miyano T, Ogushi S, Nagai T, Suzuki S, Fulka J, Fulka J., Jr. Intracytoplasmic sperm injection in the pig: where is the problem? J Reprod Dev 2003; 49: 271–273. [DOI] [PubMed] [Google Scholar]
- Beebe LFS, McIlfatrick SM, Vassiliev I, Nottle MB. Development of an improved porcine embryo culture medium for cloning, transgenesis and embryonic stem cell isolation. Clon Transgen 2013; 2: 107 doi: 10.4172/2168-9849.1000107 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.