

Abstract
Purpose
Vogt-Koyanagi-Harada (VKH) syndrome is an autoimmune disease characterized by inaugural uveomeningitidis and hearing loss and at late stages a depigmentation in eyes and skin. Melanocytes are the cells common to the four affected tissues, namely eye, brain, inner ear, and skin. Melanocytes are therefore considered as the source of self-antigens. The melanocytic proteins tyrosinase-related protein-1 (TRP1), TRP2, tyrosinase, and gp100 have been proposed as the proteins targeted by autoreactive T cells from VKH patients bearing human leukocyte antigen (HLA)-DRB1*04:05, the HLA allele classically associated with VKH disease. The objective of this work was to determine the antigens recognized by a large number of potentially autoreactive CD4 T lymphocytes obtained from the cerebrospinal fluid of one VKH patient who did not express HLA-DRB1*04:05.
Methods
T cells were isolated from the cerebrospinal fluid of a newly diagnosed HLA-DRB1*14:01,*15:03;-DPB1*01:01,*04:02 patient in the acute phase of the VKH disease and cloned by limiting dilution. Each of the 107 T cell clones, of which 90% were CD4+, was tested for its ability to secrete cytokines upon contact with autologous antigen-presenting cells loaded with either of the melanocytic proteins TRP1, TRP2, tyrosinase, gp100, Melan-A and KU-MEL-1. The sensitivity of our recombinant bacteria-based approach was validated with a CD4 T cell clone with known antigen specificity. The ability of each of the 107 clones to secrete cytokines upon nonspecific stimulation was verified.
Results
None of the 107 T cell clones was able to secrete tumor necrosis factor-α, interferon-γ, interleukin (IL)-5, or IL-17 upon contact with autologous B cells loaded with any of the six common melanocytic proteins. Nine clones secreted high-level IL-17 upon stimulation with beads coated with antibodies.
Conclusions
The self-antigens that triggered the VKH disease in this patient probably derive from proteins other than the six melanocytic proteins mentioned above. Further study of antigens that are recognized by potential autoreactive T cells from VKH patients is likely to benefit from testing a broader set of melanocytic proteins.
Introduction
Vogt-Koyanagi-Harada (VKH) disease is characterized by an inaugural uveomeningitidis and hearing loss, followed at a later stage by depigmentation of eyes and skin [1]. An association between VKH disease and human leukocyte antigen (HLA)-DR4 was described for Asian, Hispanic, or Native American patients [2-4], and in particular the HLA-DRB1*04:05 subtype was associated with VKH in Asian and Brazilian patients [5-8]. The DRB1 molecules associated with VKH disease share the motif LLEQRRA67–73 located in the peptide-binding cleft [9-11]. The HLA molecules sharing this motif may thus present to T cells a common set of peptides and by this contribute to the recognition of the ocular self-peptides [9].
VKH pathogenesis remains incompletely understood, but autoimmune T cells have nonetheless been implicated. Activated CD4 T lymphocytes are present in the cerebrospinal fluid (CSF) of VKH patients [12], usually in higher numbers than their CD8 counterparts. Interferon (IFN)-γ was found elevated in the aqueous humor of VKH patients with uveitis [13]. A few differences between blood T cells from VKH patients and control donors have been reported: a decreased expression of CD18 and AKNA transcription factors in VKH patients [14], a higher expression of transcription factor T-bet [15], and less apoptosis of T cells from VKH patients after in vitro stimulation with phytohemagglutinin [16]. Upon ex vivo nonspecific stimulation, blood CD4 T lymphocytes of VKH patients secreted slightly more IFN-γ and interleukin (IL)-2 than did cells obtained from control individuals, whereas IL-4 secretion was similar in both groups [17]. IL-17 production by CD4 T cells was stimulated by IL-23, which was suggested to be responsible for the development of uveitis seen in patients with VKH disease, and IL-17-producing CD4 T cells of VKH patients were shown to produce proinflammatory cytokines, such as tumor necrosis factor (TNF)-α [18,19].
Melanocytes can be found in the four affected tissues: choroid, inner ear, leptomeninges, and skin [20-22], and accordingly the melanocytes were proposed as the source of self-antigens. Noteworthy, skin melanocytes are destroyed (vitiligo) by some cancer patients recovering from their melanoma [23]. A patient with a metastatic melanoma developed late manifestations of VKH disease after adoptive transfer of tumor-infiltrating lymphocytes containing a high proportion of CD8 T cells specific for a peptide from melanocytic protein Melan-A [24]. In rats, injection of melanocytic proteins tyrosinase-related protein-1 (TRP1) and TRP2 induced ocular and extra-ocular inflammation, similar to human VKH disease [25].
T lymphocytes are predominant among the leucocytes present in the CSF of VKH patients, but monocytes are also present. Some of them contain melanin granules [26,27], presumably following phagocytosis of damaged melanocytes from the meninges, suggesting that melanocyte-derived antigens can activate the CSF T lymphocytes. Viruses have been proposed to be responsible for the destruction of melanocytes, thus initiating VKH disease [28]. CD4 T lymphocytes isolated from the blood or the aqueous humor of DRB1*04:05 VKH patients could be stimulated in vitro with peptides derived from melanocytic proteins TRP1, TRP2, tyrosinase, and gp100. The expanded T lymphocytes produced IFN-γ upon further stimulation with the same peptides [29-32]. Antibodies against the melanocytic protein KU-MEL-1 were detected in the blood of DRB1*04:05 VKH patients [33].
We isolated a large number of CD4 T lymphocyte clones from the CSF of a naive VKH patient in the acute phase of the disease, before any treatment. This patient expressed DRB1*14:01 and DRB1*15:03 and not DRB1*04:05, the HLA allele classically associated with VKH disease. We decided to determine the antigen specificity of these clones and stimulated each of them with autologous cells having processed either of six proteins that are potential targets for autoreactive T cells in DRB1*04:05 VKH patients, namely TRP1, TRP2, tyrosinase, gp100, Melan-A, and KU-MEL-1.
Methods
Patient
Based on the VKH Committee’s revised diagnostic criteria [1], a diagnostic of early phase VKH disease was made. The research protocol was approved by the ethics committee “Comité de Protection des Personnes CPP Ile de France III.” Written informed consent was obtained from the patient before enrolment. All research adhered to the tenets of the Declaration of Helsinki. Ophthalmic examination was performed with a Stratus OCT3 (Humphrey Instruments, Carl Zeiss Division, San Leandro, CA).
T cell clones and Epstein-Barr virus-transformed B cells
T cell clones were derived by limiting dilution from CSF mononuclear cells of this VKH patient. Briefly, the CSF was collected by lumbar puncture and directly diluted in microwells. Cells in limiting dilution were stimulated with irradiated allogenic peripheral blood mononuclear cells in 96-well U-bottom plates (104 cells/well). Irradiated LG2 Epstein-Barr virus (EBV) cells were added as feeder cells (104 cells/well). T cell clones were cultured in Iscove’s modified Dulbecco medium (IMDM; Life Technologies, Carlsbad, CA) supplemented with 0.24 mM L-asparagine, 0.55 mM L-arginine, 1.5 mM L-glutamine (AAG), 100 µg/ml streptomycin, 100 U/ml penicillin, and 30 μg/ml gentamycin (culture medium; Sigma-Aldrich, St. Louis, MO) and with 10% human serum (HS). This culture medium was supplemented with human rIL-2 (50 UI/ml; Proleukin Chiron, Amsterdam, Netherlands), human rIL-4 produced in our laboratory (5 UI/ml), IL-7 (10 ng/ml; R&D Systems Europe, Abingdon, UK), anti-CD3 antibodies (1 ng/ml; Janssen-Cilag, Berchem, Belgium), and gentamycin (15 μg/ml). After several stimulations, T cell clones were frozen. An aliquot of each clone was labeled with anti-CD3-peridinin chlorophyll protein complex (PerCP) (1/30; BD Biosciences, San Jose, CA), anti-CD8-allophycocyanin (APC) (1/40; BD Biosciences), and anti-CD4-fluorescein isothiocyanate (1/30; BD Biosciences) and analyzed on a FACSCalibur flow cytometer (BD Biosciences). Cells of the MAGEA3-specific HLA-DPB1*0401-restricted CD4 T cell clone, R12C9, hereafter the anti-MAGEA3 T cell clone, were stimulated (5×105 cells/well) every 2 weeks with HLA-DP4 EBV Ii.MAGEA3 cells (2×105 cells/well) in 24-well plates. Irradiated LG2-EBV cells were added as feeder cells (1.5×106 cells/well). An EBV-B cell line expressing DP4 molecules that was derived from the VKH patient and the HLA-DP4 EBV Ii.MAGEA3 cells were maintained in IMDM containing 10% fetal calf serum (FCS; Thermo Scientific, Cramington, UK).
Construction of recombinant protein-producing bacteria
The template expression vectors containing the cDNA sequences encoding TRP1, TRP2, tyrosinase, gp100, Melan-A, KU-MEL-1, and MAGEA3 were available in our laboratories. The cDNAs were amplified by PCR using specific primers containing an EagI restriction site and an AscI restriction site (Table 1).
Table 1. EagI and AscI restriction sites.
| Gene name | Primers (5′-3′) |
|---|---|
| Melan-A |
F: CCCTACAAGCGGCCGAATGCCAAGAGAAG |
| R: CGCTGGCTCTTAGGCGCGCCAGGTGAATAAGG |
|
| gp100 |
F: GAAGAACACACGGCCGAAATGGATCTGGTGC |
| R: TGAGAGTACTCAGGCGCGCCGACCTGCTG |
|
| tyrosinase |
F: AGAGGAAGACGGCCGAATGCTCCTGGCTGTT |
| R: TGCCTAAGCCTTTTAGGCGCGCCTAAATGGCTCTG |
|
| TRP1 |
F: TCAAGCAGACGGCCGAATGAGTGCTCCTAAA |
| R: GGGCATTTGTTAGGCGCGCCGACCACAGACTG |
|
| TRP2 |
F: TATAAAGCCCGGCCGAATGAGCCCCCTTTGG |
| R: ATGAGCACCCTAGGCGCGCCGGCTTCTTCTGT |
|
| KU-MEL-1 |
F: ACAGTTCAATCGGCCGAATGGGGGACATTC |
| R: AGCCCTGACTTAGGCGCGCCCACTGGGTA |
|
| MAGEA3 |
F: ACCAGAGTCATCGGCCGCATGCCTCTTG |
| R: CTCAGACTCAGGCGCGCCCTCTTCCCCC |
The amplification resulted in cDNAs with an EagI site at the 5′ end and an AscI site at the 3′ end, thus permitting directional cloning. After amplification, cDNAs were purified by columm chromatography (High Pure PCR product) Purification Kit ref. 1732676 (Roche Diagnostics GmbH, Mannheim, Germany) and then visualized on an ethidium bromide-stained multipurpose agarose gel. Subsequently, protein cDNAs were digested with EagI (R0505-S; Biolabs, Ipswich, MA) and AscI (R0558-S; Biolabs), purified, and directionally ligated into pKE-1 vector digested with EagI and AscI. The pKE-1 vector was kindly provided by Dr. Davis (Department of Developmental Molecular Genetics, Ben Gourion University of the Negev, Beer Sheva, Israel) [34]. This pKE-1 vector contains an inducible tac promoter and in addition to an ampicillin resistance gene, a kanamycin resistance gene. cDNAs inserted in the correct reading frame confer kanamycin resistance to the host, whereas the vector alone or cDNA inserts containing stop codons in the frame translated by the bacteria should not. The ligation products were electroporated into Escherichia coli XL1-blue bacteria. The recombinant E. coli bacteria were plated on large agar plates containing ampicillin (50 g/ml) and kanamycin (7.5 g/ml), and after overnight culture at 37 °C, the bacteria were scraped in lysogeny broth (LB) medium and frozen in LB/20% glycerol at 80 °C. To analyze the cDNA inserts of the bacterial colonies, DNA sequencing was performed using the ABI PRISM Big DyeTM Terminators v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA) and the following primers: forward 5′-CTG GCA AGC CAC GTT TGG TG-3′ and reverse 5′-AGA CGT TTC CCG TTG AAT ATG G-3′, corresponding to sequences of pKE-1 vector [34]. Sequences were determined with an ABI PRISM 310 genetic analyzer (Applied Biosystems) and evaluated using the Sequencher Analysis SoftwareTM v3.7 (Applied Biosystems). We also used another bacteria producing MAGEA3 that was previously constructed in our laboratory [35].
Western blot analysis
One milliter of bacterial cultures, induced or noninduced, were pelleted and lysed in Laemmli buffer. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE) on a 4%–12% polyacrylamide gel. The fusion proteins were analyzed by western blot using an anti-glutathione-S-transferase (anti-GST) mouse monoclonal antibody (GST [1E5]; Santa Cruz Biotechnology, Santa Cruz, CA) and alkaline phosphatase-conjugated anti-mouse immunoglobulin G (IgG) antibody. Chromogenic substrate BM Purple (ROCHE, Mannheim, Germany) was used for detection. Recombinant GST protein (GST [1–218]; Santa Cruz Biotechnology) was used as reference for quantifying the fusion proteins. The identity of the GST-fusion proteins was analyzed by western blot with antibodies directed against TRP1 (ref. NBP1 69542; Novus Biologicals, Littleton, CO), TRP2 (ref. NBP1 86893; Novus Biologicals), tyrosinase (ref. MS800P1; Thermo Scientific, Rockford, IL), gp100 (ref. MA5–13232; Thermo Scientific), Melan-A (ref. MA1–26245; Thermo Scientific), KU-MEL-1 (ref. Orb 31521; Biorbyt, Cambridge, UK), and MAGEA3 (ref. NBP1 56404; Novus Biologicals).
The complement-opsonized bacteria assay
The complement-opsonized bacteria assay procedure was described previously [35]. Briefly, after thawing, the recombinant bacteria were amplified in LB medium containing ampicillin (50 μg/ml) and kanamycin (7.5 μg/ml), at 37 °C under agitation until an optical density (OD)600 of 0.5 was reached. Isopropyl β-D-thiogalactoside (IPTG; EMD Millipore, Billerica, MA; 1 mM) was added to induce protein expression, and after 4 h at 37 °C under agitation, bacteria from 1 ml of culture were opsonized by adding 150 μl of HS containing 17% (volume [v]/v) of complement (Sigma Chemical Co., St. Louis, MO) during 1 h under agitation at 37 °C. The opsonized bacteria were pooled by transferring 345 μl of each bacteria in two different pools, and centrifuged at 1250 ×g for 15 min. The supernatant was discarded and bacteria were resuspended in 1,350 μl (pool 1: TRP1, TRP2, and gp100) or 1,800 μl (pool 2: tyrosinase, Melan-A, KU-MEL-1, and MAGEA3) of IMDM/10% FCS with gentamycin (30 μg/ml). Subsequently, 1 ml of complement-opsonized bacteria from each bacterial pool was added into the EBV-B cells of the VKH patient and plated out in three 24-well plates (1×106 cells/well) in 2 ml/well IMDM/10% FCS with gentamycin (30 μg/ml). After overnight incubation to allow uptake and processing of the bacteria-encoded proteins, EBV-B cells were used as antigen-presenting cells for the CD4 T cells.
Screening of the T cell clone specificity
The different T cell clones were usually thawed the day before and cultured overnight in 96-well U-bottom plates with IMDM/10% HS with IL-2 (25 UI/ml). Autologous EBV-B cells previously loaded with pools of opsonized bacteria were added to the microwells (60 μl/well). CD4 T cells (5×103 cells/well) were added in 100 μl IMDM/10% HS with IL-2 (100 UI/ml) and gentamycin (30 μg/ml) to the EBV-B cells. T cell clones were also stimulated by microbeads coated with anti-CD3/CD28 antibodies (one bead/three T cells; ref. 111.31D; Invitrogen, Oslo, Norway). After overnight co-culture at 37 °C, supernatants were harvested and the amount of IL-5, IFN-γ, TNF-α, and IL-17 was measured using the Bioplex multiplex bead-based assay kit (ref.197 171304000; Biorad, Hercules, CA: TNF-α, ref. 171 B5026M; IL-5, ref. 171 B5005M; IFN-γ, ref. 171 B5019M; IL-17, ref. 171 B5014M) according to the manufacturer’s procedure. A multiplex bead-based assay permits the multiplexing of up to 100 different assays within a single sample. This technique involves distinctly colored bead sets created by the use of two fluorescent dyes at distinct ratios. These beads can be further conjugated with a specific antibody. The technology enables multiplex immunoassays in which one antibody to a specific cytokine is attached to a set of beads with the same color, and the second antibody specific for the same cytokine is attached to a fluorescent reporter dye label. The use of different colored beads enables the simultaneous multiplex detection of several cytokines in the same sample by flow cytometry. Alternatively, the amount of IFN-γ was measured by the enzyme-linked immunosorbent assay using reagents from Life Technologies (Camarillo, CA).
Results
Patient
A 37-year-old black man was referred to our center because of vision loss and headaches that had started 3 weeks earlier. He had no medical history. The best decimal-corrected visual acuity was 0.3 in both eyes. Ophthalmic examination revealed 2+ chamber cells and flare in both eyes and bilateral serous retinal detachments. Due to the presence of subretinal fluid, the central retinal thickness was increased to 409 μm in the right eye and 243 μm in the left eye. The early phase of the fluorescein angiogram showed bilateral multiple hyperfluorescent pinpoints, and the late phase showed staining in the areas of the serous retinal detachments. One year later, diffuse hypopigmentary fundus changes with a moderate sunset-glow appearance were observed in both eyes on dilated funduscopic examination (Figure 1). There was no extra-ophthalmologic manifestation at the initial phase of the disease except the CSF. The CSF contained 561 white blood cells/mm3 with 95% lymphocytes and less than one red blood cell per mm3, reflecting the lack of blood–brain barrier disruption. We did not detect Herpesviridae in the CSF by PCR, and the concentration of IFN-α was normal.
Figure 1.

Ophthalmologic features of the Vogt-Konayagi-Harada (VKH) patient. A, B: Fundus shows bilateral exsudative retinal detachments. C, D: The fluorescein angiogram reveals bilateral multifocal areas of pinpoint leakage at the level of retinal pigment epithelium and optic nerve staining. E, F: Fundus photograph obtained 1 year later shows a moderate sunset-glow fundus with diffuse depigmentation of retinal pigment epithelium in both eyes.
Isolation of T cell clones from the cerebrospinal fluid
Because in VKH disease the CSF is supposed to contain an enriched population of potentially autoreactive T cells [12,20-22,26,27], CSF cells were distributed at one cell per well in five 96-well microplates, stimulated every 2 weeks with anti-CD3 and anti-CD28 antibodies in the presence of IL-2, IL-7, and irradiated allogeneic cells. One hundred forty-five clones were obtained. A third of these were phenotyped for CD3, CD4, and CD8, and as expected [12], 90% of them were CD3+CD4+CD8–.
Construction of the tools to screen for antigen specificity of the T cell clones
The HLA class II type of the patient was DRB1*14:01,*15:03; DQB1*05:03,*06:02; DPB1*01:01,*04:02. Because the HLA haplotype of this patient as well as that of many Caucasian VKH patients differed from classical HLA-DRB1*04:05 VKH patients, we decided to test the specificity of the CD4 T cell clones using a sensitive approach previously developed in our laboratory [35]. Briefly, recombinant bacteria producing each of the proteins of interest were constructed. Bacteria were incubated with complement to increase their uptake by antigen-presenting cells, namely autologous EBV-B cells. Bacteria-loaded EBV-B cells were subsequently used as stimulating cells for each of the T cell clones. On the basis of published reports, our working hypothesis was that some of these CD4 T cell clones recognized antigens derived from melanocytic proteins, and we decided to test TRP1, TRP2, tyrosinase, gp100, Melan-A, and KU-MEL-1.
Construction of bacteria producing recombinant melanocytic proteins
DNA sequences encoding any of the melanocytic proteins TRP1, TRP2, tyrosinase, gp100, Melan-A, and KU-MEL-1 were introduced into vector pKE-1, which allows the production of recombinant proteins fused at the C-terminus of GST [34]. Expression of the recombinant gene is controlled by the lacI repressor and can be induced by IPTG. Bacteria transformed with the different plasmids were selected, IPTG was added to the cultures, and the production of the recombinant proteins was analyzed in bacterial extracts by western blot with an anti-GST-tag antibody.
Full-length fusion proteins were detected for each bacterial clone, together with a large number of smaller fusion products that can represent abortive translation or degradation products (Figure 2A). The identity of the melanocytic fusion protein was confirmed using antibodies specific to each of the melanocytic proteins (Figure 3). The amount of each full-length fusion protein was estimated with a purified recombinant GST protein as a reference (Figure 2B, Table 2). This ranged from 75 to 300 pmol of protein per ml of culture.
Figure 2.

Western blot analysis of the recombinant glutathione-S-transferase (GST)-fusion proteins produced in bacteria. Recombinant GST and GST-fusion proteins were detected with a monoclonal anti-GST antibody. MW, molecular weight ladder is expressed in kDa. A: Similar amounts of proteins extracted from the bacterial cultures before (ni) and after (i) induction with Isopropyl β-D-thiogalactoside (IPTG) were loaded. The expected molecular weight of the full-length fusion protein is indicated with *. A protein of 29.9 kDa is expected for empty vector pKE-1. It contains the 26.5 kDa GST sequence with some additional amino acids at the C-terminus. Dimers of the GST protein (~60 kDa) were also observed. B: The amount of the full-length GST fusion proteins (*) produced in the bacteria was estimated using purified recombinant GST protein as a reference. Bacterial extracts from 5 and 25 µl induced culture and known amounts of a recombinant GST protein (26.5 kDa) were loaded. The amount (ng) of the GST portion of the full-length fusion protein was visually evaluated in comparison with the purified GST protein and converted into the corresponding number of moles of GST and hence of the protein fused to GST.
Figure 3.

Identity of the recombinant glutathione-S-transferase (GST)-fusion proteins produced in bacteria. Each protein of interest produced in fusion with GST was detected with a relevant specific antibody. The western blot profiles show a variety of recombinant protein fragments as previously detected by the monoclonal anti-GST antibody, as shown in Figure 2. (-): bacterial extracts from induced culture of bacteria containing the empty vector pKE-1. (+): bacterial extracts from induced cultures of recombinant bacteria containing the sequence encoding GST-fusion proteins.
Table 2. Estimation of the amount of recombinant protein produced by bacteria.
| Fusion protein | μg of GST in the GST-fusion protein | pmoles of the GST-fusion protein |
|---|---|---|
| TRP1 |
4 |
150 |
| TRP2 |
2 |
75 |
| tyrosinase |
2 |
75 |
| gp100 |
8 |
300 |
| Melan-A |
2 |
75 |
| KU-MEL-1 |
8 |
300 |
| MAGEA3 | 15 | 570 |
Amounts are expressed per milliliter of induced culture. pmoles: picomoles
Bacteria expressing recombinant protein MAGEA3 as a source of antigen for anti-MAGEA3 CD4 T cells
To assess the sensitivity of our recombinant bacteria-based approach of antigen presentation, we had no CD4 T cell clone that was specific for an antigenic peptide derived from one of the six melanocytic proteins, and we were unable to receive a functional clone from other laboratories. We had, however, a CD4 T cell clone directed against a MAGEA3 peptide presented by HLA-DP4 molecules. We constructed a pKE-1 vector containing a MAGEA3 coding sequence. Expression of the fusion protein was confirmed by western blot as described above (Figure 2A and Figure 3). The amount of the recombinant full-length GST-MAGEA3 protein was estimated to be about 570 pmol per ml of culture (Figure 2B, Table 2).
MAGEA3-producing bacteria were collected, washed, opsonized with human complement, and incubated overnight with HLA-DP4 EBV-B cells to generate antigen-presenting cells harboring on their surface HLA-DP4 molecules presenting MAGEA3 peptides. The EBV-B cell line was generated from blood cells of the patient. As a first positive control, we loaded EBV-B cells with other complement-opsonized recombinant bacteria that produced MAGEA3 [35]. As a second positive control, we used HLA-DP4 EBV-B cells transduced with a retroviral construct encoding a truncated human invariant chain (Ii) fused with the MAGEA3 protein (retro-Ii.MAGE-3) [36]. In this chimeric protein, signals within the Ii should target the MAGE-3 protein to the class II antigen-processing compartments [36]. As a third positive control, we loaded the presenting cells with the MAGEA3 peptide that is recognized by the anti-MAGE-3 T cell clone [37].
The recognition of the MAGEA3-derived antigen by T cells was assessed by measuring the amounts of IFN-γ released in the supernatant of overnight co-cultures. The anti-MAGEA3 T cell clone secreted similar amounts of IFN-γ upon contact with presenting cells loaded with either our recombinant bacteria producing MAGEA3 or the MAGEA3 control bacteria or the peptide. They secreted more IFN-γ upon stimulation with cells transduced with Ii-MAGEA3 (Figure 4). To estimate the sensitivity of this approach, our MAGEA3-producing bacteria were diluted in irrelevant bacteria, opsonized with human complement, and incubated overnight with HLA-DP4-presenting cells. Recognition by the CD4 T cells, as assessed by IFN-γ secretion, was easily detectable when MAGEA3-producing bacteria were diluted 300 times into irrelevant bacteria and still detectable with a dilution factor of 1,000 (Figure 5).
Figure 4.

Recognition of Epstein-Barr virus-transformed B cells (EBV-B cells) loaded with MAGEA3-expressing bacteria. The antigen-presenting cells (30,000/well) were EBV-B cells obtained from the Vogt-Konayagi-Harada (VKH) patient that also expressed human leukocyte antigen (HLA)-DP4 molecules. These cells were loaded with complement-opsonized bacteria, which expressed either MAGEA3 or Melan-A, as a negative control. As positive controls, we used cells loaded with another recombinant complement-opsonized bacteria producing MAGEA3 that was previously constructed in our laboratory, cells transduced with a retroviral construct encoding a truncated human invariant chain (Ii) fused with the MAGEA3 protein (retro-Ii.MAGEA3), or cells pulsed with MAGE-3.DP4 peptide ACYEFLWGPRALEVTS (24). In total, 5,000 cells of the anti-MAGE-3 T cell clone were added to each well. The amount of interferon (IFN)-γ secreted in the supernatant of the co-culture was measured by enzyme-linked immunosorbent assay (ELISA). The results shown represent an average of triplicate co-cultures. Error bars represent SD.
Figure 5.

Dilution of MAGEA3-producing bacteria in nonrelevant bacteria. A: After induction of protein expression by Isopropyl β-D-thiogalactoside (IPTG), MAGEA3-producing bacteria were mixed with Melan-A-producing bacteria, opsonized with complement, and incubated overnight with human leukocyte antigen (HLA)-DP4 Epstein-Barr virus-transformed B cells (EBV-B cells), which were generated from blood cells of the Vogt-Konayagi-Harada (VKH) patient. The anti-MAGE-3 T cell clone (5,000 cells/well) was added to the antigen-presenting cells (30,000 cells/well). The amount of interferon (IFN)-γ secreted in the supernatant of overnight co-cultures was measured by enzyme-linked immunosorbent assay (ELISA). The results shown represent an average of triplicate co-cultures. B: EBV-B.retro-Ii.MAGEA3 cells and HLA-DP4 EBV-B cells loaded with Melan-A-expressing bacteria were used as positive and negative controls, respectively. Error bars represent SD.
When 1 ml of MAGEA3-producing bacteria was opsonized with complement before loading on antigen-presenting cells, this bacterial suspension contained about 570 pmol of MAGEA3 (Table 2). Considering that our detection approach with MAGEA3 allows for a dilution factor of at least 300, a bacterial suspension containing about 1.9 pmol (570 divided by 300) should result in recognition by the anti-MAGEA3 clone. Some of the recombinant bacteria produced about 75 pmol of melanocytic protein (Table 2). This should allow for a dilution factor of 39 (75 divided by 1.9). Considering that we decided to screen the recombinant bacteria by pools of three and four different bacteria (see below), the amount of melanocytic protein produced by the bacteria should not have been a limiting factor.
Analysis of the antigen specificity of the T cell clones isolated from the cerebrospinal fluid
Cells of each of the T cell clones isolated from the CSF of the patient were cultured with antigen-presenting cells, namely autologous EBV-B cells that were previously incubated for 20 h with either of two pools of opsonized bacteria. The first pool contained three different bacteria, producing TRP1, TRP2, or gp100. The second pool contained four different bacteria, producing tyrosinase, Melan-A, KU-MEL-1, or MAGEA3. As positive control, each T cell clone was stimulated with beads coated with anti-CD3 and anti-CD28 antibodies. As another positive control, EBV-B cells loaded with MAGEA3 bacteria were tested for recognition by anti-MAGEA3 T cells. The amounts of TNF-α, IFN-γ, and IL-5 released in the supernatant of overnight co-cultures were estimated by multiplex bead-based assays. A total of 145 T cell clones were tested in monoplicates; 38 of them, upon nonspecific simulation, secreted less than 100 pg/ml of the four cytokines and were thus not taken into consideration.
The remaining 107 clones were tested in monoplicate for their ability to specifically release cytokines upon stimulation with cells loaded with each of the two bacteria pools. The criterion of positivity was to release at least twice as much of any cytokine upon stimulation with one bacterial pool versus the other. Three clones scored positive for two cytokines (Figure 6), and eight clones scored positive for one cytokine (data not shown). These 11 clones were expanded and retested in triplicate with autologous EBV-B cells loaded separately with each recombinant bacteria. None of them scored positive in this second test (Figure 7 shows the data for the three clones shown in Figure 6). We tentatively concluded that the initial positivity resulted from technical errors or monoplicate analysis.
Figure 6.

First screening of CD4 clones for cytokine secretion upon stimulation with melanocytic proteins. Bacteria coated with complement were grouped in two pools, added to autologous Epstein-Barr virus-transformed B cells (EBV-B cells), and incubated overnight. CD4 T cells (5,000/well) were added in monoplicate to EBV-B cells (30,000/well). The amount of tumor necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-5, and IL-17 in the supernatant of overnight co-cultures was estimated with a multiplex bead-based assay. As the positive control, anti-MAGE-3 T cell clone R12C9 was used.
Figure 7.

Cytokine-secretion assay with selected clones stimulated with individual proteins. After induction of protein expression by Isopropyl β-D-thiogalactoside (IPTG) and opsonization with complement, the bacteria were added to autologous Epstein-Barr virus-transformed B cells (EBV-B cells) and incubated overnight. CD4 T cells (5,000/well) were added to EBV-B cells (30,000/well). The amount of tumor necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-5, and IL-17 in the supernatant of overnight co-cultures was estimated with a multicytokine bioplex kit. As the positive control, anti-MAGE-3 T cell clone R12C9 was used. The screening of the clones was performed in triplicates. Error bars represent SD.
IL-17 secretion was also estimated by multiplex bead-based assays, and nine of the 107 clones produced significant amounts of IL-17 in response to the stimulation with beads coated with anti-CD3 and anti-CD28 antibodies (Figure 8). This is the first report of IL-17-producing CD4 T cells in the CSF of a VKH patient at the early phase of the disease.
Figure 8.
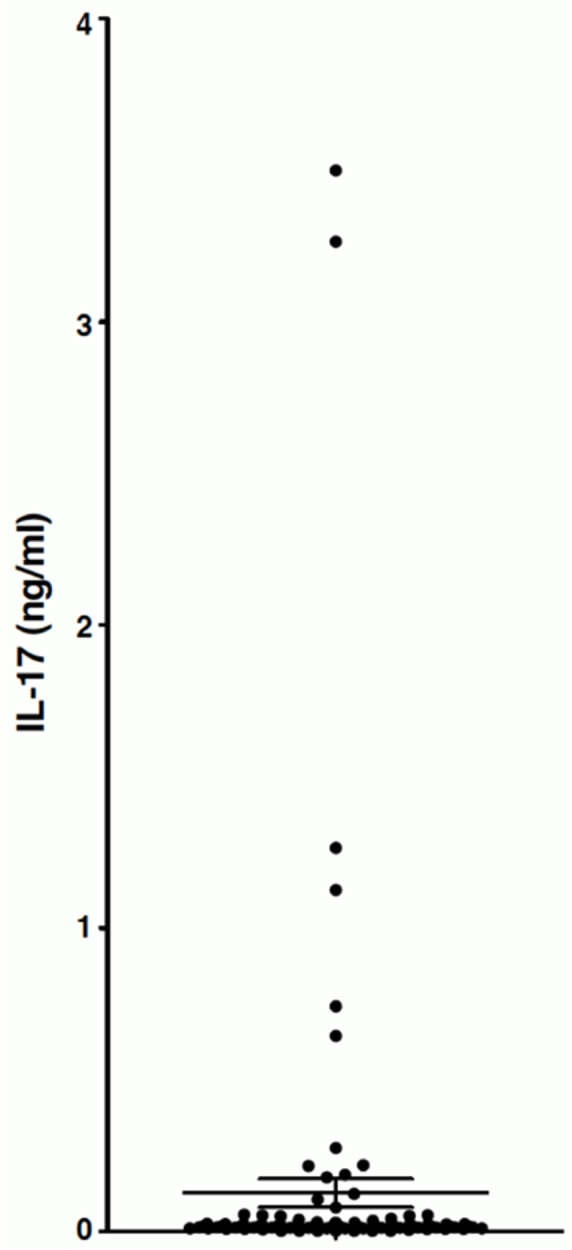
Some T cell clones obtained from the cerebrospinal fluid of the Vogt-Konayagi-Harada (VKH) patient secrete interleukin (IL)-17. T cell clones (n≥107) were stimulated with beads coated with anti-CD3 and anti-CD28 antibodies. The amount of IL-17 in the supernatant of overnight co-cultures was measured by multiplex bead-based assay. The horizontal lines represent the mean ± SEM. None of the clones shown in Figure 6 and Figure 7 secreted significant amounts of IL-17.
Discussion
It is surprising that the CD4 T cells isolated from the CSF of our patient during the acute phase of his VKH disease did not appear to recognize the melanocytic proteins TRP1, TRP2, tyrosinase, gp100, Melan-A, or KU-MEL-1. This could be explained if the CD4 T cells recognized some viral agents like cytomegalovirus (CMV), which have been presumed to be a trigger mechanism for the VKH disease [32]. Indeed, prodromal symptoms, such as a cold, usually precede the acute ocular inflammation. However, the failure to detect CMV or other Herpesviridae by PCR in the CSF of our patient does not support the hypothesis of such a viral infection.
It could also be explained if the frequency of the CSF-infiltrating CD4 T cells that recognize antigens derived from the six melanocytic proteins was lower than 1%. However, a higher frequency of autoimmune cells is expected in the case of an autoimmune disease. Indeed, in a metastatic melanoma patient who developed uveitis and vitiligo—manifestations seen at the late stage of VKH—after an adoptive transfer of in vitro-expanded tumor-infiltrating lymphocytes, 4% of the CD8 T cells detected in the aqueous humor and 2% of those detected in the blood were directed against a Melan-A peptide presented by HLA-A2 [24]. If TRP1, TRP2, tyrosinase, Melan-A, gp100, or KU-MEL-1 were to be the source of self-antigens recognized by the autoimmune CD4 T cells of our patient, we would expect that a few would recognize an antigenic peptide derived from at least one of the six different whole melanocytic proteins complexed with at least one of the six HLA class II molecules expressed by the autologous B cells.
As we failed to find T cells that recognize melanocytic antigens, we cannot exclude that we had false-negative results. We have nevertheless controlled different steps of our experimental approach. The bacteria were shown to be able to produce enough of the melanocytic protein, and the patient antigen-presenting cells were shown to be able to present a control protein, MAGEA3, to anti-MAGEA3 CD4 T cells. We showed that each T cell clone from the VKH patient was able to secrete cytokines after nonspecific stimulation. We were unfortunately unable to find a T cell clone, in particular one from an HLA-DRB1*04:05 VKH patient, specific for an antigen derived from one of the six melanocytic proteins to be able to further control our experimental procedure. However, we never experienced false-negative results with a specific CD4 T cell stimulated with protein-loaded antigen-presenting B cells. We were unable to control the recognition of autologous melanocytes by the T cell clones. Indeed, considering that a skin punch biopsy of 8 mm diameter provides less than 30,000 melanocytes and that a recognition assay with one T cell clone in triplicate necessitates 30,000 melanocytes, this important control is very difficult to perform. We did not control the recognition by the T cell clones of autologous antigen-presenting cells loaded with a lysate of allogeneic melanoma cells. We have experienced with CD4 T cell clones with known specificities that this approach is not sensitive enough.
The VKH patient analyzed in this study was typed HLA-DRB1*14:01,*15:03, an HLA haplotype that differs from the HLA-DRB1*04:05 haplotype classically associated with VKH disease. Considering that the different HLA isoforms are highly variable close to the peptide-binding cleft, the set of peptides that binds to DRB1*04:05 molecules is different from the set of peptides able to bind to DRB1*14:01 or DRB1*15:03 molecules. If we had tested in the current study recognition of peptides with the T cell clones, the lack of antigen recognition by the T cells could have been related to the genotype of the patient. However, we used six whole melanocytic proteins that were processed by autologous B cells. In analogy with the tumor immunology field, we surmised that the proteins would generate peptides able to bind to some of the molecules of our VKH patient. Indeed, a set of peptides derived from either TRP1, tyrosinase, gp100, or Melan-A have been found to stimulate anti-melanoma CD4 T cells isolated from DR4 patients. When the antigen specificity of anti-melanoma CD4 T cells was analyzed in DR15 melanoma patients, another set of peptides was identified as the target antigens, the peptides being derived from the same four above-mentioned proteins [38]. Hence, our surprise that none of the 107 T cell clones recognized even one peptide derived from one of the six melanocytic proteins.
Our negative results are in agreement with preliminary results obtained many years ago by one of the co-authors. T cells were cultured from the CSF of several VKH patients, including DRB1*04:05 individuals. The T cells were tested for their reactivity and failed to recognize antigens derived from tyrosinase, gp100, or Melan-A, despite the fact that some T cells responded to presenting cells loaded with allogeneic melanocyte lysates (Kawakami, unpublished data).
Our results seem in apparent contradiction with studies of T cells isolated from other VKH patients. In 2000, Yamaki et al. cultured for 3 days in monoplicate 200,000 blood mononuclear cells in the presence of ±30 µM of 30 amino acid-long peptides corresponding to parts of the sequences of TRP1, TRP2, and tyrosinase [31]. Proliferation was measured on day 3 by tritiated thymidine incorporation. Positive microwells were observed only with blood cells of the ten HLA-DRB1*04:05 VKH patients but not with blood cells of the three HLA-matched healthy controls [31]. One year later, the same group published similar data, but in this new set of experiments, responding T cells were cloned and tested in a proliferation assay after peptide stimulation [30]. These results suggested but did not prove that these VKH patients had melanocyte-specific T cells in their blood. Indeed, synthetic peptides are known to be contaminated with impurities, i.e., bulky chemical-protecting groups that can stimulate T cells. Thus, testing the specificity of T cells expanded with a synthetic peptide by stimulating them again with the same peptide can yield false positives. It would have been more convincing to show that T cell clones were able to specifically recognize autologous cells expressing or loaded with the relevant proteins.
In 2005, another research group reported data obtained with 34 VKH patients carrying HLA DRB1*04:05 and/or DRB1*15 alleles [29]. In this paper also, the reactivity of some microcultures to peptide stimulation was not confirmed by testing autologous presenting cells loaded with the whole relevant protein [29]. T cell clones were also established from cells infiltrating the aqueous humor of DRB1*04:05 VKH patients and found to secrete IFN- γ and chemokine (c-c motif) ligand 5 (CCL5) when tyrosinase or gp100-derived peptides were added at concentrations higher than 100 µM [32]. Noteworthy, concentrations lower than 1 μM are, in our experience, more than sufficient to induce activation of specific T cells. Controls with whole tyrosinase or gp100 proteins loaded on autologous cells were not performed. In this paper, the authors have shown that one of the peptide-specific T cell clones was able to secrete cytokines upon contact with allogeneic melanoma cells. Surprisingly, the cytokine secretion was seven times higher with melanoma cells than with cells loaded with 200 µM of peptides. In our experience, stimulation with peptide-loaded cells is stronger than stimulation with tumor cells expressing the corresponding protein. In the studies described above, the frequencies of autoreactive T cells found in the peripheral blood or aqueous humor of VKH patients were not indicated [30,32]. This is important information because in any blood donor, one can expect a frequency of CD4 T cells against most HLA–peptide combinations at about 3×10−7 of the blood CD4 T cells (based on our own unpublished observations with CD8 T cells and Chaux et al. [39]).
Thus, in apparent contradiction with data obtained with T cells from DRB1*04:05 VKH patients, none of the T cell clones of our DRB1*14:01,*15:03VKH patient was specific for a peptide derived from melanocytic proteins TRP1, TRP2, tyrosinase, gp100, Melan-A, and KU-MEL-1. Our tentative conclusion based on the analysis of this one patient is that the CD4 T lymphocytes isolated from the CSF during the early phase of the VKH disease recognize peptides derived from melanocytic proteins other than the six mentioned above.
Further study of antigen specificity of T cells from the cerebrospinal fluid of VKH patients might benefit from testing a broader set of antigen-derived melanocytic proteins. It would also be interesting to compare these results obtained at the onset of the VKH disease with results obtained with T cell clones obtained from patients with a VKH disease syndrome that is associated with melanoma or started after immune therapy for melanoma [24,40].
Acknowledgments
We thank Nathalie Krack for her administrative and secretarial assistance. This study was financially supported by grant #6000056 FF 09/05/07 from the Institut Servier (France), fellowship “Marcel Simon Year 2007” from the French Society of Internal Medicine (SNFMI), and grant #3.4514.12 from the Fonds de la Recherche Scientifique Médicale (Belgium). G. Wieërs was supported by fellowship #1.1.109.10 from the Fonds National de la Recherche Scientifique (Belgium). None of these funding sources had any role in study design; in the collection, analysis and interpretetion of data; in the writing of the report; and in the decision to submit the article for publication. The authors have no financial or non-financial competing interests.
References
- 1.Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001;131:647–52. doi: 10.1016/s0002-9394(01)00925-4. [DOI] [PubMed] [Google Scholar]
- 2.Shindo Y, Ohno S, Yamamoto T, Nakamura S, Inoko H. Complete association of the HLA-DRB1*04 and -DQB1*04 alleles with Vogt-Koyanagi-Harada's disease. Hum Immunol. 1994;39:169–76. doi: 10.1016/0198-8859(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 3.Weisz JM, Holland GN, Roer LN, Park MS, Yuge AJ, Moorthy RS, Forster DJ, Rao NA, Terasaki PI. Association between Vogt-Koyanagi-Harada syndrome and HLA-DR1 and -DR4 in Hispanic patients living in Southern California. Ophthalmology. 1995;102:1012–5. doi: 10.1016/s0161-6420(95)30920-7. [DOI] [PubMed] [Google Scholar]
- 4.Zhao M, Jiang Y, Abrahams IW. Association of HLA antigens with Vogt-Koyanagi-Harada syndrome in a Han Chinese population. Arch Ophthalmol. 1991;109:368–70. doi: 10.1001/archopht.1991.01080030070041. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg AC, Yamamoto JH, Chiarella JM, Marin ML, Sibinelli M, Neufeld R, Hirata CE, Olivalves E, Kalil J. HLA-DRB1*0405 is the predominant allele in Brazilian patients with Vogt-Koyanagi-Harada disease. Hum Immunol. 1998;59:183–8. doi: 10.1016/s0198-8859(97)00265-6. [DOI] [PubMed] [Google Scholar]
- 6.Kim MH, Seong MC, Kwak NH, Yoo JS, Huh W, Kim TG, Han H. Association of HLA with Vogt-Koyanagi-Harada syndrome in Koreans. Am J Ophthalmol. 2000;129:173–7. doi: 10.1016/s0002-9394(99)00434-1. [DOI] [PubMed] [Google Scholar]
- 7.Shindo Y, Inoko H, Yamamoto T, Ohno S. HLA-DRB1 typing of Vogt-Koyanagi-Harada's disease by PCR-RFLP and the strong association with DRB1*0405 and DRB1*0410. Br J Ophthalmol. 1994;78:223–6. doi: 10.1136/bjo.78.3.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greco A, Fusconi M, Gallo A, Turchetta R, Marinelli C, Macri GF, De Virgilio A, de Vincentiis M. Vogt-Koyanagi-Harada syndrome. Autoimmun Rev. 2013;12:1033–8. doi: 10.1016/j.autrev.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Alaez C, del Pilar Mora M, Arellanes L, Cano S, Perez-Luque E, Vazquez MN, Olivo A, Burguete A, Hernandez A, Pedroza M, Gorodezky C. Strong association of HLA class II sequences in Mexicans with Vogt-Koyanagi-Harada's disease. Hum Immunol. 1999;60:875–82. doi: 10.1016/s0198-8859(99)00024-5. [DOI] [PubMed] [Google Scholar]
- 10.Levinson RD, See RF, Rajalingam R, Reed EF, Park MS, Rao NA, Holland GN. HLA-DRB1 and -DQB1 alleles in mestizo patients with Vogt-Koyanagi-Harada's disease in Southern California. Hum Immunol. 2004;65:1477–82. doi: 10.1016/j.humimm.2004.07.236. [DOI] [PubMed] [Google Scholar]
- 11.Tiercy JM, Rathinam SR, Gex-Fabry M, Baglivo E. A shared HLA-DRB1 epitope in the DR beta first domain is associated with Vogt-Koyanagi-Harada syndrome in Indian patients. Mol Vis. 2010;16:353–8. [PMC free article] [PubMed] [Google Scholar]
- 12.Norose K, Yano A, Aosai F, Segawa K. Immunologic analysis of cerebrospinal fluid lymphocytes in Vogt-Koyanagi-Harada disease. Invest Ophthalmol Vis Sci. 1990;31:1210–6. [PubMed] [Google Scholar]
- 13.Takase H, Futagami Y, Yoshida T, Kamoi K, Sugita S, Imai Y, Mochizuki M. Cytokine profile in aqueous humor and sera of patients with infectious or noninfectious uveitis. Invest Ophthalmol Vis Sci. 2006;47:1557–61. doi: 10.1167/iovs.05-0836. [DOI] [PubMed] [Google Scholar]
- 14.Mao L, Yang P, Hou S, Li F, Kijlstra A. Label-free proteomics reveals decreased expression of CD18 and AKNA in peripheral CD4+ T cells from patients with Vogt-Koyanagi-Harada syndrome. PLoS ONE. 2011;6:e14616. doi: 10.1371/journal.pone.0014616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li B, Yang P, Zhou H, Huang X, Jin H, Chu L, Gao Y, Zhu L, Kijlstra A. Upregulation of T-bet expression in peripheral blood mononuclear cells during Vogt-Koyanagi-Harada disease. Br J Ophthalmol. 2005;89:1410–2. doi: 10.1136/bjo.2005.074062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang P, Chen L, Zhou H, Zhong H, Wang H, Huang X, Kijlstra A. Resistance of lymphocytes to Fas-mediated apoptosis in Behcet's disease and Vogt-Koyangi-Harada syndrome. Ocul Immunol Inflamm. 2002;10:47–52. doi: 10.1076/ocii.10.1.47.10331. [DOI] [PubMed] [Google Scholar]
- 17.Imai Y, Sugita M, Nakamura S, Toriyama S, Ohno S. Cytokine production and helper T cell subsets in Vogt-Koyanagi-Harada's disease. Curr Eye Res. 2001;22:312–8. doi: 10.1076/ceyr.22.4.312.5510. [DOI] [PubMed] [Google Scholar]
- 18.Chi W, Yang P, Li B, Wu C, Jin H, Zhu X, Chen L, Zhou H, Huang X, Kijlstra A. IL-23 promotes CD4+ T cells to produce IL-17 in Vogt-Koyanagi-Harada disease. J Allergy Clin Immunol. 2007;119:1218–24. doi: 10.1016/j.jaci.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Li F, Yang P, Liu X, Wang C, Hou S, Kijlstra A. Upregulation of interleukin 21 and promotion of interleukin 17 production in chronic or recurrent Vogt-Koyanagi-Harada disease. Arch Ophthalmol. 2010;128:1449–54. doi: 10.1001/archophthalmol.2010.265. [DOI] [PubMed] [Google Scholar]
- 20.Goldgeier MH, Klein LE, Klein-Angerer S, Moellmann G, Nordlund JJ. The distribution of melanocytes in the leptomeninges of the human brain. J Invest Dermatol. 1984;82:235–8. doi: 10.1111/1523-1747.ep12260111. [DOI] [PubMed] [Google Scholar]
- 21.Lohman BD, Gustafson CA, McKinney AM, Sarikaya B, Silbert SC. MR imaging of Vogt-Koyanagi-Harada syndrome with leptomeningeal enhancement. AJNR Am J Neuroradiol. 2011;32:E169–71. doi: 10.3174/ajnr.A2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugiura S. Vogt-Koyanagi-Harada disease. Jpn J Ophthalmol. 1978;22:9–35. [Google Scholar]
- 23.Houghton AN. Cancer antigens: immune recognition of self and altered self. J Exp Med. 1994;180:1–4. doi: 10.1084/jem.180.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeh S, Karne NK, Kerkar SP, Heller CK, Palmer DC, Johnson LA, Li Z, Bishop RJ, Wong WT, Sherry RM, Yang JC, Dudley ME, Restifo NP, Rosenberg SA, Nussenblatt RB. Ocular and systemic autoimmunity after successful tumor-infiltrating lymphocyte immunotherapy for recurrent, metastatic melanoma. Ophthalmology. 2009;116:981–9. doi: 10.1016/j.ophtha.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamaki K, Kondo I, Nakamura H, Miyano M, Konno S, Sakuragi S. Ocular and extraocular inflammation induced by immunization of tyrosinase related protein 1 and 2 in Lewis rats. Exp Eye Res. 2000;71:361–9. doi: 10.1006/exer.2000.0893. [DOI] [PubMed] [Google Scholar]
- 26.Kim LA, Khurana RN, Parikh JG, Rao NA. Melanin-laden macrophages in the CSF to diagnose Vogt-Koyanagi-Harada simulating ocular syphilis. Ocul Immunol Inflamm. 2008;16:59–61. doi: 10.1080/09273940801899806. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura S, Nakazawa M, Yoshioka M, Nagano I, Nakamura H, Onodera J, Tamai M. Melanin-laden macrophages in cerebrospinal fluid in Vogt-Koyanagi-Harada syndrome. Arch Ophthalmol. 1996;114:1184–8. doi: 10.1001/archopht.1996.01100140384003. [DOI] [PubMed] [Google Scholar]
- 28.Yu N, Zhang S, Sun T, Kang K, Guan M, Xiang L. Double-stranded RNA induces melanocyte death via activation of Toll-like receptor 3. Exp Dermatol. 2011;20:134–9. doi: 10.1111/j.1600-0625.2010.01208.x. [DOI] [PubMed] [Google Scholar]
- 29.Damico FM, Cunha-Neto E, Goldberg AC, Iwai LK, Marin ML, Hammer J, Kalil J, Yamamoto JH. T-cell recognition and cytokine profile induced by melanocyte epitopes in patients with HLA-DRB1*0405-positive and -negative Vogt-Koyanagi-Harada uveitis. Invest Ophthalmol Vis Sci. 2005;46:2465–71. doi: 10.1167/iovs.04-1273. [DOI] [PubMed] [Google Scholar]
- 30.Gocho K, Kondo I, Yamaki K. Identification of autoreactive T cells in Vogt-Koyanagi-Harada disease. Invest Ophthalmol Vis Sci. 2001;42:2004–9. [PubMed] [Google Scholar]
- 31.Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Tyrosinase family proteins are antigens specific to Vogt-Koyanagi-Harada disease. J Immunol. 2000;165:7323–9. doi: 10.4049/jimmunol.165.12.7323. [DOI] [PubMed] [Google Scholar]
- 32.Sugita S, Takase H, Taguchi C, Imai Y, Kamoi K, Kawaguchi T, Sugamoto Y, Futagami Y, Itoh K, Mochizuki M. Ocular infiltrating CD4+ T cells from patients with Vogt-Koyanagi-Harada disease recognize human melanocyte antigens. Invest Ophthalmol Vis Sci. 2006;47:2547–54. doi: 10.1167/iovs.05-1547. [DOI] [PubMed] [Google Scholar]
- 33.Otani S, Sakurai T, Yamamoto K, Fujita T, Matsuzaki Y, Goto Y, Ando Y, Suzuki S, Usui M, Takeuchi M, Kawakami Y. Frequent immune response to a melanocyte specific protein KU-MEL-1 in patients with Vogt-Koyanagi-Harada disease. Br J Ophthalmol. 2006;90:773–7. doi: 10.1136/bjo.2005.086520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis CA, Benzer S. Generation of cDNA expression libraries enriched for in-frame sequences. Proc Natl Acad Sci USA. 1997;94:2128–32. doi: 10.1073/pnas.94.6.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van de Corput L, Chaux P, van der Meijden ED, De Plaen E, Falkenburg JHF, van der Bruggen P. A novel approach to identify antigens recognized by CD4 T cells using complement-opsonized bacteria expressing a cDNA library. Leukemia. 2005;19:279–85. doi: 10.1038/sj.leu.2403583. [DOI] [PubMed] [Google Scholar]
- 36.Sanderson S, Frauwirth K, Shastri N. Expression of endogenous peptide-major histocompatibility complex class II complexes derived from invariant chain-antigen fusion proteins. Proc Natl Acad Sci USA. 1995;92:7217–21. doi: 10.1073/pnas.92.16.7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaux P, Vantomme V, Stroobant V, Thielemans K, Corthals J, Luiten R, Eggermont AM, Boon T, van der Bruggen P. Identification of MAGE-3 epitopes presented by HLA-DR molecules to CD4+ T lymphocytes. J Exp Med. 1999;189:767–78. doi: 10.1084/jem.189.5.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Bruggen P, Stroobant V, Vigneron N, Van den Eynde B. Peptide database: T cell-defined tumor antigens. Cancer Immun 2013; http://www.cancerimmunity.org/peptide/ [PMC free article] [PubMed]
- 39.Chaux P, Vantomme V, Coulie P, Boon T, van der Bruggen P. Estimation of the frequencies of anti-MAGE-3 cytolytic T lymphocyte precursors in blood from individuals without cancer. Int J Cancer. 1998;77:538–42. doi: 10.1002/(sici)1097-0215(19980812)77:4<538::aid-ijc11>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 40.Aisenbrey S, Luke C, Ayertey HD, Grisanti S, Perniok A, Brunner R. Vogt-Koyanagi-Harada syndrome associated with cutaneous malignant melanoma: an 11-year follow-up. Graefes Arch Clin Exp Ophthalmol. 2003;241:996–9. doi: 10.1007/s00417-003-0787-5. [DOI] [PubMed] [Google Scholar]