

ABSTRACT
The epithelium lining the epididymis has a pivotal role in ensuring a luminal environment that can support normal sperm maturation. Many of the individual genes that encode proteins involved in establishing the epididymal luminal fluid are well characterized. They include ion channels, ion exchangers, transporters, and solute carriers. However, the molecular mechanisms that coordinate expression of these genes and modulate their activities in response to biological stimuli are less well understood. To identify cis-regulatory elements for genes expressed in human epididymis epithelial cells, we generated genome-wide maps of open chromatin by DNase-seq. This analysis identified 33 542 epididymis-selective DNase I hypersensitive sites (DHS), which were not evident in five cell types of different lineages. Identification of genes with epididymis-selective DHS at their promoters revealed gene pathways that are active in immature epididymis epithelial cells. These include processes correlating with epithelial function and also others with specific roles in the epididymis, including retinol metabolism and ascorbate and aldarate metabolism. Peaks of epididymis-selective chromatin were seen in the androgen receptor gene and the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which has a critical role in regulating ion transport across the epididymis epithelium. In silico prediction of transcription factor binding sites that were overrepresented in epididymis-selective DHS identified epithelial transcription factors, including ELF5 and ELF3, the androgen receptor, Pax2, and Sox9, as components of epididymis transcriptional networks. Active genes, which are targets of each transcription factor, reveal important biological processes in the epididymis epithelium.
Keywords: epididymis epithelium, male infertility, sperm maturation, transcriptional regulation
Open chromatin in human epididymis epithelial cells reveals transcription factor networks that may coordinate normal gene expression.
INTRODUCTION
The human epididymis is a tightly coiled tube that forms a critical part of the male reproductive tract. Immature sperm leaving the testis through the efferent ducts pass through the caput, corpus, and cauda of the epididymis on their way to the vas deferens and ejaculation. During this passage they mature and undergo alterations that subsequently confer the ability to swim and fertilize an egg. The epithelium that lines the epididymis plays a pivotal role in maintenance of the luminal environment, which is required for normal sperm maturation. However, relatively little is known about the molecular basis for the coordinated function in this epithelium. Here we applied techniques of open chromatin mapping to identify cis-acting elements that regulate expression of genes encoding ion channels, ion exchangers, transporters, and solute carriers, which establish and maintain the luminal environment in the epididymis.
Any disease, whether inherited or acquired, that impacts the function of the epididymis will likely impair male fertility. An example is the common genetic disease cystic fibrosis (CF), which is associated with a high rate (97%) of male infertility due to genital duct abnormalities. The cystic fibrosis transmembrane conductance regulator (CFTR) gene, which, when mutated, causes CF, encodes a cAMP-activated chloride ion channel that is expressed in the epididymis and vas deferens. Its malfunction produces an aberrant luminal environment in these tissues with resultant obstruction of the epididymis during development and frequently loss of the vas deferens. The CFTR channel interacts with many other proteins to maintain the luminal environment in the epididymis. Here we identify trans-interacting factors that are predicted to bind to cis-regulatory elements for multiple genes encoding these interacting proteins. These transcription factor networks thus coordinate the normal function of the epididymis epithelium.
The regulatory mechanisms for many individual genes that are active in the epididymis epithelium were characterized previously by classical methods (reviewed in Kirchhoff [1]). However, other genes, particularly those that show tight cell-type-specific control involving cis-acting elements that lie outside the promoter or coding region, are less well understood. These are harder to evaluate since this requires global analysis of regulatory elements in relatively small numbers of differentiated primary epithelial cells. However, techniques developed in part by the ENCODE consortium [2], including methods to identify regions of open chromatin genome-wide such as DNase-seq (DNase I hypersensitivity mapping followed by deep sequencing [3]), have enabled these analyses. Here we describe a genome-wide map of open chromatin in primary human fetal epididymis epithelial (HEE) cells generated by DNase-seq analysis. Though these cells do not exhibit all the differentiated functions of adult epididymis, they are nevertheless a valuable resource for many biological characteristics of the epithelium. We use bioinformatic tools to identify epididymis epithelial cell-selective regions of open chromatin and determine their distribution throughout the genome. We determine that the peaks of open chromatin are more evident at the promoters of genes that are highly expressed in epididymis cells than at inactive gene promoters. Moreover, epididymis-selective peaks of open chromatin are associated with genes involved in pathways of epithelial differentiation and function with relevance to epididymal biology. Within these peaks, predicted binding sites for some epithelial-specific transcription factors are overrepresented. Next, we illustrate the power of these data to facilitate the characterization of regulatory elements for genes that are coordinately expressed in epididymis cells, such as ion channels.
MATERIALS AND METHODS
HEE Cells
HEE cells were cultured as described previously [4]. The cell cultures were established from fetal genital ducts obtained with institutional ethical permission from mid-trimester prostaglandin-induced terminations or spontaneous abortions. Cells were passaged to generate enough material for DNase digestion (1 × 107 cells) and RNA extraction but were not used if they showed any sign of dedifferentiation. CFTR expression (measured by quantitative RT-PCR [5, 6]) was used as a marker of differentiation of the HEE cells.
DNase-seq
Two DNase-seq libraries [3] were generated from three independent cultures of primary HEE cells from different donors. One DNase-seq library contained cells from a single donor, while a second DNase-seq library contained cells from two donors combined. The data from these two libraries were combined, and DNase peak calls were generated using methods described below. The DNase-seq data on five ENCODE cell types (FibroP, GM12878, K652, HepG2, and HUVEC) were generated by the ENCODE consortium [7]. All genome data coordinates refer to HG19.
DNase I Hypersensitive Sites
The peak calls for the DNase-seq data were made with the F-seq application to give a discrete number of DNase I hypersensitive sites (DHS) within the human epididymis epithelial cells [8]. These sites were determined by F-seq by fitting the data to a gamma distribution to calculate P-values. P-values below a 0.05/0.01 threshold in contiguous regions were considered significant. All DHS identified on chromosome Y were removed from the analysis to allow for comparison across both male and female cell types. After producing the number and location of DHS for these primary cells, the DHS of five nonrelated cell types were intersected with the HEE data for comparison. The five cell types included FibroP, GM12878, K562, HepG2, and HUVEC. Those sites from the HEE cells that were not overlapped by any other data sets made up the list of HEE-selective sites. The HEE sites that were overlapped by a DHS in all of the cell types composed the list of ubiquitous sites.
DHS Overlapping Gene Annotation
The genomic indices of HEE-selective DHS, ubiquitous DHS (i.e., DHS occurring in HEE cells that were overlapped by DHS from all background cell types), and all DHS were intersected with the genomic indices of all human genes, their promoters (2 kb upstream of transcriptional start sites), exons, introns, and intergenic sequence. The human gene annotation was derived from a list of all human Entrez genes downloaded from NCBI. These Entrez genes were each linked to their representative RefSeq sequences, and the RefSeq indices were downloaded from UCSC's genome browser (http://www.ncbi.nlm.nih.gov, http://genome.ucsc.edu).
In a different approach to identify RefSeq genes that were associated with HEE-selective DHS, all DHS from HEE cells and all five background cell types were individually overlapped with RefSeq genes at the base pair level (including 2 kb upstream of the TSS and 2 kb downstream of the TTS). The number of base pairs overlapping between each gene and the DHS of all cell types were tabulated. If a gene was found to overlap more total base pairs of HEE DHS than the base pair overlap count of DHS from all other cell types combined, that gene was identified as being associated with HEE-selective DHS.
Microarray Analysis
RNA was isolated by Trizol extraction from the same three primary cultures of HEE cells that were used for DNase-seq and at the same time point. Total RNA was purified by Millipore Microcon YM-100 filter centrifugation and shipped to MoGene for gene expression analysis on Nimblegen 4 × 72K HG18 60mer arrays.
DAVID Analysis
The DAVID tool can be used to read a gene list and identify within that list any statistically significant enrichment of genes from a common gene ontology (GO) term or over 40 other annotation categories, including protein-protein interactions and pathways [9, 10]. For this purpose, the human genes with HEE-selective DHS present in their promoter region were analyzed with DAVID. The background gene list used for this analysis was the same human Entrez gene list used to identify overlap between DHS and gene annotation.
Clover Analysis
The Clover application was used to search for motifs common to DHS within promoter and intergenic sequence near genes. Clover is an open-source application that identifies motifs (from an input set) that are statistically over- or underrepresented in a set of sequences with respect to a set of background sequences [11]. The promoter regions were defined as 2 kb upstream of the transcription start site of all human genes. The intergenic regions that were searched included any genomic region more than 2 kb but less then 10 kb from the start or stop of gene transcription. The set of motifs we searched for included 1422 transcription factor binding motifs from TRANS-FAC and 1316 from Jaspar [12, 13].
Nucleosome Mapping
The positions of nucleosomes within ∼230 kb spanning the CFTR locus in epididymis epithelial cells and fibroblasts was determined by BAC hybridization of mononucleosomal DNA followed by SOLiD sequencing as described previously [14].
RESULTS
Identification of DHS Genome-Wide
Regions of open chromatin in primary human epididymis epithelial cells (HEE) were identified by mapping DHS genome-wide using DNase-seq. The F-seq application, a feature density estimator for high-throughput sequence tags [8], was then used to analyze the sequence reads, which identified 132 545 DHS in the epididymis cells. These sites (peak signals) represent elements in the genome where multiple sequence reads aligned to a common region. Many cis-acting regulatory elements are found within DHS, so this open chromatin map can potentially identify elements controlling the expression of the majority of genes within the HEE cells. To distinguish between ubiquitous and cell-type-selective regulatory elements in HEE cells, we first subtracted DNase-seq data sets from five different cell types generated by the ENCODE consortium [2]. These included skin fibroblasts (FibroPark), a lymphoblastoid cell line (GM12878), an erythroleukemia cell line (K562), a liver carcinoma cell line (HepG2), and human umbilical vein endothelial cells (HUVEC). The genomic overlap between HEE DHS and the other five cell types is shown in Figure 1 and is around 40–50%. The slightly higher overlap between fibroblasts and HEE is probably an artifact of minor contamination of the primary epididymis epithelial cell cultures with epididymal fibroblasts [4]. The numbers of DHS in each cell type are shown in Table 1.
FIG. 1.
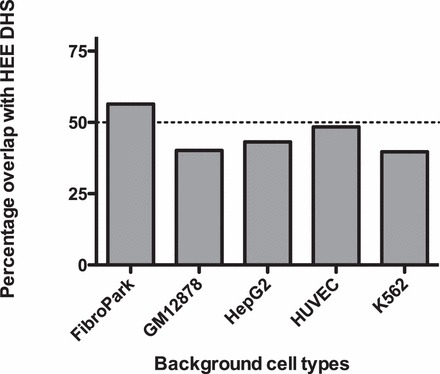
Cell-type specificity of DNase I hypersensitive sites (DHS) in HEE cells. The genomic overlap between HEE DHS and the other five cell types. Percentage of HEE DHS that overlap with the DHS of each of the individual background cell types.
TABLE 1.
Number of open chromatin sites in HTE cells and the five other cell types that were used for comparison.

Among the 132 545 HEE sites, 33 542 (25.3%) were apparently cell type selective since they did not overlap with hypersensitive sites found in any of the five other cell types, and 31 525 (23.8%) sites were ubiquitous since they intersected (at least partially) with DHS from these additional cells.
The majority (70.6%) of DHS identified in HEE cells were less than 500 base pairs (bp) in length, while the DHS that overlapped with promoter regions (2 kb upstream of a transcriptional start) were generally longer (only 21.9% were below 500 bp in length; (Supplemental Fig. S1; all Supplemental Data are available online at www.biolreprod.org). Inspection of the HEE DHS among different cell types identified 1564 NCBI RefSeq genes with a greater representation of promoter and coding region DHS than the five background cell types combined (Supplemental Table S1). Multiple genes within this list code for proteins with well-characterized or predicted functions in the epididymis epithelium. These include the CFTR gene; the solute carrier SLC26A7, which encodes a sulfate/anion transporter known to function in the kidney; and SLC9A4, the gene producing the NHE4, cation protein antiporter 4.
Distribution of DHS Across the Genome
The distribution of DHS among different genomic elements was determined using the following categories: promoter (2 kb of sequence 5′ to the transcriptional start site), exon 1, intron 1, other genic, 2 kb of sequence 3′ to the transcriptional stop site, and intergenic (Fig. 2). For this comparison, DHS were analyzed in three different categories, including all DHS, HEE-selective DHS, and ubiquitous DHS (HEE DHS overlapping with DHS from all background cell types). HEE-selective DHS are most common in other genic sites (i.e., within gene bodies [either intronic or exonic] rather that in gene promoters) and also more frequent than ubiquitous sites in intergenic regions (defined as genomic sequence located more than 2 kb from any gene). In contrast, ubiquitous sites occur more often (27.2% of ubiquitous site sequence) in promoter regions than HEE-selective (2.5%) and all (1.3%) sites. A similar distribution of DHS was evident in the first exon of genes and to a lesser extent in the first introns and 2 kb downstream of genes.
FIG. 2.
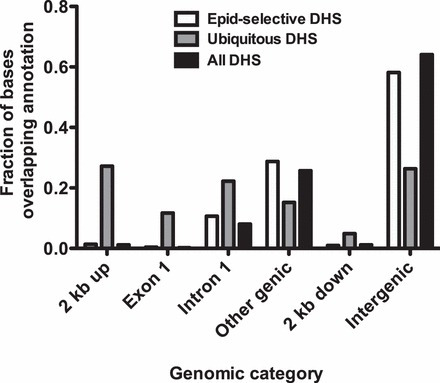
HEE-selective DHS are generally located in distal regions of genes or in intergenic sequences rather than in promoters, where ubiquitous DHS predominate. Three categories of DHS (all DHS, HEE selective DHS, and ubiquitous DHS) were overlapped with different genomic regions to determine their distribution with respect to genes. Definition: 2 kb up, including 2 kb 5′ to genes; other genic, all exons and introns of genes excluding the first; 2 kb down, including 2 kb 3′ to genes; intergenic, all genomic sequence more than 2 kb from genes.
Correlation of DNase-seq Data with Gene Expression
Gene expression profiles were generated for the same three cultures of primary epididymis that were used for DNase-seq. Total RNA was extracted and evaluated on Nimblegen 72K HG18 60mer microarrays. Gene expression values in HEE cells were divided into three groups—high expression (top 20%), medium expression (middle 20%), and low expression (bottom 20%)—and correlated with the DNase-seq signal. The DNase-seq Base Overlap Signal (the number of reads that align to each base pair position of the genome) was averaged between 1 kb 5′ and 3′ to the transcription start site of genes in each category (Fig. 3). The most abundantly expressed genes were seen to correlate with the highest DNase-seq signal.
FIG. 3.
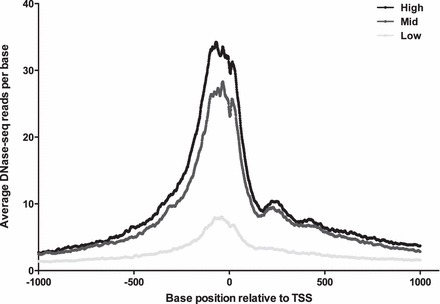
The intensity of DHS at gene promoters correlates with gene expression in HEE cells. Genes for which microarray expression data were available were separated into three categories: high expression (top 20%), middle expression (middle 20%), and low expression (bottom 20%). Then the average number of DNase-seq reads was calculated at each base between 1 kb 5′ and 1 kb 3′ to the transcription start site for each of these categories.
Pathways of Epithelial and Epididymal Structure and Function Revealed by HEE-Selective Promoter DHS
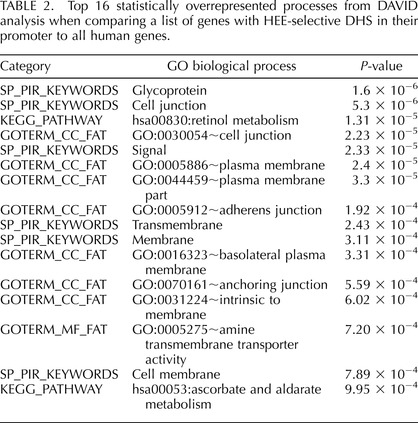
The DAVID [15] database was used to identify gene processes that were overrepresented in HEE cells in comparison to all human genes. Entrez Gene IDs for the 564 genes with one or more HEE-selective promoter DHS were combined in a text file for analysis, and the list of statistically significant DAVID ontologies/pathways (P < 0.05) are shown in Supplemental Table S2. A number of the top 16 DAVID results (Table 2) are directly related to epithelial function, including cell junction (GO:0030054 and SP_PIR key words), adherens junction (GO:0005912), basolateral plasma membrane (GO:0016323), and anchoring junction (GO:0070161). Moreover, two processes with robust P-values, retinol metabolism (Kegg pathway: hsa00830; P = 1.31 × 10−5) and ascorbate and aldarate metabolism (Kegg pathway: hsa00053; P = 9.96 × 10−4), are directly related to epididymal function [16, 17]. These data confirm the power of DNase-seq to identify cell-type-specific regulatory elements associated with open chromatin in HEE cells.
TABLE 2.
Top 16 statistically overrepresented processes from DAVID analysis when comparing a list of genes with HEE-selective DHS in their promoter to all human genes.
HEE-Selective DHS Are Enriched for Binding Sites for Relevant Epithelial Transcription Factors
The Clover application [11] was used to search for overrepresented sequence motifs in the HEE-selective DHS within promoter and intergenic regions. We expected that this analysis would identify transcription factor binding sites utilized in HEE cells. The promoter (Supplemental Table S3) and intergenic (Supplemental Table S4) sequence motifs were each analyzed in three DHS classifications: all DHS, HEE-selective DHS, and ubiquitous DHS. The comparisons between the intergenic sites are of particular interest since the representation of motifs is markedly different in the HEE-selective and ubiquitous sites. The ubiquitous sites include predicted sites for 34 different HOX transcription family members, which are overrepresented on 23/23 chromosomes, while only three motifs are overrepresented on a single chromosome on HEE-selective sites. Ubiquitous sites also encompass a high frequency of CTCF-binding motifs, which are overrepresented on 23/23 chromosomes but not in HEE-selective sites. This distribution of CTCF (CCCTC binding factor) sites is consistent with the pivotal role of this protein in maintaining higher-order chromatin structure [18–20]. Several overrepresented transcription factor binding sites that are evident within HEE-selective sites are biologically relevant. These include sites for the epithelial-specific E74-like factor (ets domain transcription factor) ELF5 (on 22 chromosomes) and ELF3/ESE-1 (on 21 chromosomes). ELF5 is known to regulate a number of epithelial specific genes in tissues containing glandular epithelium [21]. ELF3 is likewise thought to play an important role in epithelial cell differentiation and tumorigenesis. Another factor with a higher frequency of binding motifs predicted in HEE selective sites (on 10 chromosomes) is ets homologous factor EHF/ESE-3, which is a transcriptional repressor involved in epithelial differentiation.
Of particular relevance to the epididymis epithelium is the overrepresentation in HEE-selective sites of Pax2 (paired box 2) binding motifs (on all 23 chromosomes), the androgen receptor (AR half sites, on 20 chromosomes), and Sox9 (SRY [sex-determining region Y]-box 9, on 19 chromosomes). Pax2 plays a critical role in the development of the urogenital tract [22], AR is functionally important in the epididymis epithelium [23], and Sox9 is known to regulate expression of the anti-Müllerian hormone gene [24]. Other factors that are observed more frequently than expected in HEE-selective sites include hepatocyte nuclear factor 1 (HNF1A/B), though, as the motifs for these factors are also present in the “all DHS” and “ubiquitous DHS” sets, their specific role in epididymal function is less clear.
Open Chromatin at Individual Genes That Are Critical to Normal Function of the Epididymis Epithelium
We examined open chromatin at two loci encoding proteins that maintain the luminal environment in the epididymis, which is critical for normal sperm maturation. First is the CFTR gene, which was studied previously in intestinal and airway epithelial cells [6, 25] but is also expressed at relatively high levels in epididymis epithelial cells [5, 6]. In these cells, peaks of open chromatin are shown by DNase-seq at the promoter, consistent with its activity, and at important enhancer elements in intron 11 [6, 26] and intron 23 [25] (Fig. 4A). Also evident are peaks at the two enhancer-blocking insulator elements 3′ to the locus, one at +6.8 kb [27] that is epididymis selective and binds CTCF and a second at +15.6 kb that does not and is evident in many cell types [28]. Peaks at −44kb and −35 kb 5′ to the promoter are also evident in airway epithelial cells and may interact with inflammatory mediators [29]. A novel peak of epididymis-selective open chromatin that we have not observed in any other cell type to date lies in intron 20 of CFTR at chr7:117 285 528–117 285 824 (Fig. 4A, arrow). Inspection of data generated by the ENCODE consortium [2] suggests that multiple transcription factors including c-Fos (FBJ murine osteosarcoma viral oncogene homolog), c-Jun (jun avian sarcoma virus 17 oncogene homolog), JunD, and CEBPB (CCAAT/enhancer binding protein [C/EBP], beta) bind to an element at 117 285 660, coincident with the DHS (Fig. 4B). Moreover, the site corresponds to a sequence that is selectively nucleosome depleted in epididymis epithelial cells (Fig. 4C) [14] and is marked by enrichment of the histone modifications H3K4me1 and H3K27Ac (Fig. 4B), consistent with an active enhancer element.
FIG. 4.
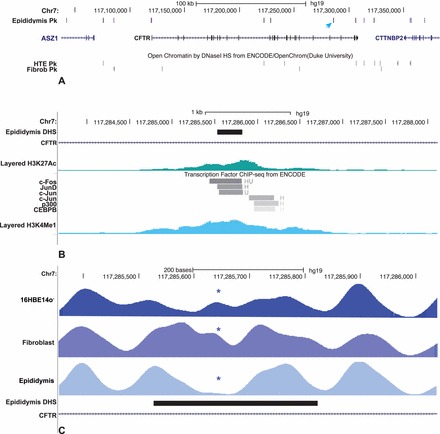
Open chromatin at the CFTR locus in HEE cells identifies a novel candidate cis-regulatory element. A) Peaks of open chromatin in HEE cells (key: blue = ubiquitous, purple = HEE selective, black = others). The novel DHS in intron 20 is marked with an arrow. HTE and fibroblast data are shown below. B) Encode ChIP-seq data for transcription factor binding and histone modification at the intron 20 DHS. C) Nucleosome positioning data across the intron 20 DHS show selective loss of a nucleosome, marked by * at the DHS core in HEE (epididymis).
Another gene that is relevant to epididymis function is the androgen receptor (AR). Comparison of DHS across this locus shows epididymis epithelial cell-selective open chromatin (Fig. 5 marked by arrows) in several introns and a site in the 3′UTR that is not present in the five ENCODE lines but is also seen in human tracheal epithelial cells [30]. These regions of open chromatin likely bind critical cell-type-selective transcription factors, which warrant further investigation. Also of interest is a DHS in the first intron of AR in both epididymis and tracheal epithelial cells but not in fibroblasts. This DHS coincides with a peak of CTCF and Rad21 occupancy in ENCODE ChIP-seq data from A549 and HepG2 cells [31]. We performed ChIP for CTCF in Calu3 (high AR expression) and 16HBE14o- (no AR expression) airway cell lines and saw substantial occupancy at this site in both cell types (Fig. 5, inset panel). Since CTCF can act as a physical barrier to interactions across the genome, it is possible that occupancy of this site plays a role in alternative splicing at the 5′ end of AR (reviewed in Dehm and Tindall [32]).
FIG. 5.
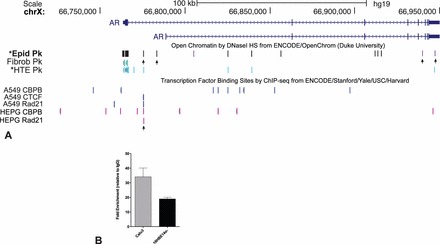
Open chromatin at the AR locus in HEE cells identifies novel candidate cis-regulatory elements. A) HEE, fibroblast (ENCODE) and HTE [30]. Transcription factor binding site in A549 and HepG2 cells from ENCODE. CTCF/RAD21 site in intron 1 is marked by arrow. B) ChIP for CTCF shows that site marked in A is enriched for the protein in Calu3 and 16HBE14o- cells.
Comparison with Open Chromatin in Other Differentiated Epithelial Cells
Since all epithelia share some common functions, such as acting as a barrier to the external environment, in addition to organ-specific functions, it was of interest to compare the epididymal (HEE) data with those we generated previously for the airway epithelium (human tracheal epithelial [HTE] cells [30]). The list of 1,564 NCBI RefSeq genes with a greater representation of promoter and coding region DHS in HEE cells than the five background cell types combined (Supplemental Table S1) was compared with the equivalent list of 974 genes from HTE cells ([30]; Supplemental Table S1). Three hundred and twenty-seven genes were found in both lists (Supplemental Table S5), suggesting that the method was quite powerful in identifying cell-type-selective processes in addition to those that are common to all epithelial cells. Among the genes that appear in both lists are Keratin 5 and Keratin 6A, both of which are expressed in epididymis and tracheal epithelial cells, particularly highly in the latter; SLC12A2, which mediates sodium and chloride transport and reabsorption and is expressed at similar levels in both cell types; and CLCA4, the calcium sensitive chloride channel that is highly expressed in tracheal epithelial cells but is inactive in the epididymis epithelial cells. Consistent with this divergent expression pattern, the location of open chromatin at the CLCA4 locus is different in these cell types. Examples of divergent cell-selective chromatin are shown for the chemokine CXCL1 (chemokine [C-X-C motif] ligand 1) and IRAK1BP1 (interleukin-1 receptor-associated kinase 1 binding protein 1) genes in Figure 6. CXCL1 is highly expressed in both HEE and HTE cells but has different profiles of open chromatin in the two cell types, implicating different regulatory mechanisms/elements for the gene. IRAK1BP1 is expressed in HEE cells but is silent in HTE cells despite open chromatin at the promoter in both cell types. A unique DHS within the first intron of the gene in HEE cells, marked by an arrow in Figure 6B, is thus a candidate for an HEE-selective intronic enhancer.
FIG. 6.

Cell-type-selective open chromatin at CXCL1 and IRAK1BP1 loci. A) CXCL1 is expressed in HEE and HTE, but the DHS are cell-type selective. B) IRAK1BP1 is expressed only in HEE cells despite the open chromatin at the gene promoter in both cell types. A unique DHS in intron 1 in HEE cells is a candidate enhancer.
DISCUSSION
Methods to decipher transcriptional networks that establish the differentiated signature of specific cell types have the power to greatly advance our understanding of biological processes. In the context of the ENCODE project [2], multiple techniques are now available to analyze regulatory elements genome-wide in primary cells. Though early data sets were generated on immortalized or tumor cell lines, an increasing number of primary cells relevant to human disease are now becoming available, including pancreatic islets [33] and tracheal epithelial cells [30]. Here we generate open chromatin data on primary human epididymis epithelial cells as a resource to further understand the molecular basis for differentiated function of this tissue. Due to the extreme difficulty of obtaining adult human epididymis epithelial cells for these analyses, we have instead used cells cultured from human fetal tissues. Clearly, in culture these cells lack the lumicrine and endocrine signaling of the intact epididymis and hence are unlikely to recapitulate all the normal functions of the postpubertal adult human epididymis epithelium. However, these cells express the androgen receptor, and immortalized derivative cell lines [34] are responsive to R1881 synthetic androgen (data not shown). Moreover, the cells express the CFTR gene, making them an ideal model for studying early events in the pathology of CF that result in obstruction and loss of the male genital ducts [5, 6, 27].
It is of interest to compare the gene expression profiles of the HEE cells used in the current analysis with others reported from adult rat and mouse epididymis tissues [35–37]. Due to interspecies differences and the different platforms used for the microarrays, these data sets are not directly comparable. However, several transcripts that were defined in the previous studies as being epididymis specific or differentially expressed in the epididymis are also highly expressed in HEE cells. These include clusterin (CLU), the adenosine A1 receptor (ADORA1), growth and differentiation factor 15 (GDF15), and glutathione peroxidase 3 (GPX3).
Identification of Novel Genes with a Role in the Epididymis Epithelium
Among the genes with a greater representation of promoter and coding region DHS in HEE cells alone are several that may have roles in normal function of the epididymis. These include spermatogenesis-associated proteins SPATA12, 16, 17, 18 (all but SPATA16 are expressed in HEE cells); inflammatory chemokines CXCL5 and CXCL8 (IL8), both of which are highly expressed; and the expressed potassium channel, subfamily K, member 1, KCNK1. Next, the list of genes with HEE-selective DHS was overlapped with a set of 205 genes that were shown to be androgen responsive in a human prostate epithelial cell line [38]. Of the 14 genes identified in both lists, seven were evident in both HTE and HEE cells and seven were seen only in HEE cells, including carbonic anhydrase 2 (CA2), delta/notch-like EGF repeat containing (DNER), and epiregulin (EREG), all of which are expressed in these cells.
Relevance to Genome-Wide Association Studies of Male Infertility
Many genome-wide association studies (GWAS), which aim to identify loci with direct relevance to human disease, identify high-P-value SNPs that lie within intergenic or intronic regions. These locations, which by default are usually defined as regulatory, often hamper studies that aim to reveal the molecular basis of the associations. However, the identification of cell-type-selective regions of open chromatin genome-wide can directly inform mechanistic studies [33]. Of relevance to epididymis function, we evaluated the list of intergenic SNPs that were found by GWAS to be in association (P < 1 × 10−4) with male fertility traits (family size) in humans [39]. Six genes with intronic or extragenic SNPs also contained HEE-selective peaks of open chromatin. These included PCDH9, MACROD2, CNRN5, ATP10N, ANKRD18, and SLC35F4. MACROD2 (MACRO domain-containing protein 2) and ANKRD18a (ankyrin repeat domain-containing protein 18A) are not well characterized, but both are expressed in HEE cells according to our microarray data, supporting their potential role in epididymis function.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. C.J. Ott and R. Yang for assistance.
Footnotes
Supported by the National Institutes of Health, R01HD068901, R01HL094585 (PI: A.H.), and the Cystic Fibrosis Foundation (PI: A.H.). These data have been deposited into GEO, accession no. GSE51003.
REFERENCES
- Kirchhoff C. Gene expression in the epididymis. Int Rev Cytol 1999; 188: 133–202. [DOI] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007; 447: 799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008; 132: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A, Coleman L. Ductal epithelial cells cultured from human foetal epididymis and vas deferens: relevance to sterility in cystic fibrosis. J Cell Sci 1989; 92 (pt 4): 687–690. [DOI] [PubMed] [Google Scholar]
- Harris A, Chalkley G, Goodman S, Coleman L. Expression of the cystic fibrosis gene in human development. Development 1991; 113: 305–310. [DOI] [PubMed] [Google Scholar]
- Ott CJ, Blackledge NP, Kerschner JL, Leir SH, Crawford GE, Cotton CU, Harris A. Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus. Proc Natl Acad Sci U S A 2009; 106: 19934–19939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, Liu Z, London D, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res 2011; 21: 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AP, Guinney J, Crawford GE, Furey TS. F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics 2008; 24: 2537–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Fu Y, Yu L, Chen JF, Hansen U, Weng Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res 2004; 32: 1372–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A. JASPAR. the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res 2008; 36: D102–D106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 2006; 34: D108–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigit E, Bischof JM, Zhang Z, Ott CJ, Kerschner JL, Leir SH, Buitrago-Delgado E, Zhang Q, Wang JP, Widom J, Harris A. Nucleosome mapping across the CFTR locus identifies novel regulatory factors. Nucleic Acids Res 2013; 41: 2857–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G., Jr., , Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003; 4: P3. [PubMed] [Google Scholar]
- Rajan N, Sung WK, Goodman DS. Localization of cellular retinol-binding protein mRNA in rat testis and epididymis and its stage-dependent expression during the cycle of the seminiferous epithelium. Biol Reprod 1990; 43: 835–842. [DOI] [PubMed] [Google Scholar]
- Chinoy NJ, Rao MV, Kumar RA. Role of ascorbic acid in metabolism of rat testis and epididymis in relation to the onset of puberty. J Biosci 1984; 6: 857–863. [Google Scholar]
- Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, de Laat W. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev 2006; 20: 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, Cobb BS, Yokomori K, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 2008; 132: 422–433. [DOI] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008; 451: 796–801. [DOI] [PubMed] [Google Scholar]
- Lapinskas EJ, Palmer J, Ricardo S, Hertzog PJ, Hammacher A, Pritchard MA. A major site of expression of the ets transcription factor Elf5 is epithelia of exocrine glands. Histochem Cell Biol 2004; 122: 521–526. [DOI] [PubMed] [Google Scholar]
- Kuschert S, Rowitch DH, Haenig B, McMahon AP, Kispert A. Characterization of Pax-2 regulatory sequences that direct transgene expression in the Wolffian duct and its derivatives. Dev Biol 2001; 229: 128–140. [DOI] [PubMed] [Google Scholar]
- O'Hara L, Welsh M, Saunders PT, Smith LB. Androgen receptor expression in the caput epididymal epithelium is essential for development of the initial segment and epididymal spermatozoa transit. Endocrinology 2011; 152: 718–729. [DOI] [PubMed] [Google Scholar]
- De Santa Barbara P, Bonneaud N, Boizet B, Desclozeaux M, Moniot B, Sudbeck P, Scherer G, Poulat F, Berta P. Direct interaction of SRY-related protein SOX9 and steroidogenic factor 1 regulates transcription of the human anti-Mullerian hormone gene. Mol Cell Biol 1998; 18: 6653–6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ott CJ, Lewandowska MA, Leir SH, Harris A. Molecular mechanisms controlling CFTR gene expression in the airway. J Cell Mol Med 2012; 16: 1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschner JL, Harris A. Transcriptional networks driving enhancer function in the CFTR gene. Biochem J 2012; 446: 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Ott CJ, Gillen AE, Harris A. An insulator element 3′ to the CFTR gene binds CTCF and reveals an active chromatin hub in primary cells. Nucleic Acids Res 2009; 37: 1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Carter EJ, Evans JR, Lawson V, Rowntree RK, Harris A. CTCF mediates insulator function at the CFTR locus. Biochem J 2007; 408: 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Leir SH, Harris A. Immune mediators regulate CFTR expression through a bifunctional airway-selective enhancer. Mol Cell Biol 2013; 33: 2843–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof JM, Ott CJ, Leir SH, Gosalia N, Song L, London D, Furey TS, Cotton CU, Crawford GE, Harris A. A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax 2012; 67: 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, Min R, Alves P, et al. Architecture of the human regulatory network derived from ENCODE data. Nature 2012; 489: 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. EndocrRelat Cancer 2011; 18: R183–R196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP, Panhuis TM, Mieczkowski P, Secchi A, Bosco D, Berney T, Montanya E, et al. A map of open chromatin in human pancreatic islets. Nat Genet 2010; 42: 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman L, Harris A. Immortalization of male genital duct epithelium: an assay system for the cystic fibrosis gene. J Cell Sci 1991; 98 (pt 1): 85–89. [DOI] [PubMed] [Google Scholar]
- Chauvin TR, Griswold MD. Androgen-regulated genes in the murine epididymis. Biol Reprod 2004; 71: 560–569. [DOI] [PubMed] [Google Scholar]
- Johnston DS, Jelinsky SA, Bang HJ, DiCandeloro P, Wilson E, Kopf GS, Turner TT. The mouse epididymal transcriptome: transcriptional profiling of segmental gene expression in the epididymis. Biol Reprod 2005; 73: 404–413. [DOI] [PubMed] [Google Scholar]
- Jelinsky SA, Turner TT, Bang HJ, Finger JN, Solarz MK, Wilson E, Brown EL, Kopf GS, Johnston DS. The rat epididymal transcriptome: comparison of segmental gene expression in the rat and mouse epididymides. Biol Reprod 2007; 76: 561–570. [DOI] [PubMed] [Google Scholar]
- Bolton EC, So AY, Chaivorapol C, Haqq CM, Li H, Yamamoto KR. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev 2007; 21: 2005–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosova G, Scott NM, Niederberger C, Prins GS, Ober C. Genome-wide association study identifies candidate genes for male fertility traits in humans. Am J Hum Genet 2012; 90: 950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.