

Abstract
The palladium-catalyzed decarboxylative allylic alkylation of diastereomeric β-ketoesters derived from 4-tert-butylcyclohexanone is described. These experiments were performed to elucidate our understanding of stereoablative enantioconvergent catalysis. A detailed analysis of the product distribution, including stereochemical outcome of the products, is included. These studies also reveal an interesting example of selectivity that is governed by competing modes of substrate and catalyst control.
Keywords: Decarboxylative, Allylic, Alkylation, Ketoesters, Palladium
1. Introduction
The concept of stereoablative enantioconvergent catalysis is illustrated by the use of α-quaternary β-ketoesters in the palladium-catalyzed decarboxylative allylic alkylation and is a subject of interest to our laboratories.1 Typical stereomutative enantioconvergent processes, such as dynamic kinetic resolution, require a pre-equilibration epimerization of starting material A followed by enantioselective conversion to product B (Pathway I, Scheme 1). Quaternary stereocenters are not typically epimerizable and, thus, in the case of our previous studies1 we believe another pathway is operative, wherein both enantiomers of the starting material A convert irreversibly to prochiral intermediate C. This prochiral intermediate C can then preferentially form one enantiomer of product B under the influence of a chiral catalyst (Pathway II, Scheme 1). This alternate pathway has been termed stereoablative enantioconvergent catalysis. The lability of the stereogenic center is illustrated with quaternary β-ketoesters, where enolate formation destroys the stereochemical information at the α-position.
Scheme 1.

Stereomutative versus stereoablative enantioconvergent catalysis
To provide evidence for stereoablative enantioconvergent catalysis, we envisioned using diastereomeric β-ketoesters 1 and 2 as substrates for the palladium-catalyzed decarboxylative allylic alkylation (Scheme 2). The stereoablative hypothesis is supported if both β-ketoesters afford similar diastereomeric product ratios as the stereochemistry at α-position of the β-ketoester is not expected to influence the outcome of the reaction.
Scheme 2.
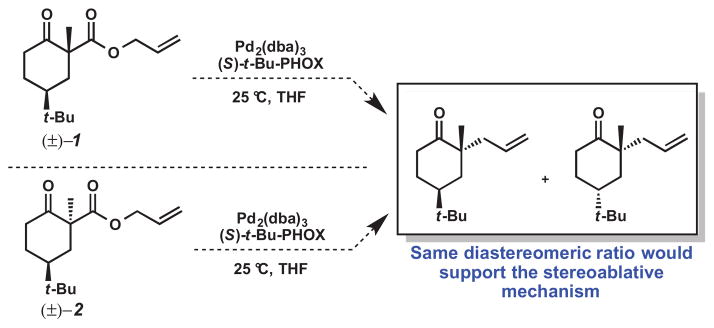
Proposed experiment to test stereoablation hypothesis
2. Results and discussion
2.1 Experimental results
Diastereomeric β-ketoesters 1 and 2 were prepared by straightforward methods previously employed in our laboratory.2 With these β-ketoesters 1 and 2 in hand, we treated each β-ketoester with Pd2(dba)3 and (S)-t-Bu-PHOX. We observed similar product yields, diastereomeric ratios and enantioselectivities for the asymmetric decarboxylative allylic alkylation of both 1 and 2 (Scheme 3). Thus, our results support the formation of an enolate wherein the stereochemistry at the α-position of the β-ketoester starting material does not influence the outcome of the reaction. The relative and absolute stereochemistry of the products 3 and 4 were determined by X-ray diffraction of their corresponding crystalline semicarbazone derivatives obtained from the asymmetric variant of the decarboxylative allylic alkylation (Scheme 4).2
Scheme 3.

Asymmetric palladium-catalyzed decarboxylative allylation of β-ketoesters
Scheme 4.

Determination of relative and absolute stereochemistry
However, two other interesting observations were made during these experiments. Minor product 4 had significantly greater enantiomeric excess than that of major product 3 (97% ee versus 39% ee). Furthermore, decarboxylative allylic alkylation of 2 was 2 times faster than the decarboxylative allylic alkylation of 1, which is counter-intuituve because stereoelectronic arguments would predict that 1 should react at a greater rate as orbital overlap between the carbonyl carbon and the α-carbon is maximized when the carboxyl is in the axial position.
To confirm the observed difference in their relative rates of reaction, 1 and 2 were treated with an achiral catalyst (Scheme 5). Decarboxylative allylic alkylation of 1 and 2 using PPh3 as ligand gave the same major and minor products as those observed in the enantioselective case. Furthermore, the difference in relative rate of reaction was more dramatic in this case; the decarboxylative allylic alkylation of 2 was 5.3 times faster than that observed for 1.3,4
Scheme 5.

Racemic palladium-catalyzed decarboxylative allylic alkylation of β-ketoester
2.2 Rationalization for lack of stereoelectronic control in decarboxylation of diastereomeric β-ketoacids
The surprising observation that β-ketoester 2, which has the ester group in an equatorial position, is more reactive for allylic decarboxylation than β-ketoester 1, which has the ester group in an axial position, contradicts stereoelectronic control arguments. However, this contradiction has also been observed in the decarboxylation of diastereomeric β-ketoacids. 5
Based on stereoelectronic arguments, β-ketoacid 7 would be predicted to be more reactive because an axially positioned carboxyl group allows for continuous overlap of the incipient p-orbital with the p-orbital on the carbonyl carbon (Figure 1). However, Pollack has reported that β-ketoacid 8 decarboxylates 3-fold faster than 7 in acidic media.5 Under basic conditions, the relative rate of reactions is more striking: β-ketoacid 8 decarboxylates 15- to 20-fold faster than 7.
Figure 1.

Kinetic experiments on rates decarboxylation of β-ketoester
Under acidic conditions, Pollack has proposed that the transition state for decarboxylation is a six-membered intermediate, in which the O–H bond rests in the same plane as the original C(3)–C(2)=O bond (Figure 2).5 The C(1)–C(3) bond that is broken sits perpendicular to this plane, which allows for the continuous overlap of the incipient p-orbital with the π* of the carbonyl bond. The proposed transition states for β-ketoacids 7 and 8 are shown in Figure 2, where the cyclohexene is shown in a half chair conformation with the tert-butyl group in the equatorial position and the methyl group in the plane of the carbon–carbon double bond. If the energies of the transition states are similar, then the relative rate of decarboxylation is determined by the relative stabilities of β-ketoacids 7 and 8. Based on A values derived from cyclohexanes (Me = 1.70 kcal/mol, COOH = 1.35 kcal/mol), β-ketoacid 8 is higher in energy; therefore, it should be more reactive. Rationalization of the relative rate of decarboxylation of β-ketoacids 7 and 8 under basic conditions is based on a similar argument. Decarboxylation of the carboxylate anions of β-ketoacids has not been investigated thoroughly, but studies on the decarboxylation of benzoylacetic acids have suggested that the transition state is late on the reaction coordinate; thus, the transition state resembles the enolate product (Figure 3).6 We can apply this observation to the decarboxylation of carboxylates 7a and 8a. If steric interactions with the axial hydrogens are ignored in the transition states shown in Figure 3, these two transition states should have similar energies, and the relative rates of reaction reflect the differences in energy of carboxylates 7a and 8a.
Figure 2.
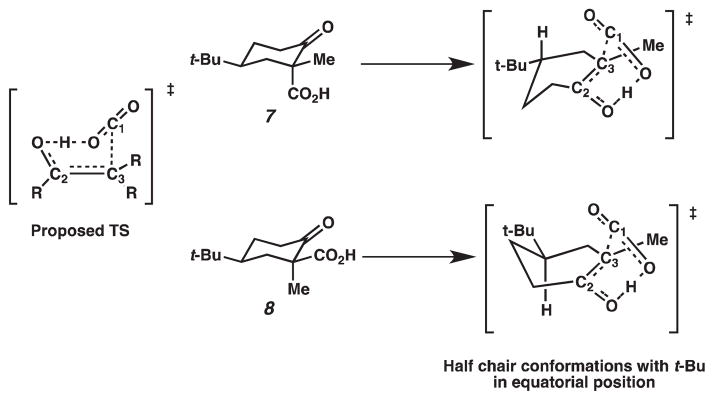
Proposed transition states under acidic conditions
Figure 3.
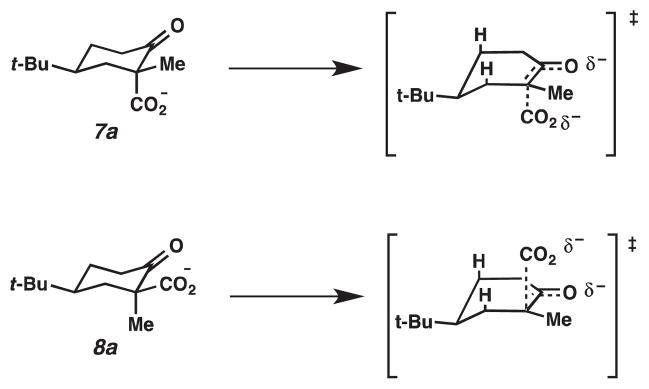
Proposed transition states under basic conditions
To predict the relative energy of 7a and 8a, the dipole-dipole repulsions between the carboxylate and the carbonyl oxygen should be considered (Figure 4). For 8a, the dipole-dipole repulsion is more significant; hence, 8a is less stable and will decarboxylate more readily. Evidence that this dipole-dipole interaction is significant can be found in the pKa values of 7 and 8. Acid 8 has a higher pKa than 7 (5.79 versus 5.29 in 70% MeOH in water at 0 °C).6
Figure 4.
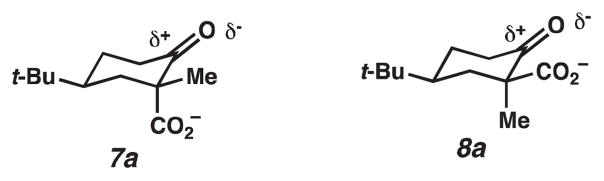
Dipole-dipole repulsions
2.3 Rationalization for faster relative rate of reaction for β-ketoesters
A possible explanation for the increased reactivity of β-ketoester 2 relative to its diastereomer assumes that the palladium π-allyl complex is formed prior to decarboxylation, and that both of these events occur in a stepwise fashion. It is proposed that formation of a carboxylate intermediate occurs and that unfavorable dipole-dipole repulsion, such as those shown in Figure 4, for the carboxylate derived from 2 would make it the more reactive diastereomer. The formation of a palladium carboxylate intermediate in our decarboxylative allylic alkylation has been supported by subsequent mechanistic studies performed by our laboratories.7 Furthermore, the possibility that Pd(II) species formed in situ facilitate decarboxylation via Lewis acid type coordination (in a manner analogous to the Brønsted acid shown in Figure 2), can not be precluded.
2.4 Rationale for greater enantioselectivity in minor product
The second interesting observation made in our asymmetric decarboxylative allylation of diastereomeric β-ketoesters is that the minor product 4 had much greater enantiomeric excess than major product 3. We believe that this observation can be attributed to competing modes of control. To help illustrate our point, we must consider all four possible products and their relative abundance for the asymmetric decarboxylative allylic alkylation of β-ketoester 1 (Scheme 3). The diastereomers and their enantiomers are shown in Figure 5, and the relative percentages of each of the products are shown in Figure 6.
Figure 5.
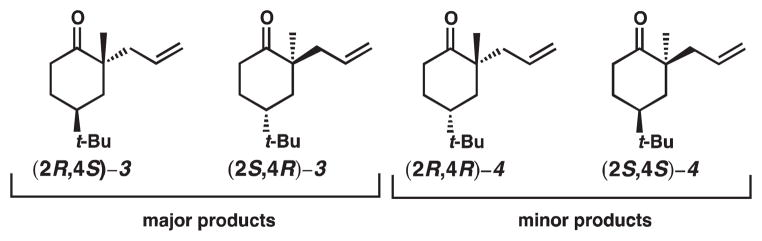
All possible products from asymmetric decarboxylative allylic alkylation of β-ketoester 1
Figure 6.

Product distribution of asymmetric decarboxylative allylic alkylation of β-ketoester 1
One type of control believed to be operative is demonstrated in the non-enantioselective alkylation of 4-tert-butylcyclohexanone enolates, which are known to have an innate selectivity for one product (i.e., substrate control, Figure 7). As shown in Scheme 6, the electrophile can be attacked by the putative enolate from either its top face or bottom face. Studies by House and coworkers have shown that the electrophile and the tert-butyl group have a trans relationship in the major product.8 Approach from the bottom face will force the cyclohexane ring into a twist boat conformation and will install the electrophile on the same face as the tert-butyl group. On the other hand, approach from the top face will install the electrophile trans to the tert-butyl group and will lead directly to the product’s chair conformation. The rationale for the favored trans relationship between the electrophile and the tert-butyl group is that the chair-like transition state is lower in energy than the transition state in a twist boat conformation. Furthermore, although the cis relationship between the electrophile and tert-butyl group affords a more thermodynamically stable product because both groups will be in equatorial positions, this reaction is under kinetic control. Thus, if we apply this model for diastereoselectivity to β-ketoesters 1 and 2, then the preferred products should have the allyl group trans to the tert-butyl group (Scheme 7), a general trend that is observed for our decarboxylative allylic alkylation with PPh3 (Scheme 5).
Figure 7.

Distribution of products as governed by two competing modes of control
Scheme 6.
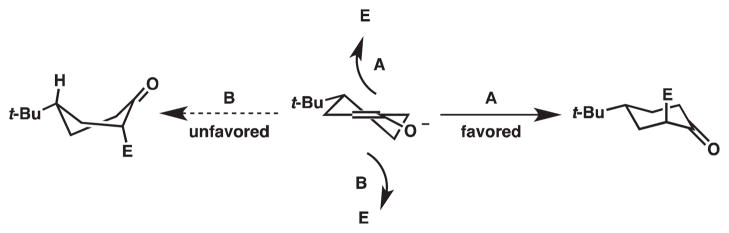
Stereoselective alkylation of t-butylcyclohexanones
Scheme 7.

Predicted products as dictated by mode of control
The other source of control arises from the catalyst (i.e., catalyst control, Figure 7). In previous experiments, we have found that (S)-t-Bu-PHOX will generally prefer to incorporate the allyl group from the Re face7; the predicted products are shown in Scheme 7. The high enantioselectivity observed for the minor product can be explained by considering the relative amount of each product and the competing modes of control (Figure 6). Ketone (2R,4S)-3 comprises 52.5% of the product mixture and is the product that is generated under doubly matched9 (catalyst and substrate) diastereocontrol. Ketone (2S,4R)-3 accounts for 22.5% of the product mixture and is formed under singly matched (substrate) diastereocontrol. Thus, only ca. 56% of the combined major diastereoisomers benefit from catalyst control and, therefore, this product exhibits depressed ee. Ketone (2R,4R)-4, the major enantiomer of the minor product, is approximately 24% of the product mixture and is the product of singly-matched catalyst control. Ketone (2S,4S)-4 is less than 1% of the product mixture and is the doubly mismatched product. The minor diastereomer, therefore, is almost exclusively under catalyst control: this is reflected in the very high ee observed (96% ee).
From our studies of the asymmetric decarboxylative allylic alkylation of β-ketoesters derived 4-tert-butylcyclohexanone, we observe a situation in which substrate and catalyst controls match or mismatch to deliver ketones 3 and 4, with varying degrees of enantiopurity. This conclusion is supported by the observation that the relative ratios of the products that are generated under singly matched diastereocontrol ((2R,4R)-4 and (2S, 4R)-3) are similar. The competition of these two types of control, one dictated by the catalyst and one dictated by the substrate, impart excellent enantioselectivity to the minor product but only moderate enantioselectivity to the major product (Figure 7).10
3. Conclusion
Our studies on the decarboxylative allylic alkylation of diastereomeric β-ketoesters derived from 4-tert-butylcyclohexanone support our hypothesis of a stereoablative enantioconvergent catalytic process. These studies also reveal an interesting example of selectivity that is governed by competing modes of substrate and catalyst control and support an interesting model for relative rates of decarboxylation for diastereomeric β-keto carboxylates.
4. Selected experimentals
A representative procedure for the decarboxylative allylic alkylation of diastereomeric β-ketoesters:
Pd2(dba)3 (67 mg, 0.07 mmol) and (S)-t-BuPHOX (73.8 mg, 0.19 mmol) were combined in a round bottom flask. The vial was evacuated for 10 minutes prior to addition of THF (88 mL). The reaction was allowed to stir for 30 minutes prior to addition of β-ketoester 2 (739 mg, 2.93 mmol) via syringe and the reaction was monitored by TLC. Once the reaction was complete, the mixture was concentrated. Isolation of products was accomplished by column chromatography (SiO2, 10 % ether in pentane).
4.1
Allyl (1R,5S)-5-(tert-butyl)-1-methyl-2-oxocyclohexane-1-carboxylate (1) was prepared according to a known procedure, see reference 1 for details. 1H NMR (300 MHz, CDCl3) δ 5.89 (m, J = 10.2 Hz, 16.2 Hz, 1H), 5.32 (m, J = 7.4 Hz, 1.2 Hz, 1H), 5.25 (m, J =10.2 Hz, 1.5 Hz, 1H), 4.62 (m, 2H), 2.49 (m, 3H), 2.02 (m, 1H), 1.57–1.18 (m, 3H), 1.29 (s, 3H), 0.90 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 208.6, 173.2, 131.7, 119.2, 66.0, 56.5, 44.4, 40.6, 39.9, 32.5, 28.6, 27.7, 21.8; IR (Neat Film, NaCl) 2961, 2865, 1717, 1229, 1140 cm−1; HRMS m/z calc’d for C15H24O3 [M]+: 252.1726, found 252.1714.
4.2 Allyl (1S,5S)-5-(tert-butyl)-1-methyl-2-oxocyclohexane-1-carboxylate (2)
1H NMR (300 MHz, CDCl3) δ 5.92 (dddd, J = 17.4 Hz, 10.5 Hz, 5.7 Hz, 5.7 Hz, 1H), 5.33 (dq, J = 17.4 Hz, 1.2 Hz, 1H), 5.23 (dq, J = 10.5 Hz, 1.2 Hz, 1H), 4.65 (dt, J = 5.7 Hz, 1.2 Hz, 2H), 2.45 (m, 2H), 2.21 (t, J = 12.6, 1H), 2.02 (m, 1H), 1.84 (dt, J =13.5 Hz, 3.3 Hz, 1H), 1.59 (m, 2H), 1.46 (s, 3H), 0.93 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 210.5, 173.2, 132.2, 118.3, 66.0, 57.4, 41.9, 38.0, 37.0, 32.6, 27.6, 26.8, 21.0; IR (Neat Film, NaCl) 2958, 2876, 1740, 1712, 1459, 1367, 1249, 1227, 1165, 1112 cm−1; HRMS m/z calc’d for C15H24O3 [M]+: 252.1726, found 252.1718.
4.3 (Trans)-2-allyl-4-(tert-butyl)-2-methylcyclohexan-1-one (3)
1H NMR (300 MHz, CDCl3) δ 5.64 (m, 1H), 5.04 (m, 2H), 2.38–2.23 (m, 4H), 2.03 (m, 1H), 1.84 (m, 1H), 1.70 (m, 1H), 1.42–1.13 (m, 2H), 1.00 (s, 3H), 0.89 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 216.0, 133.0, 118.5, 48.2, 42.0, 40.1, 38.5, 32.4, 28.3, 27.7, 22.7; IR (Neat Film, NaCl) 2962, 2870, 1709, 1366, 912 cm−1; HRMS m/z calc’d for C14H24O [M+]: 208.1827, found 208.1825; [α]D25.6 –30.00° (c 1.08, hexane).
4.4 (Cis)-2-allyl-4-(tert-butyl)-2-methylcyclohexan-1-one (4)
1H NMR (300 MHz, CDCl3) δ 5.77 (m, 1H), 5.03 (m, 2H), 2.50 (m, 1H), 2.28 (m, 2H), 2.16 (m, 1H), 2.00 (m, 1H), 1.64 (m, 2H), 1.42 (m, 2H), 1.14 (s, 3H), 0.89 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 216.2, 135.0, 117.9, 47.3, 43.3, 42.3, 38.9, 38.5, 32.5, 27.8, 27.7, 24.2; IR (Neat Film, NaCl) 2963, 2870, 1709, 1366, 912 cm−1; HRMS m/z calc’d for C14H24O [M+]: 208.1827, found 208.1836; [α]D25.7 +77.81° (c 0.105, hexane).
4.5 (E)-2-((2R,4S)-2-allyl-4-(tert-butyl)-2-methylcyclohexylidene)-N-((1S,2S,3S,5R)-2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl)hydrazine-1-carboxamide (5)
1H NMR (300 MHz, CDCl3) δ 7.83 (bs, 1H), 6.08 (d, J = 9 Hz, 1H), 5.65 (m, 1H), 5.05 (m, 2H), 4.17 (m, 1H), 2.63 (m, 2H), 2.41 (m, 1H), 2.24 (m, 3H), 1.84 (m, 6H), 1.53 (m, 2H), 1.22 (s, 3H), 1.15 (app. dd, J = 7.2 Hz, 1.0 Hz, 4H), 1.10 (s, 3H), 1.05 (s, 3H), 0.88 (d, 1H, J = 9.9 Hz), 0.85 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 156.8, 156.0, 134.1, 117.8, 48.3, 48.2, 48.0, 47.1, 42.7, 41.9, 41.8, 39.8, 38.6, 38.1, 35.5, 32.5, 28.2, 27.6, 27.0, 25.0, 23.6, 22.6, 21.0; IR (Neat Film, NaCl) 3406, 3194, 3075, 2962, 1672, 1526 cm−1, HRMS m/z calc’d for C25H43N3O [M+H]+: 402.3484, found 402.3487; [α]D25.0 +15.31° (c 0.2250, CHCl3).
4.6 (E)-2-((2R,4R)-2-allyl-4-(tert-butyl)-2-methylcyclohexylidene)-N-((1S,2S,3S,5R)-2,6,6-trimethylbicyclo[3.1.1]heptan-3 yl)hydrazine-1-carboxamide (6)
1H NMR (300 MHz, CDCl3) δ 8.42 (bs, 1H), 6.08 (d, J = 9 Hz, 1H), 5.94 (m, 1H), 5.04 (m, 2H), 4.17 (m, 1H), 2.71 (m, 2H), 2.37 (m, 3H), 1.91 (m, 5H), 1.55 (m, 3H), 1.21 (s, 3H), 1.12 (d, J = 7.5, 3H), 1.10 (s, 3H), 1.04 (s, 3H), 0.87 (d, J = 9.9 Hz, 1H), 0.84 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 157.3, 156.1, 136.4, 116.6, 48.2, 46.9, 45.1, 42.3, 41.9, 41.3, 39.4, 38.6, 38.0, 35.5, 32.5, 28.2, 27.6, 26.9, 25.6, 23.6, 22.9, 21.0; IR (Neat Film, NaCl) 3400, 3194, 2952, 2873, 1672, 1526 cm−1; HRMS m/z calc’d for C25H43N3O [M+H]+: 402.3484, found 402.3491; [α]D25.1 +29.73° (c 0.2550, hexane).
Supplementary Material
Acknowledgments
The authors wish to thank NIH-NIGMS (R01GM080269), Abbott Laboratories, Amgen, Merck, Bristol-Myers Squibb, Boehringer Ingelheim, the Gordon and Betty Moore Foundation and Caltech for financial support. CMR gratefully acknowledges the Rose Hill Foundation for pre-doctoral funding. R.A.C. gratefully acknowledges the support of this work provided by a fellowship from the National Cancer Institute of the National Institutes of Health under Award Number F31CA174359. Mr. Lawrence Henling and Dr. Michael Day are gratefully acknowledged for X-ray crystallographic structural determination. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF CRIF:MU award to the California Institute of Technology, CHE-0639094.
Footnotes
Dedicated to Professor Sarah Reisman on receipt of the Tetrahedron Young Investigator Award
NMR spectra for compounds 1–4, as well as details on the stereochemical assignment of compounds 1 and 2 can be found in the supporting information, which is available online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem Int Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 2.Please see Supporting Information for more information.
- 3.The relative rate of consumption of starting material was determined by comparing the negative slopes of Ln[β-ketoester] versus time for β-ketoesters 1 and 2.
- 4.Tsuji and coworkers previously reported the allylic alkylation of diastereomeric β-ketoesters 1 and 2 using PPh3 as ligand. Unfortunately, Their findings as presented are unclear, as the relative stereochemistry between tert-butyl and α-substituents are not explicitly delineated. See, Tsuji J, Yamada T, Minami L, Yuhara M, Nisar M, Shimizu I. J Org Chem. 1987;52:2988–2995.
- 5.Kayser RH, Brault M, Pollack RM, Bantia S, Sadoff SF. J Org Chem. 1983;48:4497–4502. [Google Scholar]
- 6.Pollack RM. In: Transition States of Biochemical Processes. Gandour RD, Schowen RL, editors. Plenum; New York: 1978. p. 467. [Google Scholar]
- 7.a) Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM. J Am Chem Soc. 2007;129:11876–11877. doi: 10.1021/ja070516j. [DOI] [PubMed] [Google Scholar]; (b) Sherden NH, Behenna DC, Virgil SC, Stoltz BM. Angew Chem Int Ed. 2009;48:6840–6843. doi: 10.1002/anie.200902575. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Behenna DC, Mohr JT, Sherden NH, Marinescu SC, Harned AM, Tani K, Seto M, Ma S, Novák Z, Krout MR, McFadden RM, Roizen JL, Enquist JA, Jr, White DE, Levine SR, Petrova K, Iwashita A, Virgil SC, Stoltz BM. Chem–Eur J. 2011;17:14199–14223. doi: 10.1002/chem.201003383. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Keith JA, Behenna DC, Sherden NH, Mohr JT, Ma S, Marinescu SC, Nielsen RJ, Oxgaard J, Stoltz BM, Goddard WA., III J Am Chem Soc. 2012;134:19050–19060. doi: 10.1021/ja306860n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.House HO, Tefertiller BA, Olmstead HD. J Org Chem. 1968;33:935–942. [Google Scholar]
- 9.Ko SY, Lee AWM, Masamune S, Reed LA, Sharpless KB, Walker FJ. Science. 1983:949–951. doi: 10.1126/science.220.4600.949. [DOI] [PubMed] [Google Scholar]
- 10.It should be noted that the percentages introduced in Figure 7 are normalized to 100, and that the actual yield of the reaction of 93%. Unreacted starting material or non-selective protonation products are assumed to account for the remaining 4R-t-Bu isomer absent from the allylic alkylation product mixture.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.