

Abstract
The first known human case of heme oxygenase-1 (HO-1) deficiency is presented in this report. The patient is a six-year-old boy with severe growth retardation. He has been suffering from persistent hemolytic anemia characterized by marked erythrocyte fragmentation and intravascular hemolysis, with paradoxical increase of serum haptoglobin and low bilirubin. An abnormal coagulation/fibrinolysis system, associated with elevated thrombomodulin and von Willebrand factor, indicated the presence of severe, persistent endothelial damage. Electron microscopy of renal glomeruli revealed detachment of endothelium, with subendothelial deposition of an unidentified material. Iron deposition was noted in renal and hepatic tissue. Immunohistochemistry of hepatic tissue and immunoblotting of a cadmium-stimulated Epstein-Barr virus–transformed lymphoblastoid cell line (LCL) revealed complete absence of HO-1 production. An LCL derived from the patient was extremely sensitive to hemin-induced cell injury. Sequence analysis of the patient's HO-1 gene revealed complete loss of exon-2 of the maternal allele and a two-nucleotide deletion within exon3 of the paternal allele. Growth retardation, anemia, iron deposition, and vulnerability to stressful injury are all characteristics observed in recently described HO–1 targeted mice. This study presents not only the first human case of HO-1 deficiency but may also provide clues to the key roles played by this important enzyme in vivo.
Introduction
Heme oxygenases (HO) are rate-limiting enzymes that catalyze the conversion of heme into carbon monoxide and biliverdin (1). They consist of two different isoforms, HO-1 and HO-2. They have antioxidant capacity and therefore act as potent anti-inflammatory proteins whenever oxidative injury takes place. HO-2 is constitutively produced within the brain and testes, whereas HO-1 is produced ubiquitously, but only marginally in the resting state (2). Rapid induction of HO-1 follows various stresses (3, 4). Recent studies have suggested protective roles of HO-1 in various inflammatory conditions (5). However, no human disease has so far been attributed to primary deficiency of HO-1. We describe a six-year-old boy with HO-1 deficiency. The patient has been suffering marked growth retardation and developmental delay associated with erythrocyte fragmentation and persistent intravascular hemolysis. Marked abnormality of coagulation/fibrinolysis system, paradoxical increase of haptoglobin concentration, and low level of serum bilirubin were intriguing findings in this patient. Low serum bilirubin concentration in the presence of continuing hemolysis led us to suspect an abnormality in the hemoglobin metabolic pathway, and we wondered if any defect in HO activity was present in this patient. Immunoblotting analysis of Epstein-Barr virus–transformed lymphoblastoid cell line (LCL) clearly showed that he has complete deficiency of HO-1 production after stimulation by stress-inducing factors. Mutational analysis revealed abnormalities in both alleles. Complete deletion of exon2 was found in the maternal allele, and a 2 nucleotide deletion was present within exon3 of the paternal allele. The combination of these two mutations in the patient resulted in the failure of production of functional HO-1 protein. Lack of this important enzyme may explain the wide variety of clinical symptoms and laboratory findings noted.
Methods
Case presentation.
The patient was 26 months old when he first sought medical advice at a hospital because of recurrent fever and generalized erythematous rash. His brother and sister were healthy. The mother had experienced two intrauterine fetal deaths. Growth retardation was apparent and marked hepatomegaly was noted, but the spleen was impalpable. Asplenia was confirmed by abdominal ultrasonography and isotope image scanning. Hematuria and proteinuria were constantly present. The white cell count was 51,600/μl (normal 3,300–8,800). Platelet count was 2,260,000/μl (normal 130,000–350,000). Microcytic, hypochromic anemia was marked with erythrocyte count 1.48 × 106/μl (normal 4.30–5.50) and hemoglobin concentration 4.9 g/dl (normal 13.5–17.0). Serum iron concentration was 64 μg/dl (normal 50–170). Lactate dehydrogenase was 17,470 IU/L (normal 196–355). Aspartate aminotransferase was markedly elevated at 442 IU/l (normal 9–42), but alanine aminotransferase was within normal limits. Ferritin was 780 ng/ml (normal 26–280). Marked abnormalities of the coagulation fibrinolysis system were noted. Fibrinogen was 109 mg/dl (normal, 196–356), fibrin degradation product was 300.1 μg/ml (normal <5), d-dimer was 186.1 μg/ml (normal <2.5), thrombin-antithrombin complex was 202.2 μg/l (normal <3), and plasmin-α2 plasmin inhibitor complex was 22.3 μg/ml (normal <0.8). Thrombomodulin was 12 ng/ml (normal <3.5) and Von Willebrand factor was 580% (normal 60–170). Hyperlipidemia was another prominent finding. Triglyceride was 638 mg/dl (normal 32–115) and total cholesterol was 552 mg/dl (normal 132–220). Low-density lipoprotein (LDL) cholesterol was predominant. Total bilirubin levels were, on the contrary, constantly low at 0.1–0.3 mg/dl (normal 0.2–1.3). Serum haptoglobin concentration was 878 mg/dl (normal 30–252). Hemopexin was undetectable by immunoelectrophoresis. Serum heme concentration was extremely high at 490 μM (normally not detectable or <1 μM). Repeated direct and indirect Coombs’ tests were always negative. Serum concentration of interleukin (IL)-6 was 4 pg/ml, tumor necrosis factor-α was <5 pg/ml, IL-2 was 0.8 U/ml, and interferon-γ was <0.1 IU/ml. Kidney biopsy revealed mild mesangial proliferation and endothelial detachment from the glomerular basement membrane. Liver biopsy showed mild inflammatory changes with minimal lymphocyte infiltration. Rudimentary splenic tissue was found on this occasion, but no histopathological confirmation was done. He has been on oral steroid and various nonsteroidal anti-inflammatory medications since he was referred to our hospital 3 years ago. The patient is now 6 years old and still requires regular transfusions of erythrocytes because of progressive hemolytic anemia.
Iron staining of the liver and the kidney biopsy specimen.
Paraffin-embedded liver and kidney sections from the patient were dewaxed and rehydrated. Prussian-blue staining was performed to detect ferric iron deposition within the tissue.
Cell preparation.
Peripheral blood mononuclear cells (PBMC) were isolated from the patient and three normal controls. LCLs were prepared by transforming lymphocytes from normal controls and the patient using supernatant from B95-8. LCL from the patient was further cloned by a limiting dilution method (6).
Immunohistochemistry of HO-1–producing cells.
Paraffin-embedded liver sections from the patient and a normal control were dewaxed and rehydrated. The section was heat-treated in a microwave oven and rinsed in Tris buffer. After blocking with normal goat serum, the sections were stained with appropriate dilutions of anti–HO-1 rabbit antiserum (Affinity BioReagents Inc., Golden, Colorado, USA) for 1 h at room temperature. After washing the slides in Tris buffer, alkaline phosphatase–conjugated goat anti–rabbit antiserum (TAGO Inc., Burlingame, California, USA) was reacted for 30 min at room temperature. Alkaline phosphatase activity was visualized using Fast Red TR salt (Sigma Chemical Co., St. Louis, Missouri, USA) after further washing of the slides in Tris buffer.
Immunoblotting of HO-1 and HO-2.
PBMC or LCLs were suspended in Roswell Park Memorial Institute medium (RPMI)-1640 containing 10% FCS and antibiotics and cultured for 6 h with cadmium chloride (10 μM). Control cultures were performed with medium alone. PBMC (2 × 106) or LCLs (1 × 107) were washed in PBS, and the cell pellets were treated with 10% TCA for 10 min on ice. The cells were spun down and suspended in 100 μl of lysis solution containing 9 M urea, 2% Triton X-100, and 10% 2-mercaptoethanol. The mixture was disrupted by sonication and neutralized with 1M Tris solution. The lysates (25 μl per lane) were applied without boiling to 5%–20% polyacrylamide gradient gels (ATTO, Tokyo, Japan), and electrophoresis was carried out in SDS. The proteins were blotted onto nitrocellulose filters using a HORIZBLOT apparatus (ATTO). Blots were blocked in 5% skim milk in PBS for 1 h and reacted with rabbit anti–HO-1 or anti–HO-2 antiserum (Affinity BioReagents Inc.) diluted at 1:1,000 in PBS–0.1% Tween-20 (PBS-T) for 1 h. The membranes were washed and further incubated with horseradish peroxidase–conjugated donkey anti–rabbit antibody (Chemicon International Inc., Temecula, California, USA) at a 1:20,000 dilution in PBS-T for 1 h. Immunoblots were developed by using an ECL Western blotting detection system (Amersham International, Amersham, United Kingdom) (7).
Cytotoxicity assay.
LCLs were used to evaluate the sensitivity to stress-inducing agents. The viability of all these cell lines or clones was >90% before the culture, as determined by the trypan blue dye exclusion test. Cells were suspended in RPMI-1649 culture medium supplemented with 10% FBS, 25 mM HEPES, 5 × 10–5 M 2-mercaptoethanol, 100 U/ml penicillin, and 10 μg/ml gentamicin at 105/ml. The cell suspension was plated in 48-well culture plates (Costar No. 3548, Corning-Costar Corp., Cambridge, Massachusetts, USA) and was cultured for 24 h in the presence of various concentrations of hemin (Sigma Chemical Co.). Cell viability was analyzed by flow cytometry after the culture. Dead cells were detected by decreased forward light scatter, high annexin-V binding and uptake of propidium iodide (8). Briefly, washed cells were resuspended in annexin-V binding buffer, and FITC-conjugated annexin-V (Bender MedSystems, Vienna, Austria) was added to the cell suspension at 1:500 dilution. Cells were reacted for 15 min in the dark and diluted 1:10 with the binding buffer. Propidium iodide was added at 2 μg/ml to the cell suspension before the analysis. Cell size, annexin-V binding, and propidium iodide uptake were evaluated by an Ortho Cytoron Absolute flow cytometer (Ortho Clinical Diagnostic Systems, Tokyo, Japan). Cells with normal forward light scatter were regarded as alive when annexin-V binding or propidium iodide uptake was negative. Relative percentages of cell survival were determined compared with the cells cultured with medium alone. LCLs from the patient and the controls were cultured with 50 μM hemin for various time periods, and the supernatant was harvested at each time point.
Measurement of heme concentration.
Serum or supernatant heme concentration was measured by the pyridine-hemochromogen method as described previously (9).
Analysis of HO-1 mRNAs.
RNA was isolated with a standard acid guanidinium phenol chloroform method from the patient, his parents, and control LCLs cultured with 10 μM of cadmium chloride for 6 h. Reverse transcription was performed with random hexamer primer (Takara Shuzo Co., Tokyo, Japan) and RAV-2 reverse transcriptase (Takara Shuzo Co.). PCR amplification of the entire open reading frame of the HO-1 gene was performed with primers HO-1 5N sense (5′-CTCCCCTCGAGCGTCCTC-3′) and HO-1 3N antisense (5′-CCTTCAGTGCCC ACG GTAA-3′). The PCR conditions were 95°C for 30 s, 55°C for 30 s, and 72°C for 90 s, for 35 cycles. After agarose gel electrophoresis and ethidium bromide staining, each band was cut out from the gel, and the PCR products were collected by the standard electroelution method. Samples were sequenced with the ABI Prism Dye Terminator sequencing kit (Perkin-Elmer Applied Biosystems Foster City, California, USA) according to the manufacturer's instructions.
DNA extraction and paternal mutation–specific PCR.
Genomic DNA was extracted from the patient, his parents, and control peripheral blood leukocytes according to the standard techniques. PCR specific for the paternal mutation was performed on genomic DNA with primers HO-1 del5 (5′-GCCCTGG AGGAGGAGATTG-3′) and HO-1 del3 (5′-GCGCTGCAT GGCTGGTGTA-3′) for 35 cycles at 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s. For the DNA control, HO-1 3 (5′-GGCTGGTGTGTAGGGGATG-3′) was used instead of HO-1 del3 in combination with HO-1 del5, and PCR was performed under the same conditions.
Results
Plasma and peripheral blood smears.
The patient's plasma, centrifuged immediately after venipuncture, contained large amounts of lipid in the upper layer. The plasma was a dark maroon color and extremely turbid. Hematocrit was always low (Fig. 1a). May-Grünwald and Giemsa staining of the peripheral blood smear revealed abundant fragmented or dysmorphic, bizarre-shaped erythrocytes (Fig. 1b, arrows). Erythroblasts and Howell-Jolly bodies were occasionally seen, and platelets were numerous.
Figure 1.

Plasma and peripheral blood smears. (a) Centrifuged peripheral venous blood from control (left) and patient (right). (b) Peripheral blood smear was stained by May-Grünwald and Giemsa. Arrows indicate fragmented erythrocytes.
Microscopic examination of renal biopsy specimen.
Mild mesangial proliferation and thickening of the capillary loop were observed within the glomeruli. Scattered lymphocyte infiltration was also seen (Fig. 2a). By transmission electron microscopy, endothelial swelling and detachment were prominent throughout the glomerular capillary (Fig. 2b, arrow). An unidentifiable material was deposited between the detached endothelium and glomerular basement membrane (Fig. 2b, asterisk). A normal glomerular capillary is shown in Fig. 2c.
Figure 2.
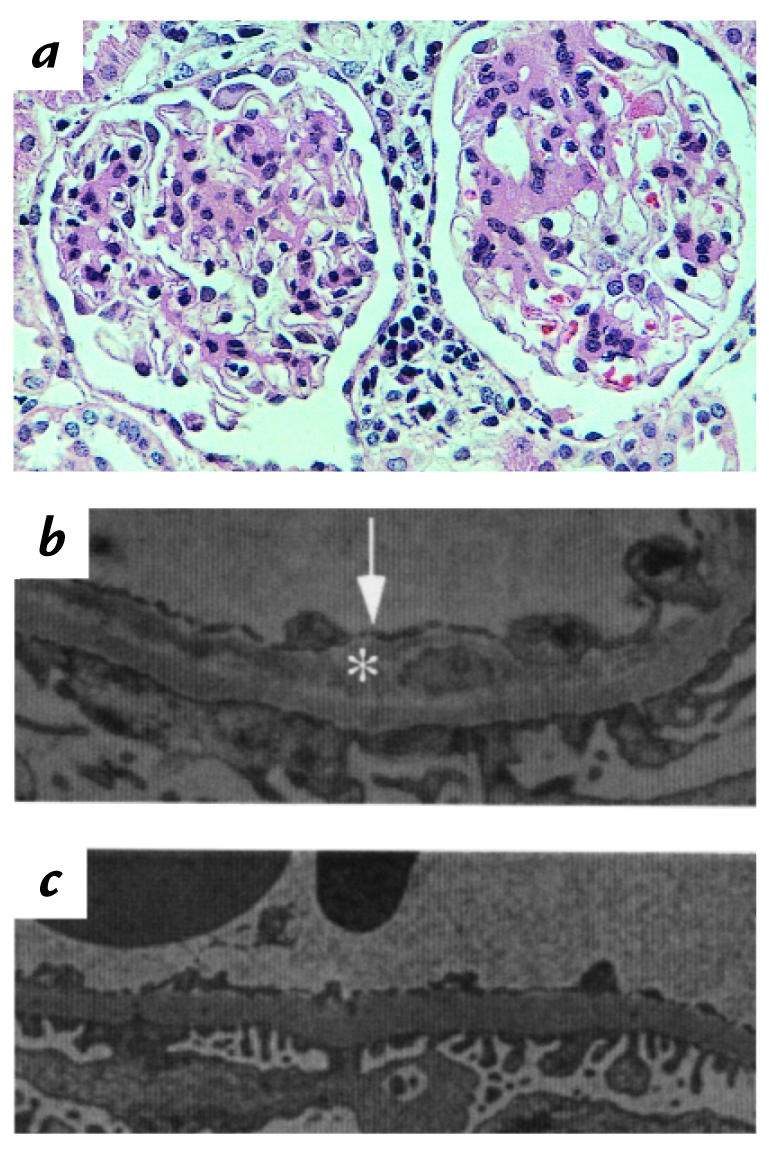
Pathology of renal glomerular loop. (a) Hematoxylin and eosin staining of the renal glomeruli. (b) Electron microscopy of glomerular capillary of the patient. Arrow indicates the detached endothelium and asterisk indicates subendothelial deposit. (c) Electron microscopy of control glomerular capillary.
Iron staining of paraffin-embedded specimens.
Iron staining was performed by a standard Prussian-blue staining method. Both kidney (Fig. 3a) and liver (Fig. 3b) sections contained multiple foci of iron deposition. Proximal tubules were the major site of iron deposition within the kidney. Iron deposition was very rare within the glomeruli (Fig. 3a, asterisk). Both hepatic parenchymal and Kupffer cells contained ferric iron.
Figure 3.

Iron staining of the kidney and liver. Paraffin-embedded specimens of the kidney and liver were stained for iron. (a) Kidney. ×200. (b) Liver. ×200. Asterisk indicates a glomerulus.
Immunohistochemistry of HO-1–producing cells.
Immunohistochemistry of paraffin-embedded liver sections is shown in Fig. 4. Intense HO-1 staining was detected within Kupffer cells of the control liver (Fig. 4a, arrow). Parenchymal cells, however, did not produce HO-1. In contrast, no HO-1 staining was detected in parenchymals or Kupffer cells of the patient's liver (Fig. 4b, arrow).
Figure 4.

Immunohistochemical staining of HO-1 in the liver. Paraffin-embedded specimen of the liver was stained with anti–HO-1 antiserum. (a) Control. (b) Patient. Arrows indicate Kupffer cells. HO-1, heme oxygenase-1.
HO production by PBMC and LCL.
PBMC, either from the patient or three controls, did not produce HO-1 when they were cultured alone (Fig. 5a, lanes 1, 3, 5, and 7). PBMC from the patient did not produce any detectable HO-1 after cadmium stimulation (Fig. 5a, lane 2). In contrast, significant levels of HO-1 were induced by cadmium within PBMC from the controls (Fig. 5a, lanes 4, 6, and 8). Control LCL produced both HO-1 and HO-2 after cadmium stimulation (Fig. 5b, lane 1). LCLs from the father (Fig. 5b, lane 3) and the mother (Fig. 5b, lane 4) also produced comparable levels of HO-1 and HO-2. LCL from the patient, however, produced only HO-2 and no detectable HO-1, even after cadmium stimulation (Fig. 5b, lane 2). Different stimulations, including sodium arsenite or hemin did not induce HO-1 production by the patient's LCL, either (data not shown).
Figure 5.

Immunoblotting of HO-1 induced in cadmium-stimulated cells. (a) PBMC from patient (lanes 1 and 2) and control 1 (lanes 3 and 4). Control 2 (lanes 5 and 6), and control 3 (lanes 7 and 8) were cultured alone (lanes 1, 3, 5, and 7) or with cadmium (lanes 2, 4, 6, and 8). Lysate samples were prepared as shown in Methods. Blotted membrane was reacted simultaneously with anti–HO-1 antiserum and anti–HO-2 antiserum, followed by the reaction with HRP-conjugated anti–rabbit IgG. (b) LCLs from patient, parents, and control were cultured with cadmium and treated similarly. Lane 1, control; lane 2, patient; lane 3, father; and lane 4, mother. HRP, horseradish peroxidase; LCL, lymphoblastoid cell line, PBMC, peripheral blood mononuclear cells.
Cytotoxicity analysis.
The sensitivity of the patient cell lines to oxidative injury was tested by a cytotoxicity analysis. Five different clones from patient LCL and LCLs from five normal controls were selected for analysis. Hemin, a substrate of HO-1, at different concentrations did not show any cytotoxicity against control LCLs even after 24 hours of culture (Fig. 6, open circles). Marked cell death was induced in the patient culture after 24 hours (Fig. 6, closed circles). More than 60% of the cells were dead at 50 μM of hemin, and the survival rate declined dose-dependently. Kinetic study revealed that patient LCL started to die after only 12 hours of culture (data not shown).
Figure 6.

Cytotoxicity assay. LCLs from patient (closed circles) and control (open circles) were cultured for 24 h with different concentrations of hemin. Dead or apoptotic cells were identified by lowered forward light scatter and high annexin-V binding using a flow cytometer. Percent survival was determined compared with cells cultured with medium. Each datum represents the mean ± SD of five independent experiments. *P < 0.001.
Heme concentration in culture supernatant.
In both control and patient cultures, heme concentrations in the supernatant did not decrease significantly during 24 hours of culture (Table 1).
Table 1.
Changes in supernatant heme concentration in LCL cultures

Mutational analysis of HO-1 gene.
Structural organization of HO-1 cDNA and construction of primers are shown in Fig. 7. Reverse transcriptase-PCR for HO-1 mRNA was performed using the primers as described in Methods. Although the control expressed HO-1 mRNA of single size (Fig. 8a, lane 1), both the patient and mother expressed HO-1 mRNA of two different sizes (lane 2 and lane 4, respectively). Cells from the father expressed a single band of mRNA (lane 3). In the patient, the smaller size was predominant, and only a minor fraction revealed a near normal size. Direct sequencing of each band was performed. The larger, or paternal, band showed a two-nucleotide deletion within exon3 (Fig. 8b). Sequencing of the smaller, or maternal, band revealed a complete loss of exon2 (Fig. 8c). Mutation-specific PCR to detect the two-nucleotide deletion within exon3 was performed using DNA preparations from the control (Fig. 8d, lane 1), patient (lane 2), father (lane 3), and mother (lane 4). The specific mutation was detected only in the patient and father. Control PCR product was detected similarly in each sample (Fig. 8e).
Figure 7.

Structural organization of HO-1 gene and construction of PCR primers for mutational analysis.
Figure 8.

Mutational analysis of HO-1 gene. (a) Reverse transcriptase-PCR for HO-1 mRNA. M denotes molecular weight marker. Lane 1, control; lane 2, patient; lane 3, father; and lane 4, mother. (b) Sequence analysis of paternal allele and normal control allele. (c) Sequence analysis of maternal allele and normal control allele. (d) Mutation- specific PCR of genomic DNA. Data from control (lane 1), patient (lane 2), father (lane 3), and mother (lane 4) are shown. (e) Control PCR.
Discussion
HO-1 is a stress-induced isoform of heme oxygenases and is not produced constitutively at a significant level (1, 2, 10). HO-1 is induced rapidly after oxidative stress and is thought to act as a potent endogenous factor for the resolution of stress-induced inflammatory injury (11–14). The detection of this particular isoform can, therefore, be used as an indicator of oxidative injury in a particular tissue, and modulation of the enzyme levels may alter the antioxidative state of living organisms (15). Inhibition of HO-1 activity may result in extreme vulnerability of an individual to common stressful stimuli, including infections and environmental toxic substances (5). A cascade of inflammatory reactions and vascular endothelial damage may rapidly result, and various complications may follow with sustained oxidative injury. However, no such human disease with low or absent HO-1 activity has been reported to date. In this regard, it is intriguing that Poss and Tonegawa (16) recently reported on HO-1–targeted mice, showing that they lack the ability to reuse iron and are characterized by progressive anemia, tissue iron deposition, chronic inflammation, and delayed growth. Furthermore, these mice are extremely sensitive to oxidative injury and are prone to death with lipopolysaccharide (LPS) administration (17). Table 2 shows the summary of characteristics of the HO-1–targeted mice in comparison with those of our patient.
Table 2.
Comparison of findings between human HO-1 deficiency and HO-1 targeted mouse

This study represents the first reported case of human HO-1 deficiency. It is recently reported that Kupffer cells are the major source of HO-1 production in rat liver (18). Immunohistochemical study in this report also shows that HO-1 is primarily produced by Kupffer cells in the liver. Absence of HO-1 staining in the patient clearly indicates that he lacks HO-1 production in vivo upon physiological stresses. PBMC from the patient did not produce HO-1 even when they were stimulated by an optimum concentration of cadmium, further indicating that the patient has a genetic abnormality in HO-1 gene. Lack of HO-1 production by LCLs from the patient argues against the possibility that HO-1 production by Kupffer cells or PBMC was secondarily downregulated by the sustained inflammatory state in this patient. In addition, mutations were detected in both alleles of HO-1 gene, further confirming that the HO-1 defect in the patient is the primary abnormality.
Hemoglobin and its metabolites are known to be potent inducers of HO-1 (13, 14, 19). It was clearly shown by the cytotoxicity assay that HO-1 provides efficient protection against certain oxidative stress, such as hemin. HO-2, which is expressed normally and constitutively within the patient's cells, does not provide protection against hemin-induced cell injury. Furthermore, the cell protection provided by HO-1 is not simply through the degradation of heme added to the culture, because heme was added in excess in the form of hemin, and there was no significant decrease in the heme concentration even in the culture of control LCLs. The precise mechanism of hemin-induced death of HO-1–deficient cells is currently being studied extensively. These in vitro studies and the clinical symptoms presented by the patient demonstrate the critical importance of HO-1, rather than HO-2, in iron metabolism and cell protection from oxidative damage. Many of the clinical features and laboratory findings are compatible with those found in HO-1–targeted mice, as shown in Table 2. It is not clear, however, if the spectrum of clinical findings and laboratory data observed in this patient can be explained solely by the lack of HO-1. Certainly, asplenia makes some contribution to the modulation of the patient's condition, either by prolonging the intravascular survival of damaged cells and therefore augmenting the heme-mediated oxidative stress, or by minimizing the red cell destruction and preventing the progression of anemia. The mechanism of hyperlipidemia, with high cholesterol and triglyceride levels, is not clear. Prolonged corticosteroid therapy alone may explain the levels seen in this patient. Bilirubin is a known antioxidant (20), and it also acts as a scavenger for degraded LDL cholesterol (21–24). Impaired bilirubin production in this patient might also be responsible for the abnormally elevated serum cholesterol. Furthermore, a high LDL cholesterol level itself, coupled with free heme, may accelerate the oxidative injury of the vascular endothelium (25). High thrombomodulin concentration indicates that the patient's endothelium suffers constant injury (26, 27). Electron microscopic study of the patient's kidney shows peculiar detachment of the endothelium within the glomeruli. These changes may lead to the constant destruction of erythrocytes and platelets. Fragmented, exhausted erythrocytes and platelets may persist within the circulation for a long time in the absence of a spleen, and they may in turn damage the endothelium and act as potent procoagulants (28). Iron deposition was detected within renal proximal tubular epithelium, hepatic parenchymal cells, and Kupffer cells in HO-1–targeted mice. Our patient showed similar iron deposition, and the finding is again compatible with the mouse model. Cellular infiltration in the liver or kidney was much less impressive in the patient samples than the findings reported in HO-1–targeted mice. The age of the patient and history of stress exposures may alter the course of the illness and pathological findings significantly. Although the existence of renal damage in HO-1–targeted mice has not been described, heme and its derivatives are known to cause renal tubular damage, and a recent report indicated that HO-1 plays an important role in protecting against epithelial injury of renal tubules (29). HO-1 defect is certainly responsible for the renal tubular injury and subsequent proteinuria and hematuria seen in this patient.
Although there is no positive family history or consanguinity of the parents, abnormality of HO-1 gene was confirmed by the mutational analysis of both alleles of HO-1 gene. Milder cases, with unexplained chronic inflammation, may be found in the future if careful analysis of HO-1 gene is performed. Our patient may represent an extreme end and a prototype of HO-1 deficiency that has a wide spectrum of clinical presentations. Analysis of the complicated features of the patient may lead to a further understanding of the key roles played by HO-1 in vivo.
Acknowledgments
We thank N. Taniguchi, H. Hashimoto, N. Okuda, and K. Konishi for helpful discussions. We also thank M. Nagai, A. Kanzaki, and T. Okazaki for analyzing serum haptoglobin and hemoglobin, and Y. Tsunematsu for providing biopsy materials. We are grateful to our young patient and his family for their help in pursuing the study, and to our nursing staff for their dedication to the care of this particular patient. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture in Japan.
References
- 1.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanism, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 2.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 3.Okinaga S, et al. Regulation of human heme oxygenase-1 gene expression under thermal stress. Blood. 1996;87:5074–5084. [PubMed] [Google Scholar]
- 4.Cantoni L, Rossi C, Rizzardini M, Gardina M, Ghezzi P. Interleukin-1 and tumor necrosis factor induce hepatic haeme oxygenase. Biochem J. 1991;279:891–894. doi: 10.1042/bj2790891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med. 1996;2:87–90. doi: 10.1038/nm0196-87. [DOI] [PubMed] [Google Scholar]
- 6.Thorley-Lawson DA, Mann KP. Early events in Epstein-Barr virus infection provide a model for B cell activation. J Exp Med. 1985;162:45–59. doi: 10.1084/jem.162.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwai K, et al. Differential expression of bcl-2 and susceptibility to anti-Fas-mediated cell death in peripheral blood lymphocytes, monocytes, and neutrophils. Blood. 1994;84:1201–1208. [PubMed] [Google Scholar]
- 8.Martin SJ, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of bcl-2 and abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Duve C. A spectrophotomeric method for the simultaneous determination of myoglobin and hemoglobin in extracts of human muscle. Acta Chem Scand. 1948;2:264–289. doi: 10.3891/acta.chem.scand.02-0264. [DOI] [PubMed] [Google Scholar]
- 10.Polla BS, Stubbe H, Kantengwa S, Maridonneau-Parini I, Jacquier-Sarlin MR. Differential induction of stress proteins and functional effects of heat shock in human phagocytes. Inflammation. 1995;19:363–377. doi: 10.1007/BF01534393. [DOI] [PubMed] [Google Scholar]
- 11.Taketani S, Kohno H, Yoshinaga T, Tokunaga R. The human 32-kDa stress protein induced by exposure to arsenite and cadmium ions is heme oxygenase. FEBS Lett. 1989;245:173–176. doi: 10.1016/0014-5793(89)80215-7. [DOI] [PubMed] [Google Scholar]
- 12.Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA. 1989;86:99–103. doi: 10.1073/pnas.86.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vercellotti GM, et al. Heme and vasculature: an oxidative hazard that induces antioxidant defenses in the endothelium. Artif Cells Blood Substit Immobil Biotechnol. 1994;22:207–213. doi: 10.3109/10731199409117415. [DOI] [PubMed] [Google Scholar]
- 14.Balla J, Jacob HS, Balla G, Nath K, Vercellotti GM. Endothelial cell heme oxygenase and ferritin induction by heme proteins: a possible mechanism limiting shock damage. Trans Assoc Am Physicians. 1992;105:1–6. [PubMed] [Google Scholar]
- 15.Kutty RK, et al. RT-PCR assay for heme oxygenase-1 and heme oxygenase-2: a sensitive method to estimate cellular oxidative damage. Ann NY Acad Sci. 1994;738:427–430. doi: 10.1111/j.1749-6632.1994.tb21833.x. [DOI] [PubMed] [Google Scholar]
- 16.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci USA. 1997;94:10919–10924. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poss KD, Tonegawa S. Reduced stress defence in heme oxygenase 1-deficient cells. Proc Natl Acad Sci USA. 1997;94:10925–10930. doi: 10.1073/pnas.94.20.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goda N, et al. Distribution of heme oxygenase isoforms in rat liver. Topographic basis for carbon monoxide-mediated microvascular relaxation. J Clin Invest. 1998;101:604–612. doi: 10.1172/JCI1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balla J, et al. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci USA. 1993;90:9285–9289. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 21.Stocker R, Glazer AN, Ames BN. Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci USA. 1987;84:5918–5922. doi: 10.1073/pnas.84.16.5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frei B, Stocker R, Ames BN. Antioxidant defences and lipid peroxidation in human blood plasma. Proc Natl Acad Sci USA. 1988;85:9748–9752. doi: 10.1073/pnas.85.24.9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neuzil J, Stocker R. Free and albumin-bound bilirubin are efficient co-antioxidants for a-tocopherol, inhibiting plasma and low density lipoprotein lipid peroxidation. J Biol Chem. 1994;269:16712–16719. [PubMed] [Google Scholar]
- 24.Wagner JR, Motchnik PA, Stocker R, Sies H, Ames BN. The oxidation of blood plasma and low density lipoprotein components by chemically generated singlet oxygen. J Biol Chem. 1993;268:18502–18506. [PubMed] [Google Scholar]
- 25.Belcher JD, et al. Vitamin E, LDL, and endothelium. Brief oral vitamin supplementation prevents oxidized LDL-mediated vascular injury in vitro. Arterioscler Thromb. 1993;13:1779–1789. doi: 10.1161/01.atv.13.12.1779. [DOI] [PubMed] [Google Scholar]
- 26.Seigneur M, et al. Plasma thrombomodulin: new approach of endothelium damage. Int Angiol. 1993;12:355–359. [PubMed] [Google Scholar]
- 27.Wada H, et al. Increased levels of vascular endothelial cell markers in thrombotic thrombocytopenic purpura. Am J Hematol. 1993;44:101–105. doi: 10.1002/ajh.2830440206. [DOI] [PubMed] [Google Scholar]
- 28.Zwaal RFA, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–1132. [PubMed] [Google Scholar]
- 29.Vogt BA, et al. Glomerular inflammation induces resistance to tubular injury in the rat. A novel form of acquired, heme oxygenase-dependent resistance to renal injury. J Clin Invest. 1996;98:2139–2145. doi: 10.1172/JCI119020. [DOI] [PMC free article] [PubMed] [Google Scholar]