

Abstract
Significance: Quiescin sulfhydryl oxidase 1 (QSOX1) is an enzyme that oxidizes thiols during protein folding, reducing molecular oxygen to hydrogen peroxide. Tumor cells may take advantage of oxidative environments at different stages of tumorigenesis, but QSOX1 may also serve additional functions in tumors. Recent Advances: Several groups have reported the over-expression of QSOX1 in breast, pancreas, and prostate cancers. A consensus is building that QSOX1 over-expression is important during tumor cell invasion, facilitating tumor cell migration at the tumor-stroma interface. As such, QSOX1 may be considered a prognostic indicator of metastatic potential or even indicate that cancer is present in a host. Critical Issues: However, some controversy exists between QSOX1 as a marker of poor or favorable outcome in breast cancer. More studies are required to reveal what advantage QSOX1 provides to breast and other types of cancer. More specifically, it is critical to learn which tumor types over-express QSOX1 and use its enzymatic activity to their advantage. Future Directions: As interest increases in understanding the mechanisms of tumorigenesis within the extracellular matrix and how tumor cells influence fibroblasts and other stromal cells, QSOX1 may be revealed as an important player in cancer detection and prognosis. Defining the mechanism(s) of QSOX1 activity in tumors and in in vivo models will provide important insights into how to target QSOX1 with anti-neoplastic agents. Antioxid. Redox Signal. 21, 485–496.
Introduction
This review concerns the emerging role of a sulfhydryl oxidase (quiescin sulfhydryl oxidase 1 [QSOX1]) as a potentially important target in the continuum of tumorigenesis. In the next few sections, we will re-illuminate the cancer problem for readers by briefly discussing cancer incidence, diagnosis, treatment, and outcome. Pathogenesis will be discussed with an emphasis on tumor microenvironment, extracellular matrix (ECM), and cell-cell interactions. The discovery, structure, enzymology, biochemistry, and function of QSOX1 will also be reviewed and discussed. Distinct connections will be made among recent reports on the biology of QSOX1 and how it relates to cancer. We will compare and contrast conflicts in the literature and supplement the discussion with data from our own studies in an attempt to clarify these controversies.
The Cancer Problem
Cancer is the second leading cause of death in the United States after cardiovascular disease. It accounts for 23% of all deaths, but is primarily a disease of the elderly, peaking in incidence in the 8th decade. A person's lifetime risk of cancer in the United States is ∼40%. The most common causes of cancer death in men are lung, prostate, colorectal, pancreas, and liver. For women, the most common causes of cancer death are lung, breast, colorectal, pancreas, and ovary (1). As will be discussed next, QSOX1 is overexpressed in several malignancies, especially breast and pancreatic carcinoma.
Diagnosis of the most common types of solid malignant tumors is based on detection through imaging and biopsy. Radiological imaging includes X-rays tests (e.g., computerized tomography scan, mammography), magnetic resonance imaging, or ultrasound (e.g., transrectal ultrasound of the prostate) and FluoroDeoxyGlucose-Positron Emission Tomography scanning (24). Alternatively, cancers may be detected by visual inspection such as through endoscopy (colorectal cancer) or physical examination (e.g., melanoma) (32). Confirmation of diagnosis, ultimately, requires tissue sampling such as via surgical excision, endoscopic biopsy, or image-guided needle biopsies. The diagnosis of cancer is made histologically by morphologic characteristics of the cells, their nuclei, staining pattern, production of certain protein antigens (demonstrated by immunohistochemistry [IHC]), or, even more recently, direct demonstration of certain gene mutations (11).
The treatment of cancer relies on the elimination of malignant cells from the body. For a contained neoplasm, this may be accomplished by surgical resection. The complexity of the surgical procedure is very much dependent on the location and size of the tumor (64). Even when the primary tumor is resected, individual tumor cells may have already circulated hematogenously and seeded other tissues, resulting in microscopic unknown metastases. This aspect of tumor biology makes cure unlikely and recurrences common. Many patients at presentation are not surgical candidates usually due to metastasis, observed by imaging. Lymph nodes, liver, lung, brain, and bones are common sites of metastasis. For these patients, options include chemotherapy and radiation therapy, which may be given preoperatively or as sole therapy. The most commonly used chemotherapeutic agents target rapidly dividing cells and induce breaks or other damage to tumor cell DNA, including oxidative stress (54). Radiotherapy kills cancer cells by damaging DNA and cellular proteins with high energy photons or particles (3). Both chemo and radiotherapy may be given to treat unresectable or recurrent disease, or be used in conjunction with surgery to increase the chance of cure (37). Although many chemotherapeutic drugs induce oxidative stress, the anti-oxidant response in tumor cells, and especially cancer stem cells, may make them less sensitive to radiotherapy (26, 45, 66). In addition, chemo and radiotherapy have suboptimal specificity for tumor cells, and many normal cells are damaged, especially the rapidly dividing cells of the bone marrow, gastrointestinal (GI) tract, and hair follicles. This fact leads to the toxicities of these modalities, limiting their effectiveness.
More recently, new therapies have been developed that are designed to inhibit specific tumor enzymes (11) or block the action of growth factors (50). Approaches to targeting specific proteins have included the rational development of enzyme inhibitors or the generation of monoclonal antibodies. Examples of small chemical inhibitors include the tyrosine kinase inhibitors, which were initially developed to treat leukemias. The initial example was imatinib mesylate, which competes for the ATP binding site on the kinase enzyme (e.g., bcr-abl, cKIT), thus preventing phosphorylation of tyrosines on the target protein (25). The tyrosine kinase inhibitors have revolutionized the treatment of not only leukemia, but also renal cell carcinoma, neuroendocrine tumors, and GI stromal tumors. Other examples of targeted enzyme therapy for cancer include the mammalian target of rapamycin inhibitors for breast and renal cell cancers (everolimus, temsirolimus) (2, 55), the tyrosine kinase inhibitor regorafenib for colorectal cancer (33), and aromatase inhibitors, which decrease the production of estrogen and are used to treat estrogen receptor (ER)-positive breast cancer (61). Examples of monoclonal antibodies used to target a specific protein include bevacizumab, which targets vascular endothelial growth factor (VEGF), cetuximab and panitumumab, which bind epidermal growth factor receptor (EGFR) (colorectal cancer) (83), and trastuzamab, which targets human EGFR-2 and is used in HER-2 positive breast cancer (75).
It is apparent that cancers arise due to a series of genomic DNA mutations, leading to the expression of pro-tumor genes (oncogenes) or the inhibition of tumor suppressor genes (4). Some of the genes are heritable, leading to kindreds with high rates of malignancies such as of the breast (BRCA1, BRCA2), colon (APC), and others (28). However, the vast majority of cancer occurs due to spontaneous mutations in DNA and is not heritable. Some of these mutations are in DNA repair genes. When these genes are silenced through mutations, other mutations may rapidly accumulate in the cell, resulting in genetic instability (65). Another recently described mechanism is the methylation of DNA in tumor suppressor gene promoter regions, leading to the silencing of tumor suppressor genes and resulting in carcinogenesis (4).
Although cancer is a genetic disease, the downstream result of mutations in DNA affects the types and quantities of proteins made. These proteins may confer advantages to the cancer cell, resulting in tumorigenesis. For example, if promoter activation results in the overexpression of genes and the over-production of growth factor receptors such as EGFR, this may result in rapid cellular proliferation of the cancer cells (77). If proteins are produced that affect cell adherence, extracellular environment, and it affects orientation compared with neighboring cells, an epithelial cell may become more mesenchymal (epithelial to mesenchymal transition [EMT]) and migrate from its proper location (8, 13, 15, 16, 69) All of this results in the cancer phenotype: uncontrolled growth, lack of contact inhibition, immortalization, invasion of adjacent tissues, and metastasis. Nonmalignant cells are contact inhibited; they will stop growing once the surface on which they are growing becomes confluent. Cancer cells will continue to grow piling up on each other. Nonmalignant cells in culture also are also subject to the Hayflick limit, or the number of times a cell will divide before undergo programmed cell death (apoptosis). Cancer cells will divide indefinitely and are, thus, immortal.
Recently, the ECM has been recognized to be critically important for the function of both normal and cancer cells. The ECM consists of a mixture of structural and functional proteins forming a protecting and nurturing lattice around the cells (86). In normal tissues, fibroblasts produce these proteins, which include collagens, cadherins, integrins, and others. Cancer cells also produce and rely on the ECM for protection from the immune response and for cellular signaling by functionally modifing local micro-environments (48). Cancer cells migrate through the ECM in the process of local invasion. They induce the deposition of ECM proteins and adhesion molecules to form fibrous capsules around them in a process called desmoplasia. Adhesion molecules in the ECM may have a role in clinical chemotherapeutic drug resistance (14). The composition of the ECM may be different in different tissues, explaining why some cancers spread (metastasize) to certain organs but not others (colon cancer spreads to the liver but rarely to the bones, while prostate cancer spreads to the bones but rarely to the liver) (14). Cancer cells may alter their local environment through the direct secretion of proteins, enzymes, and (directly or indirectly) growth factors. This extracellular environment has increasingly been recognized as a potential target for cancer therapy. For example, in metastatic colorectal cancers, VEGF is secreted, which stimulates the growth of new blood vessels feeding the tumor. Thus, the development of the VEGF-inhibitor bevacizumab is considered vital for cancer therapy (50). It is apparent that by targeting the ECM, including enzymes secreted into the ECM, new effective cancer treatments may be developed (80).
In this review, we will describe the enzyme QSOX1 and discuss its emerging role in carcinogenesis. We will review the evidence that QSOX1 is a key enzyme involved in a tumor's ability to modify the ECM and that these functions enable the tumor to communicate with and/or modify its environment at the tumor-stroma interface, resulting in invasion and subsequent growth of the tumor. Finally, we will propose QSOX1 as a potential therapeutic target to treat cancer. In considering the role of QSOX1, several oncological principles should be kept in mind. These include the shortcomings of current nonselective chemo and radiotherapeutic modalities, the importance of the ECM in carcinogenesis, the role of oxidative stress in both carcinogenesis and resistance to therapy, and the importance of the rational development of targeted inhibitors of key enzymes as new treatments for cancer.
Brief History of QSOX1
In 1993, Coppock et al. identified two novel cDNA clones from the WI38 fibroblast cell line that were up-regulated as the cells transitioned from logarithmic growth to quiescence (21). Coppock named the two clones, quiescins “Q6” and “Q10.” Two related mRNAs represented Q6 and are now called QSCN6 or QSOX1 long (QSOX1-L, also called QSOX1a) and short (QSOX1-S, also called QSOX1b) splice variants. Coppock reported that QSOX1 expression correlated with (i) exit from the cell cycle into G0, (ii) cell:cell contact, and (iii) disruption of cell adherence. Interestingly, he did not find that the transformation of fibroblasts with SV-40 increased QSOX1 expression. This conflicts with more recent data, including our own in which another tumorigenic virus, Epstein-Barr virus (EBV), transformed B cells, causing them to over-express QSOX1, while the same donor's nonadherent lymphocytes do not. In light of recent findings that QSOX1 is over-expressed in multiple tumor types, it is remarkable that QSOX1 was first identified in quiescent fibroblasts, as tumor cells are anything but quiescent.
Subsequently, Coppock determined the sequence and genomic location for QSOX1 in 1998 (20). QSOX1 is located on chromosome 1q24 and is an ancient gene fusion from thioredoxin (TRX) and ERV1, a yeast sulfhydryl oxidase (20). Bioinformatic searches revealed a signal sequence-suggesting secretion, but no KDEL endoplasmic reticulum-retention sequence, despite subsequent findings of QSOX1 localization in the ER and, more recently, in the Golgi (44, 82, 85). QSOX1 homology domains with sequence similarity to protein disulfide isomerase (PDI) were identified at the N-terminus of the protein followed by one functional and one nonfunctional TRX domain. ERV1/ALR (73) catalytic domains near the C-terminus drive the catalytic function of QSOX1 that participate in disulfide shuttling between QSOX1 and its client proteins. Evolutionary trees constructed by Coppock et al. suggest that the QSOX1 gene fusion occurred over a billion years ago. Importantly, the sequence analysis demonstrated CxxC motifs in the TRX1 and ERV1/ALR domains that are critical for QSOX1 enzyme to oxidize thiol groups in proteins. The presence of CxxC and ERV1/ALR domains provided the first clue that QSOX1 was a sulfhydryl oxidase involved in protein folding and led Lisowsky's group to find that ERV1, similar to QSOX1, was a flavin-linked sulfhydryl oxidase (20)
While Coppock's group was determining the nucleotide sequence of QSOX1, in 1996, Colin Thorpe's laboratory began to isolate and enzymatically characterize a flavin adenine dinucleotide (FAD)-dependent sulfhydryl oxidase from chicken egg white (35, 38–42, 70, 82). In an elegant series of experiments, Hoober et al. purified an FAD-containing sulfhydryl oxidase (QSOX1) from a large quantity of chicken egg white. Hoober et al. demonstrated that QSOX1 oxidizes small molecules such as dithiothreitol and glutathione as well as larger proteins with multiple thiols such as ovalbumin, insulin, RNAse A, and lysozyme. Using a combination of anti-Q6 antibodies from Coppock's group and early MALDI-TOF studies, Hoober and Thorpe's egg white sulfhydryl oxidase was shown to be immunologically related and, ultimately, identical to quiescin Q6 (QSOX1) Even at this early stage in the characterization of QSOX1, Hoober et al. suggested the involvement of QSOX1 in the ECM (38). Sequence analyses and biochemical studies then probed the roles of CxxC and the domains in QSOX1 catalysis, leading to the identification of active domains and (5, 22, 35, 38, 82).
QSOX1 Structure and Enzymatic Activity
Crystal structures of a truncated human (6) and intact murine and trypanosomal QSOX1 (5) were solved by the Fass group. These structural studies revealed a PDI-like oxidoreductase region containing TRX domains at the N-terminus of the protein. Near the C-terminus, the isoalloxazine ring in FAD was shown to bind to QSOX1 sandwiched between α3 and α4 helices, just distal to an active CxxC disulfide site, similar to Erv1/ALR. The primary difference between FAD binding to QSOX1 and ERV is that the binding site is more accessible in QSOX1.
Structural studies often reveal clues for potential protein function. Since QSOX1 is the only known sulfhydryl oxidase with both disulfide-generating and disulfide-transferring capabilities, it is instructive to learn how the enzyme accomplishes disulfide relays. From the QSOX1 structure reported by Alon et al. (5), major conformational changes in the enzyme likely occur during disulfide relay. Alon et al. reported that the CxxC sequence in the first TRX domain at the N-terminus moves into close proximity to the second CxxC in the ERV1 domain during disulfide shuttling using DTT as a substrate, providing a structural basis for the benefit of gene fusion between TRX and ERV domains and enhancing the redox activity and efficiency in a single catalytic protein (5). As predicted by Heckler et al. (35), mutagenesis studies have shown that the third CxxC motif in QSOX1 is not essential for sulfhydryl oxidase activity (49). It would be highly informative to perform structural studies using native reduced protein as a substrate to learn how QSOX1 generates and transfers disulfides as a protein is folded.
Enzymatically, QSOX1 was shown to introduce disulfide bonds into proteins with a high efficiency (Km=110–330 μM/thiol) and to prefer protein substrates rather than small mono- and di-thiol containing molecules (40–42). QSOX1 also cooperates with PDI during protein folding. A provocative model proposed by the Thorpe group that continues to hold is that QSOX1 indiscriminately oxidizes unfolded protein thiols in the ER (and Golgi), while partially reduced PDI iteratively re-folds proteins into a functional, native state via redox shuffling (30, 40). This cooperative interplay between PDI and QSOX1 also likely involves reduced glutathione in the ER (71) to keep PDI partially reduced. Ultimately, disulfide exchanges among the CxxC motifs reduces FAD, producing dihydroflavin, followed by a reduction of molecular oxygen to form hydrogen peroxide as the protein is folded (Fig. 1).
FIG. 1.

Cartoon of quiescin sulfhydryl oxidase 1 (QSOX1)-L, a 747 amino acid sulfhydryl oxidase. Thioredoxin domains are labeled TRX1 and TRX2. TRX2 does not contain CxxC. ERV/ALR domains (amino acids 396–503) contain 2 CxxC sequences and an FAD-binding domain. FAD binds between the α3 and α4 helices in the QSOX1 protein structure. NEQEQPLGQWHLS peptide is shown (amino acids 631–643) along with the transmembrane domain (amino acids 710–730). QSOX1-S is 604 amino acids and identical in sequence except for amino acids 603 (leucine) and 604 (isoleucine). Molecular oxygen is shown as being reduced by QSOX1 to H2O2 as disulfides are formed in the protein. FAD, flavin adenine dinucleotide. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
QSOX1-S and–L are composed of identical sequences until the middle of the 12th exon in which the short form splices out 733 base pairs in exon 12 and splices into exon 13 (also called exon 12c), terminating after two amino acids, encoding a 604 amino acid protein without a transmembrane region contained in the 733 bp region. The long form is a 747-amino-acid protein that includes most of exon 12, including the transmembrane region. Soloviev et al. recently reported the existence of seven additional RNA splice variants (QSOX1c-i), but it is not known whether any of them are translated (78). QSOX1 variants f, g, h, and i truncate very early, making them unlikely to have function if translated. However, if QSOX1c, d, and e are translated, it is possible that they would have sulfhydryl oxidase activity because they contain the ERV/ALR-FAD binding domain for the reduction of molecular oxygen to hydrogen peroxide.
QSOX1 Protein Expression: Tumor Versus Normal
Experiments by Bulleid's group have recently shown that QSOX1-L is proteolyzed and secreted into the ECM, despite the existence of a transmembrane domain in QSOX1-L (74). QSOX1-L was proteolytically cleaved within the ectodomain of the protein, leaving the transmembrane domain associated with the cell while the ectodomain was released extracellularly into the medium. Further, Bulleid and colleagues suggest secreted QSOX1-L and–S form dimers such that QSOX1-L regulates the amount of QSOX1-S secreted by the cell. It should be noted that QSOX1-S is highly over-expressed compared with QSOX1-L in all tumor cell lines tested to date (see Fig. 3, below). Therefore, some excess QSOX1-S that is not dimerized with cleaved QSOX1-L should be secreted from tumor cells and into the ECM or into circulation. In addition, it is unclear how the NEQEQPLGQWHLS peptide from QSOX1-L reported by our group (7) is cleaved from QSOX1-L just upstream of the cleavage products reported by Bulleid and colleagues.
FIG. 3.

Western blot analysis of QSOX1 expression in tumor cell lines. Cytoplasmic lysates were made from ∼5 million cells growing in culture. 20 μg of whole cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by transfer to polyvinylidene fluoride (PVDF) membrane and probed with polyclonal anti-QSOX1 antibodies (Proteintech, catalog # 12713-1-AP). Starting left to right from the molecular weight ladder, the first 4 cell lines (V19I-sA2) were immortalized by human papilloma virus E6 and E7 oncogenes (kind gift from Dr. Melissa Herbst-Kralovetz). Tissue origins of the cell lines are as follows. V19I is a vaginal epithelial; 3ECI is an ectocervical epithelial; and A2 &sA2 are endocervical epithelial tumor cell lines. CaCo is colon cancer; HEC-1A is uterine epithelial; and INT 407 is an embryonic intestinal cell line. EBV ND111 and ND111 (PBMC) are from the same donor, indicated by arrows except that “EBV ND111” is an EBV-transformed lymphoblastoid cell line (B-LCL). MCF-10A is a nontumorigenic breast cell line, and BxPC3 is a pancreatic cell line from a malignant ductal adenocarcinoma. 4T1, 3LL, and B16 are murine breast, lung, and melanoma tumor cell lines, respectively. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In 2004, Tury et al. demonstrated QSOX1 protein expression in rats in the anterior lobe of the pituitary (adenohypophysis) and that its expresson was regulated by estrogens. RNA expression profiling showed that QSOX1 mRNA was very high in placenta and comparatively less high in the lungs (22). More recent histologic studies in the embryonic mouse suggest that QSOX1 protein expression appears in developing fetal tissue, which is consistent with the idea presented later in this review that QSOX1 is involved in tissue remodeling, invasion, and metastasis (68). In 2009, we performed IHC using anti-QSOX1 antibodies (Proteintech) to determine whether QSOX1 was over-expressed in pancreatic tumor tissues resected from patients with pancreatic cancer. We found that QSOX1 protein was highly over-expressed in tumor cells, but not in adjacent normal tissue, including stromal fibroblasts, macrophages, and infiltrating lymphocytes (7, 47). Others have performed IHC on noncancer tissues and reported QSOX1 expression in tissues and cells with high secretory loads such as apocrine glands in skin, pancreatic islets, parotid glands, and intestine in columnar epithelia (82).
The apparent differences in QSOX1 protein expression between normal and tumor tissues may simply lie in the fact that normal tissues were antibody stained without the presence of tumor in the tissue section or slide. When staining tissue sections containing tumors, normal tissue containing stromal fibroblasts, macrophages, and infiltrating lymphocytes is almost always present in a section. Therefore, QSOX1 protein expression can be compared in tumor cells and adjacent normal stroma. For example, we have never seen pancreatic islets express QSOX1 when staining a tissue section containing ductal adenocarcinoma of the pancreas (Fig. 2). Since tumor cells express QSOX1 at such high levels, the stain is removed after a few minutes, leaving any cells that express relatively low levels of QSOX1 unstained. In contrast, one might leave the substrate to stain a tissue section without tumor for a longer period of time if expression is weak. Figure 2 illustrates lack of expression of QSOX1 from a tissue microarray (TMA) of the pancreas without tumor cells (left) compared with another tissue punch containing tumor cells from the same TMA slide. Further support of this idea comes from Soloviev et al., who found that QSOX1 was one of four highest ranked genes expressed in SAGE cDNA databases from breast cancer patients that was not expressed in normal breast SAGE databases (78). Therefore, while QSOX1 transcript may be present, the level of QSOX1 protein produced in nonmalignant cells—even in cells with a high secretory load such as islet cells in the pancreas—is much less than what tumor cells produce.
FIG. 2.
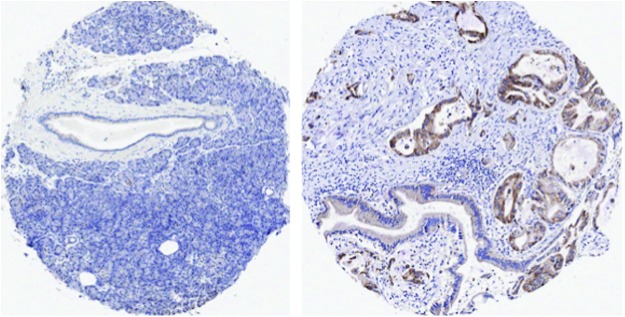
QSOX1 expression in normal pancreatic tissue (left) and pancreatic tissue containing tumor from a patient with ductal adenocarcinoma of the pancreas. Immunohistochemical staining for QSOX1 protein expression was performed on the same tissue microarray slide using the same reagents, same incubation, and same development times. Two tissue punches were stained for QSOX1 as reported in Antwi et al. (7). If a cell produces QSOX1 protein in a tissue, anti-QSOX1 antibodies will detect it (stain it). The bound anti-QSOX1 antibodies are then detected with goat anti-rabbit IgG coupled to horseradish peroxidase enzyme. When a substrate (3-3′-diaminobenzidine, DAB) is added, QSOX1 expression is observed as brown staining. QSOX1 expression was uniformly associated with tumor cell cytoplasm.
Tumor Biology of QSOX1
One of the first reports associating QSOX1 and cancer was indirect. Hellebrekers et al. were studying epigenetically silenced genes in endothelial cells derived from tumors (36). They found that QSOX1, clusterin, and fibrillin 1 were suppressed during angiogenesis. In this study, knockdown of QSOX1 using short hairpin RNA resulted in an increase in endothelial cell sprouting. If Hellebreker's findings are correct, an unanswered question is how QSOX1 expression is connected to the suppression of angiogenesis, especially when QSOX1 appears to be involved in invasion (44, 46, 47). It is likely that different mechanisms are required for invasion and growth at a metastatic site, so one explanation could be as follows. QSOX1 may help tumor cells invade through a basement membrane and gain access to the bloodstream, but once circulating tumor cells intravasate back into a tissue and begin to grow at a metastatic site, QSOX1 is no longer required and so its expression is turned off, enabling an angiogenic phenotype to emerge. No group has followed up on this finding to date.
Another puzzling study was reported by Morel et al. (56). Although breast cancer cells naturally over-express QSOX1, Morel et al. engineered MCF-7 to over-express guinea pig QSOX1. After treating MCF-7 cells engineered to over-express guinea pig QSOX1 with hydrogen peroxide, QSOX1 was reported to be elevated and protected the cells against oxidative stress-induced apoptosis. This report is curious for two reasons. First, malignant breast cancer cell lines and tumor tissue already over-express human QSOX1 (47, 78), and second, hydrogen peroxide is naturally produced as a result of the enzymatic activity of QSOX1 as shown by Thorpe and colleagues (40, 41, 82). It is not known that QSOX1 is involved in a positive feedback loop in which hydrogen peroxide induces QSOX1 to be expressed, which produces more hydrogen peroxide via the enzymatic activity of QSOX1.
In gene expression profiling experiments using laser capture microdissection of human prostate cancer tissue, Song et al. showed that RNA expression of QSOX1 and clusterin were elevated when Nkx3.1 was down-regulated (79). Nkx3.1 is a homeodomain transcriptional regulator and tumor suppressor gene in prostate cancer (10, 53). Nkx3.1 expression is decreased during prostatic hyperplasia and prostatic intraepithelial neoplasia (PIN) in mouse models of prostate cancer and in humans (34). This inverse relationship between the Nkx3.1 transcriptional regulator and QSOX1 could suggest that QSOX1 plays an important role early in driving human prostate tumorigenesis from prostatic hyperplasia to PIN to stromal invasion. Hence, over-expression of QSOX1 may play a role earlier than previously thought in prostate cancer. Experiments to follow up on this observation are warranted.
QSOX1 may also contribute to genetic instability. Since QSOX1 produces hydrogen peroxide as a result of its enzymatic activity, the over-expression of QSOX1 makes the cellular microenvironment highly oxidative, containing reactive oxygen species (ROS). This oxidative cellular environment triggers the production of glutathione/glutathione reductase, thioredoxin reductase, and superoxide dismutatse anti-oxidant responses. It is well known that ROS cause adducts in DNA and that these adducts may lead to genomic mutations, especially if DNA repair mechanisms are not functional (12). However, ROS also affects a multitude of cellular functions [reviewed in (63)] including transcription factor activation (31, 59, 76), intracellular signaling (18, 81, 84), cell cycle (19), motility (62), and anoikis (63).
Since ROS are toxic, cells should detoxify them to function and survive. Tumor cells, however, can utilize this oxidative environment. Oxidation of pro-growth signaling molecules such as Src, Akt, and Erk kinases drives cell survival and helps escape death pathways. For example, oxidatively activated Src enables ligand-independent phosphorylation of EGFR, leading to downstream activation of ERK and Akt, pro-survival signals. Activated ERK and Akt constitutively phosphorylate Bim, a pro-apoptotic protein, leading to its degradation by the proteasome (63). It remains to be seen which pathways and specifically how the activity of QSOX1 affects tumor cell growth, invasion, and metastasis. Since the subjects of tumor cell growth and metastasis through redox signaling have been extensively reviewed by Pani et al. (63), we will not provide an in-depth review of redox mechanisms and tumor cell growth. Rather, we will continue to review the potential role of QSOX1 in cancer.
In 2009, we reported finding a 13-mer peptide from QSOX1-L, residues 631–643 (NEQEQPLGQWHLS), in plasma from patients with pancreatic ductal adenocarcinoma that was not present in plasma from control donors (7). This peptide was found to be stable in whole blood for 24 h. Levels of the QSOX1-L peptide ranged between 7.1 and 156.6 ng/ml, while peptide levels in plasma from control individuals ranged from 3.6 to 13.4 ng/ml (7). QSOX1-L peptide has not yet been reported to be present in plasma from patients with other malignancies; however, if the tumor biology of QSOX1 is similar among different cancers, one would expect to find this peptide in plasma. Since this tumor-derived peptide circulates in blood from cancer patients, it could have utility as a blood-based biomarker.
In the same report by Antwi et al., IHC of pancreatic cancer patients' tumors demonstrated that QSOX1 parent protein was over-expressed, making a direct connection between the presence of peptide in plasma and over-expression of the parent protein in tumors. Expression was typically confined to the cytoplasm or organelles within the cytoplasm of tumor cells, while QSOX1 protein was not detected in adjacent stromal cells and infiltrating lymphocytes. QSOX1-S and–L over-expression was also observed in pancreatic cancer cell lines, CFPac-1, Panc-1, and BxPC3, but not expressed in peripheral blood mononuclear cells (PBMC) by Western blot analysis (7).
Subsequent studies on the spectrum of QSOX1 expression among cell lines representing different histological types of tumors suggest that both long and short splice variants of QSOX1 are over-expressed in multiple tumor cell types, including vaginal, uterine and cervical, colon, intestinal epithelial, and even common mouse tumor cell lines such as 4T1 (breast), 3LL (lung), and B16 (melanoma), shown in Figure 3. Although PBMC do not show QSOX1 protein expression, B cells from the same donor PBMC transformed with EBV show strong QSOX1 expression (arrows in Fig. 3, arrows). Since EBV-transformed B cells can form tumors in immunodeficient animals, it is tempting to hypothesize that QSOX1 over-expression is a marker of tumorigenesis.
On finding that QSOX1 was over-expressed in multiple types of cancers, but not in resting PBMC, the question arose as to whether QSOX1 was simply a marker of proliferation similar to Ki-67. To address this question, we exposed PBMC to phytohemagglutinin (PHA), a lectin that causes lymphocytes to rapidly proliferate into leukemia-like blasts. As shown in Figure 4, PHA-treated PBMC only weakly express QSOX1 compared with the pancreatic tumor cell line BxPC3. Moreover, QSOX1 is very weakly expressed in cell lines that are unable to form tumors in immunodeficient mice (MCF-10A and HPDE6) compared with malignant tumor cell lines MCF-7, BT549 (breast), and BxPC3 (pancreas) (Fig. 4). Taken together, QSOX1 expression in EBV-transformed B lymphoblastoid cells along with weak or lack of QSOX1 expression strongly suggest that QSOX1 is likely to be a marker of tumorigenesis, not simply proliferation.
FIG. 4.

QSOX1 is a marker of tumorigenesis, not proliferation. One million PBMCs were either untreated or stimulated with phytohemagglutinin (PHA) for 3 days. Cell lysates were prepared and protein concentrations were normalized such that 20 μg of each cell lysate was added to each lane of an SDS-PAGE gel. Electrophoresed proteins were transferred to PVDF membrane and probed with Protein Tech polyclonal anti-QSOX1 antibodies as described in Figure 3.
In 2011 and 2013, we reported that QSOX1 plays a role in pancreatic and breast tumor cell growth and invasion in vitro (46, 47). We also demonstrated that QSOX1 affects the proteolytic activity of matrix metalloproteins (MMP-2 and -9) secreted by tumor cells. Using short hairpin RNA in a lentiviral vector, QSOX1 was stably knocked down in pancreatic and breast tumor cell lines. Tumor cells in which QSOX1 was silenced grew at less than 30% the rate of controls. Furthermore, QSOX1-silenced tumor cells were prevented from invading through the Matrigel basement membrane in modified Boyden chamber assays. As proof that QSOX1 played an important role in invasion, the addition of recombinant QSOX1 protein to the media rescued the invasive phenotype of shQSOX1-transduced breast tumor cell lines (47). Mechanistically, silencing QSOX1 decreased MMP proteolytic activity, but not MMP mRNA in both pancreatic and breast tumor cell lines.
Affymetrics gene expression data from 1881 molecularly typed cases of breast cancer was used to create a web-based database called Gene expression-based Outcome for Breast cancer Online (GOBO). We also utilized this publically available tool to demonstrate that luminal B breast cancer patients whose tumors overexpress QSOX1 have very poor overall and relapse-free survival (47). Pathologically, stronger QSOX1 protein expression was associated with higher grade tumors that were ER-positive and Her-2 and cytokeratin 5/6 negative. These data suggest that QSOX1 is an independent predictor of poor clinical outcome. Collectively, our pancreas and breast publications suggest that QSOX1 is associated with and may produce poorer outcomes in patients with cancer.
Controversy Over Tumor Biology
One report by Pernodet et al. (67) is contrary to our findings in breast and pancreas cancer as well as contrary to other groups that share our position (44, 78). Pernodet et al. performed quantitative RTPCR on mRNA from an internal set of invasive ductal carcinomas of the breast and concluded that QSOX1 expression was lower in patients with “perjorative prognostic factors.” Other results from this report suggest that silencing QSOX1 expression enhances growth in vivo and invasion in vitro of MB-231 breast carcinoma, while over-expressing QSOX1 enhances growth and invasion in vitro in MCF-7 cells. The data in this report are difficult to interpret and the experiments not well controlled. For example, IHC figures in Pernodet are very difficult to interpret due to the lack of staining tumor and normal tissue on the same slide. Furthermore, the IHC pictures are out of focus. Pernodet et al. also did not also consider molecular classifications of tumors in their mRNA analysis, nor were their cell lines certified, which may explain some of the disparate findings. Since experimental results from Pernodet et al. and Katchman et al. are nearly polar opposites, an editorial drew attention to the preponderance of data favoring Katchman et al., but clearly called for clarification of the discrepancy in the literature (23).
Two other publications support QSOX1 as an indicator of poor relapse-free and overall survival in cancer. First, Soloviev et al. reported that QSOX1 mRNA is up-regulated in grades 2 and 3 breast cancer, but decreased in grade 1 breast carcinomas (78). They also corroborated that QSOX1 is independent of Her-2 expression as a predictor of aggressive disease and poor outcome. Second, Ilani et al. recently reported that QSOX1 activity is required for the proper incorporation of laminin into the ECM of fibroblasts (44). As a result, the migration of lung tumor cells across a layer of fibroblasts was attenuated when fibroblasts were silenced from expressing QSOX1. These findings support and complement our pancreas and breast cancer reports that QSOX1 plays an important role in tumor cell invasion. Given the importance of the ECM in cancer and the interplay between tumors and stroma, a study of the role of QSOX1 in the ECM promises to yield important findings in the near future.
Why Do Tumor Cells Over-Express QSOX1?
Our laboratory and the Fass laboratory have begun answering the question of why tumor cells over-express QSOX1. The Fass group recently reported that QSOX1 was required for the incorporation of laminin into the ECM of fibroblasts. Moreover, lung tumor cell invasion across a fibroblast monolayer was attenuated when QSOX1 protein was blocked with an antibody or if it was silenced with shRNA. These findings indicate that QSOX1 is active in the ECM, likely at the tumor-stroma interface. Our data demonstrate that silencing QSOX1 in tumor cells suppresses their ability to invade across Matrigel, an artificial basement membrane. The exogenous addition of recombinant QSOX1 to the QSOX1-silenced tumor cells rescued their invasive phenotype. Mechanistically, we demonstrated that QSOX1 post-translationally activates MMPs. Since MMPs degrade basement membranes during the metastatic cascade, QSOX1 is likely to play a major role in invasion and metastasis.
In addition to providing tumor cells with the advantages of enhanced growth and invasion, QSOX1 may be up-regulated in tumors- because they produce excess protein for the reasons diagrammed in Figure 5 under “Nuclear Damage.” Excess protein production requires enzymes involved in protein folding such as heat shock proteins [reviewed in (57)] and PDIs. PDIs are particularly interesting with regard to QSOX1, because the enzymes perform similar redox functions. As mentioned earlier, QSOX1 is considered as generating disulfide bonds indiscriminately in proteins as they are produced in the ER, while PDI corrects and re-shuffles mispaired disulfides resulting in proper disulfide pairings. While QSOX1 and PDI cooperate to fold a wide range of proteins, PDI is not a substrate of QSOX1 despite the fact that PDI contains multiple CxxC motifs with free thiols in reduced form (71). Neither QSOX1 nor PDI are efficient in folding proteins independent of each other. This finding is surprising, because (i) both proteins reside in the ER and presumably shuttle disulfides in the same proteins, and (ii) it suggests that QSOX1 prefers some substrates compared with others. In addition, PDI and QSOX1 expression do not significantly correlate with each other in any cell line from the NCI-60 (72).
FIG. 5.

Proposed model for why QSOX1 is over-expressed. Point mutations may cause kinases to be overactive, resulting in increased transcription of multiple genes, many of which contain cysteine residues. Hypo- or hypermethylation of cancer genomes causes areas of the genome that would usually be silent to be transcribed and translated. Chromosomal instability may occur from defects in centromeres, chromosomal segregation, telomere, and microsatellite instability and defects in DNA repair. Gene amplification can be caused by DNA polymerase slippage, retrotransposition, and homologous recombination. Translocations and inversions are caused by aberrant expression of recombinase activating genes, double-stranded breaks due to environmental factors (reactive oxygen species [ROS]), and improper homologous pairing during mitosis. Aneuploidy results from multiple cell divisions in which chromosomes are improperly replicated and unstable such that chromosomes are improperly segregated during mitosis. The transcription of multiple splice variants may occur as a result of a dysregulated aneuploid genome, which is hypomethylated, and genes are amplified, or a protein encoding gene is translocated downstream of an actively transcribed region of the genome (58). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
As mentioned earlier and depicted in Figure 5, most tumors have genetic aberrations ranging from point mutations in signaling molecules or growth factor receptors to chromosomal rearrangements in which large sections of chromatin from different chromosomes break during cell division and reattach in the wrong places (translocations and inversions). This sometimes juxtaposes growth factors or kinases downstream of highly active promoter/enhancer regions in chromatin. The Philadelphia chromosome translocation t(9:22) serves as one example in which the Abl1 gene on chromosome 22 breaks during cell division and translocates (fuses) to the bcr gene from chromosome 9. This results in constitutive activation of the Abl1 tyrosine kinase and causes chronic myelogenous leukemia as well as other types of leukemias. In nonmalignant cells with diploid chromosomes, normal levels of transcription and translation occur, resulting in the expression of about 10,000 genes, depending on the cell type. However, most tumors are aneuploid with highly unstable chromosomes exposed to transcriptional machinery, resulting in aberrant RNA transcripts. This relaxed transcriptional and translational control provides ample opportunity for the tumor cell to translate many missense irregular proteins. If gene amplification occurs in a region of DNA containing a growth factor receptor, the number of growth factor receptors per cell would be increased, resulting in aberrant cellular proliferation. Hypo- or hypermethylation may also increase the transcription of oncogenes or decrease levels of tumor suppressor genes, leading to tumor growth and the inability of the cell to die. Unlike nonsense mediated decay, which degrades RNA transcripts with premature stop codons (43), missense transcripts would be translated into protein and require disulfide bond formation similar to any other protein. The extent of missense proteins produced in tumor cells is not known, but p53 alone has 6 missense mutations (29). In addition, spliceosome machinery is also known to be dysregulated in cancer (60). This could lead to thousands of mis-spliced transcripts that produce proteins which QSOX1 may attempt to fold.
The cause of most genetic mutations is unknown, but ROS, as mentioned earlier, can damage DNA, leading to mutations (Fig. 6). Although ROS may induce apoptosis, they have also been shown to activate oncogenic transcription factors, [reviewed in (51)], affect cell cycle by elevating factors that bind p53 to prevent cell cycle arrest (17), and even mediate post-translational modifications such as phosphorylation-dephosphorylation reactions driving second-messenger signaling (27). Importantly, QSOX1-produced ROS may drive EMT involving inhibition of glycogen synthase kinase-3β and nuclear translocations of SNAIL (zinc finger transcription factor) and β-catenin (62, 63). Unlike epithelial cells, mesenchymal cells lack apical-basolateral polarity, stable tight junctions, do not form monolayers, and exhibit migratory and invasive properties. The invasive and migratory nature of mesenchymal cells is primarily due to loss of cell surface expression of E-cadherin, up-regulation of N-cadherin, and modification of cytoskeletal components such as vimentin and α-smooth muscle actin, which drive cell motility.
FIG. 6.

Model for QSOX1-mediated tumor cell invasion. QSOX1 activates matrix metalloproteinases (MMPs) that are capable of digesting basement membranes (46) and incorporates laminin into fibroblasts, which facilitates the migration of tumor cells into the fibroblast layer (44). The production of H2O2 by QSOX1 may also elicit vascular endothelial growth factor (VEGF) production by tumor cells, stimulating blood vessel growth into the tumor (62). Taken together, these events may facilitate tumor cell migration through stroma and invasion of tumor cells into blood circulation. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Inflammatory cells produce ROS as a part of their killing mechanisms. Tumor cells may escape this danger by activating a motility/invasion program. If QSOX1 is active in the ECM and producing hydrogen peroxide, as we and others have reported, ROS produced in the tumor microenvironment could induce motility in tumor cells. The production of ROS by QSOX1 in the ECM may mimic this inflammatory effect.
In addition, Arbiser et al. (9) reported that Nox1, a catalytic subunit of the superoxide-generating NADPH-oxidase, causes malignant transformation of nontumor NIH 3T3 cells and increases tumorigenicity of the human prostate cell line, DU-145. Hydrogen peroxide produced by Nox1 also up-regulated VEGF and VEGF receptor, leading to increased angiogenesis and MMP activity in tumors in vivo as shown in Figure 6. Although we have not determined whether H2O2 produced by QSOX1 elicits this activity, this is further evidence that tumor-derived ROS drives important features of tumorigenesis.
It is logical to hypothesize that other proteins related to the function of QSOX1 might be elevated in cancer. Using commercially available antibodies, we did not observe an over-expression of Erv1/ALR or Ero1 in tumor cells.
Conclusions
It is not surprising that the connection between QSOX1 and cancer was not published until 2007–2008. cDNA expression arrays performed on the NCI-60 panel of tumor cells show elevated QSOX1 mRNA in some cell lines, but variable levels of QSOX1 expression among the same and different histological types of tumors. QSOX1 RNA levels did not uniformly stand out among other genes in each of the NCI-60 cell lines (72). This is curious, because nearly every tumor cell line and fresh tumor tissue we have stained with anti-QSOX1 antibodies over-expresses QSOX1; while nonmalignant cells, such as lymphocytes and MCF-10A, do not. Further investigation into QSOX1 expression and its effects on other cellular proteins is warranted.
In addition, tumor biology studies on the ECM and stroma are very difficult to perform, because it is challenging to separate the contribution of the ECM from tumor cells and fibroblasts. Therefore, if the benefit that QSOX1 provides to tumor cells resides in the ECM, there is more QSOX1-related tumor biology to discover. Thankfully, early work on the enzymology was established by Colin Thorpe's group (71) and crystal structures are known for QSOX1, reported by the Fass group (6). If QSOX1 plays an important role in tumor cell invasion and subsequent metastasis, these early studies provide a firm foundation on which to continue studying not only the tumor biology of QSOX1, but also the study of QSOX1 as an anti-neoplastic target.
Abbreviations Used
- DAB
3-3′-diaminobenzidine
- EBV
Epstein-Barr virus
- ECM
extracellular matrix
- EGFR
epidermal growth factor receptor
- EMT
epithelial to mesenchymal transition
- ER
estrogen receptor
- FAD
flavin adenine dinucleotide
- GI
gastrointestinal
- IHC
immunohistochemistry
- MMP
matrix metalloproteins
- PBMC
peripheral blood mononuclear cells
- PDI
protein disulfide isomerase
- PHA
phytohemagglutinin
- PIN
prostatic intraepithelial neoplasia
- PVDF
polyvinylidene fluoride
- QSOX1
quiescin sulfhydryl oxidase 1
- ROS
reactive oxygen species
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TMA
tissue microarray
- TRX
thioredoxin
- VEGF
vascular endothelial growth factor
References
- 1.2013. American Cancer Society American Cancer Society. www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2013/index Accessed July102013
- 2.Agarwala SS. and Case S. Everolimus (RAD001) in the treatment of advanced renal cell carcinoma: a review. Oncologist 15: 236–245, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmad SS, Duke S, Jena R, Williams MV, and Burnet NG. Advances in radiotherapy. BMJ 345: e7765, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Al-Sohaily S, Biankin A, Leong R, Kohonen-Corish M, and Warusavitarne J. Molecular pathways in colorectal cancer. J Gastroenterol Hepatol 27: 1423–1431, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Alon A, Grossman I, Gat Y, Kodali VK, DiMaio F, Mehlman T, Haran G, Baker D, Thorpe C, and Fass D. The dynamic disulphide relay of quiescin sulphydryl oxidase. Nature 488: 414–418, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alon A, Heckler EJ, Thorpe C, and Fass D. QSOX contains a pseudo-dimer of functional and degenerate sulfhydryl oxidase domains. FEBS Lett 584: 1521–1525, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Antwi K, Hostetter G, Demeure M, Decker G, Ruiz Y, Sielaff T, Koep L, and Lake D. Analysis of human plasma peptidome reveals potential biomarker for pancreatic cancer. J Proteome Res 8: 4722–4731, 2009 [DOI] [PubMed] [Google Scholar]
- 8.Aokage K, Ishii G, Ohtaki Y, Yamaguchi Y, Hishida T, Yoshida J, Nishimura M, Nagai K, and Ochiai A. Dynamic molecular changes associated with epithelial-mesenchymal transition and subsequent mesenchymal-epithelial transition in the early phase of metastatic tumor formation. Int J Cancer 128: 1585–1595, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy S, Arnold RS, and Lambeth JD. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc Natl Acad Sci U S A 99: 715–720, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asatiani E, Huang WX, Wang A, Rodriguez Ortner E, Cavalli LR, Haddad BR, and Gelmann EP. Deletion, methylation, and expression of the NKX3.1 suppressor gene in primary human prostate cancer. Cancer Res 65: 1164–1173, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Ashman LK. and Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opin Investig Drugs 22: 103–115, 2013 [DOI] [PubMed] [Google Scholar]
- 12.Berquist BR. and Wilson DM 3rd., Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett 327: 61–72, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blick T, Widodo E, Hugo H, Waltham M, Lenburg ME, Neve RM, and Thompson EW. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis 25: 629–642, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Box C, Rogers SJ, Mendiola M, and Eccles SA. Tumour-microenvironmental interactions: paths to progression and targets for treatment. Semin Cancer Biol 20: 128–138, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Cannito S, Novo E, Compagnone A, Valfre di Bonzo L, Busletta C, Zamara E, Paternostro C, Povero D, Bandino A, Bozzo F, Cravanzola C, Bravoco V, Colombatto S, and Parola M. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis 29: 2267–2278, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Cannito S, Novo E, di Bonzo LV, Busletta C, Colombatto S, and Parola M. Epithelial-mesenchymal transition: from molecular mechanisms, redox regulation to implications in human health and disease. Antioxid Redox Signal 12: 1383–1430, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Cano CE, Gommeaux J, Pietri S, Culcasi M, Garcia S, Seux M, Barelier S, Vasseur S, Spoto RP, Pebusque MJ, Dusetti NJ, Iovanna JL, and Carrier A. Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer Res 69: 219–226, 2009 [DOI] [PubMed] [Google Scholar]
- 18.Chiarugi P. and Buricchi F. Protein tyrosine phosphorylation and reversible oxidation: two cross-talking posttranslation modifications. Antioxid Redox Signal 9: 1–24, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Chiu J. and Dawes IW. Redox control of cell proliferation. Trends Cell Biol 22: 592–601, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Coppock DL, Cina-Poppe D, and Gilleran S. The quiescin Q6 gene (QSCN6) is a fusion of two ancient gene families: thioredoxin and ERV1. Genomics 54: 460–468, 1998 [DOI] [PubMed] [Google Scholar]
- 21.Coppock DL, Kopman C, Scandalis S, and Gilleran S. Preferential gene expression in quiescent human lung fibroblasts. Cell Growth Differ 4: 483–493, 1993 [PubMed] [Google Scholar]
- 22.Coppock DL. and Thorpe C. Multidomain flavin-dependent sulfhydryl oxidases. Antioxid Redox Signal 8: 300–311, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Das P, Siegers GM, and Postovit LM. Illuminating luminal B: QSOX1 as a subtype-specific biomarker. Breast Cancer Res 15: 104, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeGrado TR, Turkington TG, Williams JJ, Stearns CW, Hoffman JM, and Coleman RE. Performance characteristics of a whole-body PET scanner. J Nucl Med 35: 1398–1406, 1994 [PubMed] [Google Scholar]
- 25.Deininger MW. and Druker BJ. Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacol Rev 55: 401–423, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, and Clarke MF. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458: 780–783, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esposito F, Ammendola R, Faraonio R, Russo T, and Cimino F. Redox control of signal transduction, gene expression and cellular senescence. Neurochem Res 29: 617–628, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med 359: 2143–2153, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Freed-Pastor WA. and Prives C. Mutant p53: one name, many proteins. Genes Dev 26: 1268–1286, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilbert HF. Protein disulfide isomerase and assisted protein folding. J Biol Chem 272: 29399–29402, 1997 [DOI] [PubMed] [Google Scholar]
- 31.Go YM. and Jones DP. Redox control systems in the nucleus: mechanisms and functions. Antioxid Redox Signal 13: 489–509, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldman L. and Schafer AI. (Eds). Goldmans's Cecil Medicine. Philadelphia: Saunders, 2012 [Google Scholar]
- 33.Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, Lenz HJ, Goldberg RM, Sargent DJ, Cihon F, Cupit L, Wagner A, and Laurent D. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381: 303–312, 2013 [DOI] [PubMed] [Google Scholar]
- 34.He WW, Sciavolino PJ, Wing J, Augustus M, Hudson P, Meissner PS, Curtis RT, Shell BK, Bostwick DG, Tindall DJ, Gelmann EP, Abate-Shen C, and Carter KC. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics 43: 69–77, 1997 [DOI] [PubMed] [Google Scholar]
- 35.Heckler EJ, Alon A, Fass D, and Thorpe C. Human quiescin-sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis. Biochemistry 47: 4955–4963, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hellebrekers DM, Melotte V, Viré E, Langenkamp E, Molema G, Fuks F, Herman JG, Van Criekinge W, Griffioen AW, and van Engeland M. Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res 67: 4138–4148, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Hernandez-Aya LF. and Gonzalez-Angulo AM. Adjuvant systemic therapies in breast cancer. Surg Clin North Am 93: 473–491, 2013 [DOI] [PubMed] [Google Scholar]
- 38.Hoober KL, Glynn NM, Burnside J, Coppock DL, and Thorpe C. Homology between egg white sulfhydryl oxidase and quiescin Q6 defines a new class of flavin-linked sulfhydryl oxidases. J Biol Chem 274: 31759–31762, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Hoober KL, Joneja B, White HB, 3rd, and Thorpe C. A sulfhydryl oxidase from chicken egg white. J Biol Chem 271: 30510–30516, 1996 [DOI] [PubMed] [Google Scholar]
- 40.Hoober KL, Sheasley SL, Gilbert HF, and Thorpe C. Sulfhydryl oxidase from egg white. A facile catalyst for disulfide bond formation in proteins and peptides. J Biol Chem 274: 22147–22150, 1999 [DOI] [PubMed] [Google Scholar]
- 41.Hoober KL. and Thorpe C. Egg white sulfhydryl oxidase: kinetic mechanism of the catalysis of disulfide bond formation. Biochemistry 38: 3211–3217, 1999 [DOI] [PubMed] [Google Scholar]
- 42.Hoober KL. and Thorpe C. Flavin-dependent sulfhydryl oxidases in protein disulfide bond formation. Methods Enzymol 348: 30–34, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Huang L, Lou CH, Chan W, Shum EY, Shao A, Stone E, Karam R, Song HW, and Wilkinson MF. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol Cell 43: 950–961, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ilani T, Alon A, Grossman I, Horowitz B, Kartvelishvily E, Cohen SR, and Fass D. A Secreted disulfide catalyst controls extracellular matrix composition and function. Science 341: 74–76, 2013 [DOI] [PubMed] [Google Scholar]
- 45.Iwao-Koizumi K, Matoba R, Ueno N, Kim SJ, Ando A, Miyoshi Y, Maeda E, Noguchi S, and Kato K. Prediction of docetaxel response in human breast cancer by gene expression profiling. J Clin Oncol 23: 422–431, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Katchman BA, Antwi K, Hostetter G, Demeure MJ, Watanabe A, Decker GA, Miller LJ, Von Hoff DD, and Lake DF. Quiescin sulfhydryl oxidase 1 promotes invasion of pancreatic tumor cells mediated by matrix metalloproteinases. Mol Cancer Res MCR 9: 1621–1631, 2011 [DOI] [PubMed] [Google Scholar]
- 47.Katchman BA, Ocal IT, Cunliffe HE, Chang YH, Hostetter G, Watanabe A, Lobello J, and Lake DF. Expression of quiescin sulfhydryl oxidase 1 is associated with a highly invasive phenotype and correlates with a poor prognosis in Luminal B breast cancer. Breast Cancer Res 15: R28, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim SH, Turnbull J, and Guimond S. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol 209: 139–151, 2011 [DOI] [PubMed] [Google Scholar]
- 49.Kodali VK. and Thorpe C. Quiescin sulfhydryl oxidase from Trypanosoma brucei: catalytic activity and mechanism of a QSOX family member with a single thioredoxin domain. Biochemistry 49: 2075–2085, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lambrechts D, Lenz HJ, de Haas S, Carmeliet P, and Scherer SJ. Markers of response for the antiangiogenic agent bevacizumab. J Clin Oncol 31: 1219–1230, 2013 [DOI] [PubMed] [Google Scholar]
- 51.Landriscina M, Maddalena F, Laudiero G, and Esposito F. Adaptation to oxidative stress, chemoresistance, and cell survival. Antioxid Redox Signal 11: 2701–2716, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Lee J, Hofhaus G, and Lisowsky T. Erv1p from Saccharomyces cerevisiae is a FAD-linked sulfhydryl oxidase. FEBS Lett 477: 62–66, 2000 [DOI] [PubMed] [Google Scholar]
- 53.Macoska JA, Trybus TM, Benson PD, Sakr WA, Grignon DJ, Wojno KD, Pietruk T, and Powell IJ. Evidence for three tumor suppressor gene loci on chromosome 8p in human prostate cancer. Cancer Res 55: 5390–5395, 1995 [PubMed] [Google Scholar]
- 54.Michl P. and Gress TM. Current concepts and novel targets in advanced pancreatic cancer. Gut 62: 317–326, 2013 [DOI] [PubMed] [Google Scholar]
- 55.Mohamed A, Krajewski K, Cakar B, and Ma CX. Targeted therapy for breast cancer. Am J Pathol 183: 1096–1112, 2013 [DOI] [PubMed] [Google Scholar]
- 56.Morel C, Adami P, Musard JF, Duval D, Radom J, and Jouvenot M. Involvement of sulfhydryl oxidase QSOX1 in the protection of cells against oxidative stress-induced apoptosis. Exp Cell Res 313: 3971–3982, 2007 [DOI] [PubMed] [Google Scholar]
- 57.Murphy ME. The HSP70 family and cancer. Carcinogenesis 34: 1181–1188, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nambiar M. and Raghavan SC. How does DNA break during chromosomal translocations? Nucl Acids Res 39: 5813–5825, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oliveira-Marques V, Marinho HS, Cyrne L, and Antunes F. Role of hydrogen peroxide in NF-kappaB activation: from inducer to modulator. Antioxid Redox Signal 11: 2223–2243, 2009 [DOI] [PubMed] [Google Scholar]
- 60.Padgett RA. New connections between splicing and human disease. Trends Genet: TIG 28: 147–154, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palmieri C, Patten DK, Januszewski A, Zucchini G, and Howell SJ. Breast cancer: current and future endocrine therapies. Mol Cell Endocrinol 382: 695–723, 2013 [DOI] [PubMed] [Google Scholar]
- 62.Pani G, Galeotti T, and Chiarugi P. Metastasis: cancer cell's escape from oxidative stress. Cancer Metastasis Rev 29: 351–378, 2010 [DOI] [PubMed] [Google Scholar]
- 63.Pani G, Giannoni E, Galeotti T, and Chiarugi P. Redox-based escape mechanism from death: the cancer lesson. Antioxid Redox Signal 11: 2791–2806, 2009 [DOI] [PubMed] [Google Scholar]
- 64.Papavasiliou P, Chun YS, and Hoffman JP. How to define and manage borderline resectable pancreatic cancer. Surg Clin North Am 93: 663–674, 2013 [DOI] [PubMed] [Google Scholar]
- 65.Patel SG. and Ahnen DJ. Familial colon cancer syndromes: an update of a rapidly evolving field. Curr Gastroenterol Rep 14: 428–438, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pelicano H, Carney D, and Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat 7: 97–110, 2004 [DOI] [PubMed] [Google Scholar]
- 67.Pernodet N, Hermetet F, Adami P, Vejux A, Descotes F, Borg C, Adams M, Pallandre JR, Viennet G, Esnard F, Jouvenot M, and Despouy G. High expression of QSOX1 reduces tumorogenesis, and is associated with a better outcome for breast cancer patients. Breast Cancer Res 14: R136, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Portes KF, Ikegami CM, Getz J, Martins AP, de Noronha L, Zischler LF, Klassen G, Camargo AA, Zanata SM, Bevilacqua E, and Nakao LS. Tissue distribution of quiescin Q6/sulfhydryl oxidase (QSOX) in developing mouse. J Mol Histol 39: 217–225, 2008 [DOI] [PubMed] [Google Scholar]
- 69.Radisky ES. and Radisky DC. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J mammary Gland Biol Neoplasia 15: 201–212, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raje S, Glynn NM, and Thorpe C. A continuous fluorescence assay for sulfhydryl oxidase. Anal Biochem 307: 266–272, 2002 [DOI] [PubMed] [Google Scholar]
- 71.Rancy PC. and Thorpe C. Oxidative protein folding in vitro: a study of the cooperation between quiescin-sulfhydryl oxidase and protein disulfide isomerase. Biochemistry 47: 12047–12056, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, Doroshow J, and Pommier Y. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res 72: 3499–3511, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Riemer J, Bulleid N, and Herrmann JM. Disulfide formation in the ER and mitochondria: two solutions to a common process. Science 324: 1284–1287, 2009 [DOI] [PubMed] [Google Scholar]
- 74.Rudolf J, Pringle MA, and Bulleid NJ. Proteolytic processing of QSOX1A ensures efficient secretion of a potent disulfide catalyst. Biochem J 454: 181–190, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanchez-Munoz A, Perez-Ruiz E, Jimenez B, Ribelles N, Marquez A, Garcia-Rios I, and Alba Conejo E. Targeted therapy of metastatic breast cancer. Clin Transl Oncol 11: 643–650, 2009 [DOI] [PubMed] [Google Scholar]
- 76.Sekhar KR, Rachakonda G, and Freeman ML. Cysteine-based regulation of the CUL3 adaptor protein Keap1. Toxicol Appl Pharmacol 244: 21–26, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, and McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177–182, 1987 [DOI] [PubMed] [Google Scholar]
- 78.Soloviev M, Esteves MP, Amiri F, Crompton MR, and Rider CC. Elevated transcription of the gene QSOX1 encoding quiescin Q6 sulfhydryl oxidase 1 in breast cancer. PloS One 8: e57327, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Song H, Zhang B, Watson MA, Humphrey PA, Lim H, and Milbrandt J. Loss of Nkx3.1 leads to the activation of discrete downstream target genes during prostate tumorigenesis. Oncogene 28: 3307–3319, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sounni NE. and Noel A. Targeting the tumor microenvironment for cancer therapy. Clin Chem 59: 85–93, 2013 [DOI] [PubMed] [Google Scholar]
- 81.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, and Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270: 296–299, 1995 [DOI] [PubMed] [Google Scholar]
- 82.Thorpe C, Hoober KL, Raje S, Glynn NM, Burnside J, Turi GK, and Coppock DL. Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes. Arch Biochem Biophys 405: 1–12, 2002 [DOI] [PubMed] [Google Scholar]
- 83.Tol J. and Punt CJ. Monoclonal antibodies in the treatment of metastatic colorectal cancer: a review. Clin Ther 32: 437–453, 2010 [DOI] [PubMed] [Google Scholar]
- 84.Truong TH. and Carroll KS. Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry 51: 9954–9965, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tury A, Mairet-Coello G, Poncet F, Jacquemard C, Risold PY, Fellmann D, and Griffond B. QSOX sulfhydryl oxidase in rat adenohypophysis: localization and regulation by estrogens. J Endocrinol 183: 353–363, 2004 [DOI] [PubMed] [Google Scholar]
- 86.Wong GS. and Rustgi AK. Matricellular proteins: priming the tumour microenvironment for cancer development and metastasis. Br J Cancer 108: 755–761, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]