

Abstract
Bioorthogonal chemistry has become one of the main driving forces in current chemical biology, inspiring the search for novel biocompatible chemospecific reactions for the past decade. Alongside the well-established labeling strategies that originated the bioorthogonal paradigm, we have recently proposed the use of heterogeneous palladium chemistry and bioorthogonal Pd0-labile prodrugs to develop spatially targeted therapies. Herein, we report the generation of biologically inert precursors of cytotoxic gemcitabine by introducing Pd0-cleavable groups in positions that are mechanistically relevant for gemcitabine’s pharmacological activity. Cell viability studies in pancreatic cancer cells showed that carbamate functionalization of the 4-amino group of gemcitabine significantly reduced (>23-fold) the prodrugs’ cytotoxicity. The N-propargyloxycarbonyl (N-Poc) promoiety displayed the highest sensitivity to heterogeneous palladium catalysis under biocompatible conditions, with a reaction half-life of less than 6 h. Zebrafish studies with allyl, propargyl, and benzyl carbamate-protected rhodamines confirmed N-Poc as the most suitable masking group for implementing in vivo bioorthogonal organometallic chemistry.
Introduction
Bioorthogonal chemistry is an emerging field of research that focuses on the development and application of selective chemical reactions susceptible to occurring in a biological environment without interfering with the normal function of its components.1,2 These reactions are used as labeling methods to study cell constituents in their native state, including proteins and biomolecules that cannot be monitored by genetically encoded reporters (e.g., glycans, lipids, DNA, etc.).1−4 Looking beyond the well-established labeling strategies that originated the bioorthogonal field, the induction of local chemotherapy by biologically inert means could allow for the reduction of adverse pharmacological effects in distant organs and tissues. This therapeutic paradigm has so far reached the clinic with the so-called photodynamic therapy, a method based on the local generation of cytotoxic reactive oxygen species by spatially controlled excitation (and subsequent energy transfer to the molecular oxygen present in the tissue) of a nontoxic systemically administered photosensitizer with a benign light source.5 Following the same principle of bioorthogonality, novel strategies are being currently investigated by several groups to enable local conversion of chemically masked prodrugs into cytotoxic small molecules using either harmless electromagnetic radiations6,7 or biocompatible heterogeneous catalysts.8
Technically speaking, for a bioorthogonal reaction to be optimal to living systems, the corresponding nonbiotic reactive partners need to display (i) high chemical stability to enzymatic processing and (ii) selective reciprocal reactivity in biocompatible conditions (water, pH 7.4, isotonicity, and 37 °C). Importantly, the reactants and the resulting product/s should be nontoxic, unless this is the intended outcome of the process. The Staudinger ligation and the copper-free azide alkyne cycloaddition, originally developed by the Bertozzi group,9−11 are representative examples of such chemistry. In addition to catalyst-free ligations, the use of transition metals to functionalize biomolecules or activate fluorogenic probes in living cells has recently emerged as a new alternative in the field.8,12,13 These biocompatible processes, also known as bioorthogonal organometallic (BOOM) reactions,14 are mediated by nonbiotic transition metals such as copper(I),15 ruthenium(II),16−18 or palladium species8,14,18−23 and have expanded the diversity of chemical groups and transformations suitable for use bioorthogonally in cell culture.
Aiming to modulate drug activity by an internal self-renewable control element rather than an external radiation source (e.g., light in photopharmacology7), we have recently proposed the development of nonbiological transition metals into biocompatible catalytic devices to allow the activation of systemically administered prodrugs at the location determined by the device.8 This bioorthogonally activated chemotherapeutic strategy would be optimal to implement focalized treatment of unresectable primary or metastatic tumors and could also find application in the treatment of other disorders such as local infections, Parkinson’s disease, cardiovascular disorders, etc. In the seminal investigation of such an approach,8 we reported the development of a selective metallo-substrate (5-fluoro-1-propargyluracil) and a solid-phase palladium catalyst (Pd0-resins) as a highly efficient bioorthogonal prodrug/activator system. On the basis of their complementary chemical roles, cytotoxic 5-fluorouracil was efficiently generated via extracellular BOOM catalysis in cell culture.8 Importantly, when each of these reagents was cultured separately, neither of them exhibited cytotoxicity, demonstrating the feasibility of the method.
To explore this paradigm further, herein we report a comprehensive study on the development and evaluation of novel Pd0-labile prodrugs of gemcitabine (deoxycytidine analogue clinically used to treat several types of cancers24) using two masking strategies: (i) carbonate formation at the 5′-OH group of the sugar moiety and (ii) carbamate masking of the 4-NH2 group of the cytosine base (Figure 1).
Figure 1.
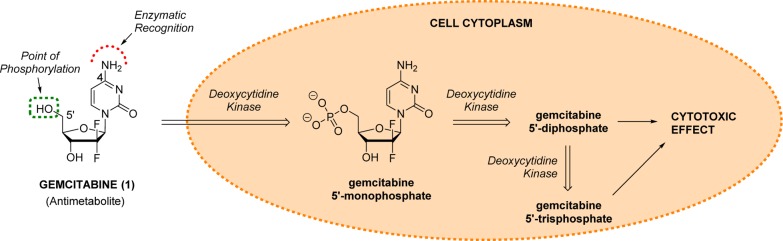
Gemcitabine and its cytotoxic mode of action. Following cell entry, gemcitabine is successively phosphorylated by deoxycytidine kinase at its 5′-OH position to generate cytotoxic metabolites.
Results and Discussion
Design and Synthesis of Pd0-Labile Gemcitabine Prodrugs
Gemcitabine (marketed as Gemzar by Eli Lilly and Co.) is an antimetabolite antineoplastic agent widely employed, in combination or alone, to treat several difficult-to-cure cancerous processes (e.g., nonsmall-cell lung carcinoma, metastatic breast cancer, and ovarian cancer),24 including being a first-line therapy in the treatment of pancreatic cancer.25 It displays a narrow therapeutic index, with its most severe side effects being myelosuppression, pulmonary toxicity, and renal failure.24 Gemcitabine is intracellularly converted by deoxycytidine kinase into its therapeutically active metabolites, the 5′-diphosphate and 5′-triphosphate derivatives (Figure 1). Gemcitabine 5′-diphosphate inhibits ribonucleotide reductase, a strictly conserved enzyme among all living organisms responsible for regulating the total rate of DNA synthesis, while the triphosphorylated derivative can become incorporated into the DNA, thereby inhibiting nuclear replication.26
On the basis of gemcitabine’s mode of action, masking the 5′-OH group of its sugar moiety would chemically block the generation of its cytotoxic metabolites (Figure 1). In a different manner but similarly relevant, modification of the 4-amino group of the cytosine base would result in a reduction of the enzyme–substrate recognition. The absence of a free NH2 group in that position would hinder the formation of essential hydrogen bonding interactions with the amino acid Asp133 located within the activation site of the enzyme, thus reducing the efficacy of the phosphorylation process.27 Furthermore, the main detoxification route of gemcitabine is via hydrolytic deamination of the 4-NH2 group. This is mediated in the liver by cytidine deaminase, which irreversibly converts gemcitabine into its inactive metabolite difluoro-deoxyuridine.28 Consequently, masking the 4-NH2 group would result in a doubly beneficial effect to the bioorthogonal strategy: it would reduce the cytotoxic properties of the resulting prodrug and protect the circulating drug precursor from premature liver deactivation before reaching the target tissue.
Allyl and propargyloxycarbonyl groups (Alloc and Poc, respectively) have been widely employed as OH and NH2 protection strategies29−32 and have shown sensitivity to palladium catalysis in biocompatible conditions.8,14,19 Therefore, both groups were chosen to derivatize gemcitabine 1 into palladium-labile prodrugs. Because of the requirement of an additional hydrogen source to undergo palladium-mediated cleavage, the carboxybenzyl group (Cbz) was used to generate control prodrugs resistant to oxidative catalysis.29,33 Prodrugs were synthesized using a semisynthetic strategy by functionalization of gemcitabine, 1 (Scheme 1). Treatment of 1 with allyl, propargyl, or benzyl chloroformate in the presence of DBU formed the corresponding carbonate prodrugs 2a,p,b in moderate yields. To synthesize the 4-carbamate derivatives, the primary alcohol in position 5′ was first silylated using TBS-Cl and imidazole, followed by treatment with the corresponding alkyl chloroformates. TBAF-mediated deprotection yielded the final carbamate prodrugs 5a,p,b in moderate to good yields.
Scheme 1. Synthesis of Gemcitabine Prodrugs.

Reagents and conditions: (i) DBU (2.5 equiv), DMF, addition of alkylchloroformate (1.5 equiv) at 4 °C, then r.t. overnight (47–53%); (ii) TBS-Cl (1.1 equiv), imidazole (3.5 equiv), DMF, r.t., 2.5 h (88%); (iii) pyridine (3.2 equiv), DMF or THF, addition of alkylchloroformate (1.5 equiv) at 4 °C, then r.t. overnight 12–48h (42–69%); (iv) TBAF (2.5 equiv), THF, r.t., overnight (62–92%).
Bioorthogonality Study: Carbonate vs Carbamate Gemcitabine Prodrugs
Although, in principle, the masking of either the 5′-OH or the 4-NH2 group of gemcitabine could both have a significant impact on the drug’s antiproliferative properties, the limiting factor in the development of any bioorthogonally activated prodrug approach is the stability of the protecting group to enzymatic metabolism. To determine the robustness of each deactivation strategy in cell culture, dose–response studies with gemcitabine (1) and prodrugs 2a,p,b and 5a,p,b were carried out in two human cancer cell lines. Pancreas adenocarcinoma BxPC-3 and Mia PaCa-2 cells were chosen as cell models due to the clinical relevance of gemcitabine as a first-line drug in pancreatic cancer therapy.24,25 As shown in Figure 2, carbamate-protected prodrugs 5a,p,b showed significant reduction of cytotoxicity relative to that of gemcitabine (EC50 (5a,p,b)/EC50 (1) > 23) in both cell lines, verifying the efficacy of the deactivation strategy. On the contrary, prodrugs 2a,p,b displayed cytotoxicity similar to that of gemcitabine (1), suggesting that these prodrugs are rapidly bioactivated inside cells. These results are in accordance with the weaker nature of the carbonate bond32 and strongly indicate that carbonate-based masking strategies are not suitable for bioorthogonal applications. Consequently, only carbamate prodrugs 5a,p,b were moved forward in this study.
Figure 2.
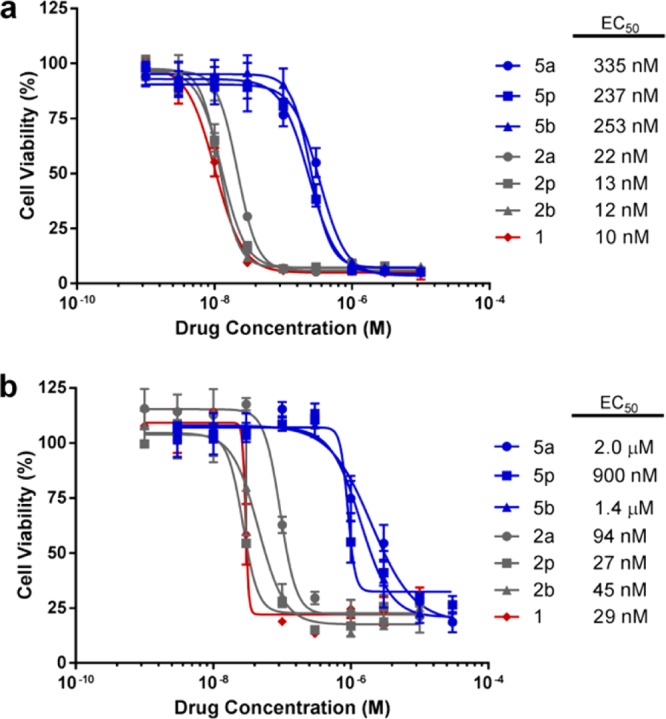
Study of the prodrugs’ bioorthogonality. Semilog dose–response curves and calculated EC50 values of prodrugs 2a,p,b (in gray) and 5a,p,b (in blue) in comparison to those of unmodified gemcitabine (1, in red) in (a) BxPC-3 and (b) Mia PaCa-2 cells. Cell viability was measured at day 4 using PrestoBlue reagent. Error bars: ± SD from n = 3.
Pd0-Mediated Conversion of Prodrugs 5a,p,b into Gemcitabine
Polystyrene-supported reagents have been employed for a wide range of cell-based applications and have shown excellent biocompatibility both in vitro and in vivo.8,34−36 As catalytically active Pd0 nanoparticles can be readily generated and trapped in an amino-functionalized polystyrene matrix,8,14 Pd0-functionalized resins (150 μm in average diameter, Figure 3a) were prepared from NovaSyn TG amino resin HL following the procedure previously reported8 and used as the heterogeneous palladium source (4.4% w/w in Pd). To test the susceptibility of the different carbamates to Pd0-mediated catalysis in a biocompatible environment, compounds 5a,p,b (100 μM) and Pd0-resins (1 mg/mL, [Pd0] = 400 μM) were dispersed in PBS (300 mOsm/kg, pH 7.4) and incubated at 37 °C for 24 h (Figure 3b). Reactions were analyzed by HPLC at different time points (0, 6, and 24 h), with prodrugs 5a,p,b being retained for 2.56, 2.45, and 2.96 min, respectively, and the gemcitabine peak appearing at 0.93 min (Figure 3c–f). Treatment of Cbz-protected compound 5b with the palladium source produced negligible levels of gemcitabine (1) after 24 h (Figure 3f), which corresponds with the need for an additional hydrogen source to achieve reductive cleavage of Cbz groups.29,33 On the contrary, incubation of compounds 5a and 5p with Pd0-resins led to the generation of significant levels of gemcitabine (1) in less than 24 h (Figure 3d,e). Interestingly, deprotection of 5p proceeded in a faster and cleaner manner, with a reaction half-life of <6 h (Figure 3e). These results are in accordance with previous observations8,37,38 that have reported the efficient cleavage of various propargylated compounds by palladium species in biocompatible conditions. A potential mechanism for this process and for the reported formation of nontoxic 1-hydroxyacetone8,39 (6) as a reaction byproduct is proposed in Figure 3g.
Figure 3.

Palladium-mediated conversion assay in cell-free biocompatible conditions. (a) SEM image of Pd0-resins. Scale bar = 150 μm. (b) Pd0-mediated carbamate cleavage of compounds 5a,p,b. (c) HPLC chromatogram of unmodified gemcitabine (1). (d–f) HPLC chromatograms (UV detector 280 nm) of 100 μM PBS solutions of compounds 5a (left panel), 5p (central panel), and 5b (right panel) treated with Pd0-resins at 37 °C for 0 h (top), 6 h (central), and 24 h (bottom). (g) Proposed mechanism for the Pd0-catalyzed cleavage of O/N-propargyl groups in water.
Pd0-Mediated Prodrug Activation in Cell Culture
Bioorthogonal in situ generation of gemcitabine (1) from the carbamate prodrugs was first investigated in standard cell culture conditions with BxPC-3 cells using Pd0-resins as the extracellular activating device. Prodrugs 5a,p,b and Pd0-resins were incubated independently (negative controls) or in combination (BOOM activation assay) and unmodified gemcitabine (1) used as the positive control. While neither the prodrugs nor the Pd0-resins exhibited cytotoxicity, a combination of 5a and 5p with Pd0-resins displayed a strong toxigenic effect (Figure 4), confirming the in situ bioorthogonal synthesis of cytotoxic gemcitabine (1). In the context of the present study, “toxigenic effect” describes the gemcitabine levels generated by each prodrug/Pd0-resin combination in cell culture which in turn result in a cytotoxic phenotype. This effect is indirectly quantified by determining the antiproliferative activity caused by each prodrug/Pd0-resin combination, which is directly proportional to the concentration of cytotoxic drug generated, and qualitatively proved by the verification of its mode of action. Consistent with the anticipated resistance of the N-Cbz functional group to palladium-mediated oxidative cleavage, cells treated with prodrug 5b and Pd0-resins did not produce any negative effect on cell viability.
Figure 4.
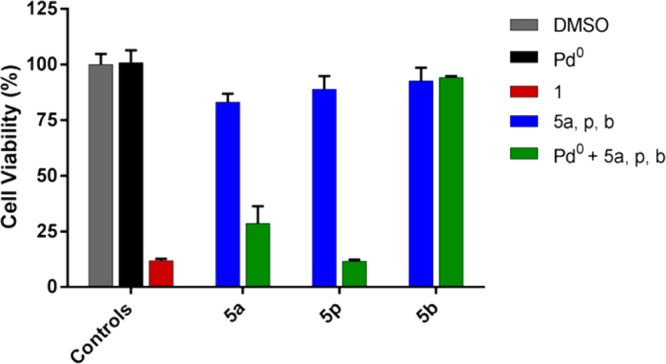
Palladium-mediated conversion of prodrugs 5a,p,b into gemcitabine (1) in pancreatic cancer BxPC-3 cells. Experiments: 0.1% (v/v) DMSO (untreated cell control, in gray); 0.67 mg/mL of Pd0-resins (negative control, in black); gemcitabine (100 nM, positive control, in red); prodrug 5a,p,b (100 nM, negative control, in blue); and 0.67 mg/mL of Pd0-resins + prodrug 5a,p,b (100 nM, BOOM activation, in green). Cell viability was measured at day 5 using PrestoBlue reagent. Error bars: ± SD from n = 3.
Dose–response studies were subsequently carried out to compare the treatment of 5a and 5p/Pd0-resin combinations at different concentrations in two cell lines: BxPC-3 and Mia PaCa-2 cells. As shown in Figure 5a,b, reduction of cell viability was observed with compounds 5a and 5p only when incubated with Pd0-resins. Analysis of the toxigenic effect displayed by each prodrug/catalyst combination clearly ranked the N-Poc-protected prodrug 5p as the most effective gemcitabine-generating precursor in the presence of Pd0-resins, exhibiting analogous cytotoxic activity to that of the unmodified drug. Importantly, neither the palladium source nor the prodrugs exhibited antiproliferative activity when separately incubated at any of the concentrations tested, thus supporting the bioorthogonality of the strategy. It is important to note that the combination of N-Alloc-protected prodrug 5a and Pd0-resins resulted in significantly higher toxigenic effect against BxPC-3 cells than that in Mia PaCa-2 cells. Since both cell lines display similar sensitivity to unmodified gemcitabine, these results show that the palladium-mediated cleavage of the N-Alloc group of prodrug 5a is significantly affected by either the cell type or the culture conditions (BxPC-3 cells are grown in RPMI, while Mia PaCa-2 cells are cultured in DMEM medium). On the contrary, the combined treatment of 5p and Pd0-resins displayed comparable toxigenic effects in both cell lines, indicating that the biological environment has little or no influence on the N-Poc deprotection process, as would be expected from a truly bioorthogonal reaction.
Figure 5.
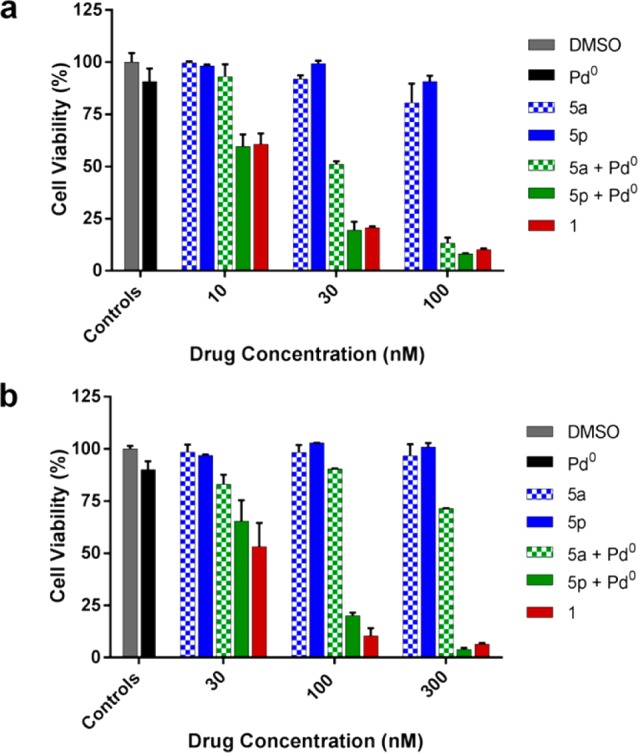
Bioorthogonally activated toxigenic effect in cancer cell culture: (a) BxPC-3 and (b) Mia PaCa-2 cells. Dose–response study: 0.1% (v/v) DMSO (untreated cell control, in gray); 0.67 mg/mL Pd0-resins (negative control, in black); gemcitabine (positive control, in red); prodrug 5a,p (negative control, in blue); and 0.67 mg/mL of Pd0-resins + prodrug 5a,p (BOOM activation, in green). Cell viability was measured at day 5 using PrestoBlue reagent. Error bars: ± SD from n = 3.
Real-time monitoring of BxPC-3 cell proliferation using an IncuCyte ZOOM microscope was performed to compare the effect of each prodrug/catalyst combination upon the kinetics of tumor cell growth. Time-lapse imaging of the negative controls 5a and 5p showed a standard growth curve up to 100% cell confluence level by day 4–5 (light and dark blue curves, respectively; Figure 6), while gemcitabine treatment efficiently suppressed cell proliferation within 24 h. In accordance with the cell viability studies, the toxigenic effect mediated by either prodrug 5a or 5p incubated with Pd0-resins (light and dark green curves, respectively; Figure 6) were patently different (see Supporting Information for a motion picture of the IncuCyte experiment). The prodrug 5p/catalyst combination inhibited cell proliferation with the same efficacy and kinetic response as unmodified gemcitabine (1), proving that cytotoxic levels of the drug were rapidly generated. Although treatment with the 5a/catalyst combination markedly delayed BxPC-3 cell growth relative to the negative controls, its antiproliferative effect was significantly inferior to either the 5p/Pd0-resins combination or unmodified gemcitabine (1), indicating quantitatively lower levels of active drug being generated.
Figure 6.
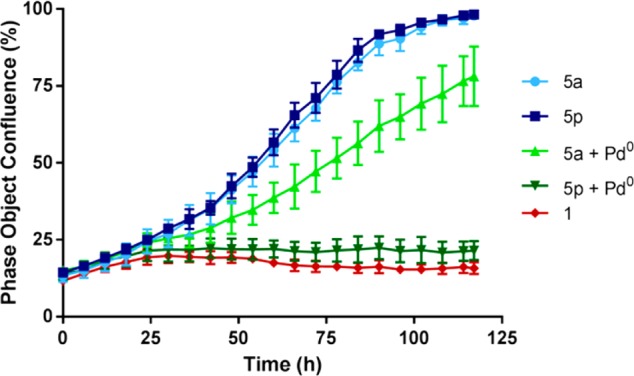
Real-time cell confluence study in pancreatic cancer BxPC-3 cells. The cell population was monitored for 120 h using an IncuCyte ZOOM system in an incubator (5% CO2 and 37 °C). Drug/prodrug concentration: 30 nM. Error bars: ± SD from n = 3.
Study of Cytotoxic Mode of Action
Double-stranded breaks caused by DNA damage induce phosphorylation of the variant histone of the H2A protein family, γ-H2AX. Phosphorylated γ-H2AX is responsible for recruiting and localizing the DNA repair mechanism.40 Gemcitabine damages DNA through partial DNA chain termination and by stalling of replication forks.41 In order to analyze the toxigenic effect caused by the prodrug/catalyst combination at the molecular level, immunofluorescence studies were carried out to probe for phosphorylated γ-H2AX as a marker of DNA damage in Mia PaCa-2 cells (Figure 7). While the negative controls (Pd0-resins or prodrug 5p separately incubated; Figure 7a,b) did not show the presence of phosphorylated γ-H2AX in the cell nuclei, cells treated with either gemcitabine (Figure 7c) or the Pd0-resins + 5p combination (Figure 7d) expressed significant levels of phospho-γ-H2AX. This study further illustrates that the cytotoxic activity generated from the prodrug/catalyst combination and unmodified gemcitabine is equivalent.
Figure 7.
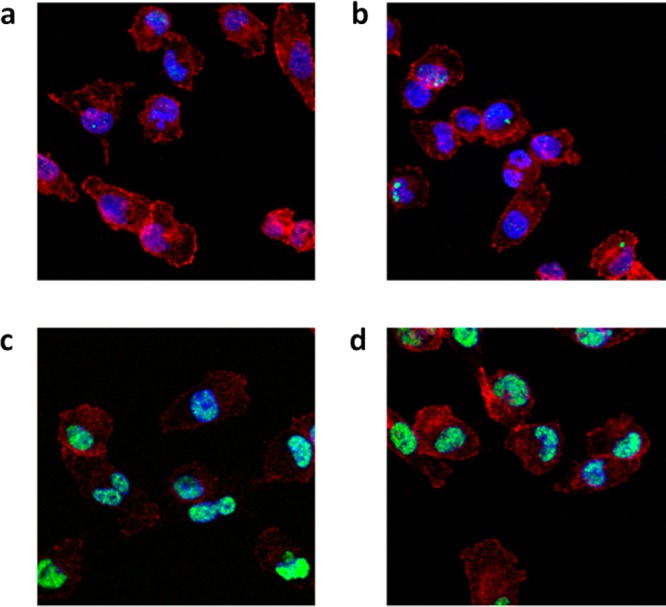
DNA damage study. Merge fluorescent images of Mia PaCa-2 cells 24 h after treatment with (a) 0.67 mg/mL Pd0-resins (negative control); (b) prodrug 5p (300 nM, negative control); (c) gemcitabine (300 nM, positive control); and 0.67 mg/mL of Pd0-resins + prodrug 5p (300 nM, BOOM activation assay). Fluorescent labels: Hoechst 33342 for cell nuclei (blue), Alexa Fluor 594 phalloidin for F-actin (red), and anti-phospho-histone γ-H2AX + Alexa Fluor 488 secondary antibody for phosphorylated γ-H2AX (green).
Pd0-Mediated Probe Activation in Zebrafish
We have recently shown that catalytically functional Pd0-resins can be implanted in the yolk sac of zebrafish embryos without inducing toxicity or affecting the embryo development.8 To compare the in vivo bioorthogonality and palladium sensitivity of each of the carbamate protecting groups under study, N,N′-bis(alkyloxycarbonyl) rhodamine derivatives 8a,p,b were prepared and used as off-on fluorescent probes (Figure 8a). Compounds 8a,p,b were synthesized from rhodamine 110, 7, by reaction with the corresponding alkylchloroformate and a base (triethylamine or pyridine) in dry DMF. The resulting nonfluorescent rhodamine precursors were incubated with 2-dpf zebrafish embryos containing either a nonfunctionalized resin (nonactive resin) or a Pd0-resin (active) in the yolk sac for 24 h and subsequently analyzed by fluorescence microscopy. Zebrafish embryos treated with Alloc-protected compound 8a showed high levels of fluorescence emission regardless of the presence or absence of palladium, particularly from the yolk sac and the digestive system (Figure 8b,c, top panel). Because of the high levels of biochemically generated fluorescence background, local palladium-mediated activation of 8a within the zebrafish yolk was not detectable, an indication of the low bioorthogonality of the N-Alloc group in zebrafish. On the contrary, compounds 8p and 8b showed improved biochemical stability, with the fluorescent background signal mostly being observed from the digestive system, which is in accordance with our previous observations.8 As expected, Cbz-protected compound 8b did not exhibit sensitivity to palladium catalysis (Figure 8b,c, lower panel), while local palladium-mediated generation of rhodamine 110 was clearly observed from 8p in the zebrafish yolk sac containing a Pd0-resin (Figure 8b,c, middle panel). As shown in Figure 8d, image analysis demonstrated up to a 4-fold increase in fluorescence intensity within close vicinity of the Pd0-resin (blue line). On the contrary, apart from the autofluorescence intensity typically observed from the nonfunctionalized resin, this inactive resin did not lead to an increment of fluorescence levels in the area surrounding it (red line). This study suggests that, from the carbamate groups herein studied, only the N-Poc masking strategy would allow generating locally increased concentrations of a functionally active small molecule by palladium heterogeneous catalysis in vivo.
Figure 8.

Pd0-mediated carbamate cleavage of rhodamine precursors in zebrafish. (a) BOOM conversion of nonfluorescent lipophilic compounds 8a,p,b into highly fluorescent hydrophilic rhodamine 110. (b) 3-dpf zebrafish embryos (n = 5) containing a nonfunctionalized resin (indicated with white arrows) after incubation with 5 μM of compounds 8a (top), 8p (middle), and 8b (bottom) for 24 h at 31 °C. (c) 3-dpf zebrafish embryos (n = 5) containing a Pd0-resin (indicated with purple arrows) after incubation with 5 μM of compounds 8a (top), 8p (middle), and 8b (bottom) for 24 h at 31 °C. Fish were imaged by phase contrast and fluorescent microscopy (ex., 470/40; em., 525/50). (d) Analysis of fluorescence intensity/pixel across a horizontal line of approximately 300 μm drawn in the yolk sac of the zebrafish embryos and encompassing both the resin and the area surrounding it. Note: the red and blue lines represent the areas of fluorescence intensity profile measured over the inactive resin (b, middle right panel) and the Pd0-resin (c, middle left panel), respectively.
Conclusions
The chemical protection of gemcitabine’s 4-amino group as a carbamate led to a significant reduction of the drug’s cytotoxic activity. Among the carbamate derivatives investigated, the N-Poc group exhibited the highest sensitivity to palladium-mediated cleavage at 37 °C in both buffered solution and cell culture. This is noteworthy since, to date, our group14,19 and others12,13,16−18,42−44 have favored the use of N-Alloc groups over N-Poc to implement coupling and decoupling strategies45 for BOOM studies. Cell viability, time-lapse microscopy, and DNA damage assays confirmed the bioorthogonal generation of cytotoxic gemcitabine from the combined treatment of extracellular Pd0-resins and the N-Poc-protected precursor in pancreatic cancer cell culture. Unlike the N-Alloc- and N-Cbz-protected derivatives, the bis-N,N′-Poc-rhodamine 1108 became locally activated by a Pd0-resin implanted in the yolk sac of zebrafish embryos, further reinforcing its nomination as the carbamate group of choice for developing bioorthogonal studies based on heterogeneous palladium catalysis. Parallel independent studies from Chen et al. (intracellular protein activation by palladium-mediated homogeneous catalysis), published just before the submission of this article,46 reported analogous findings with the N-Poc group. Finally, the simplicity of the masking strategy together with the biocompatibility and efficacy of the palladium-mediated deprotection process underlines the potential of this method to modulate the pharmacodynamics and pharmacokinetics of amino-containing drugs.
Experimental Procedures
Synthetic Procedures
General Methods
Chemicals and solvents were purchased from Fisher Scientific, Sigma-Aldrich, or VWR International Ltd. Gemcitabine HCl was purchased from Shandong Boyuan Pharmaceutical Co. Ltd. NMR spectra were recorded at ambient temperature on a 500 MHz Bruker Avance III spectrometer. Chemical shifts are reported in parts per million (ppm) relative to the solvent peak. Rf values were determined on Merck TLC Silica gel 60 F254 plates under a 254 nm UV source. Purification was carried out by flash column chromatography using commercially available silica gel (220–440 mesh, Sigma-Aldrich). All compounds used in the biological experiments were >95% pure, as measured by HPLC using an evaporative light scattering detector. Method: eluent A, water and formic acid (0.1%); eluent B, acetonitrile and formic acid (0.1%); A/B = 95:5 to 5:95 in 3 min, isocratic 1 min, 5:95 to 95:5 in 1 min, and isocratic 1 min.
Synthesis of Pd0-Resins
Pd0-functionalized resins were prepared from NovaSyn TG amino resin HL (0.39 mmol NH2/g) as previously described.8
Synthesis of Carbonate-Protected Derivatives 2a,p,b
Gemcitabine HCl (150 mg, 0.5 mmol) was dissolved in dry DMF (3 mL) with DBU (194 μL, 1.30 mmol) under N2 atmosphere. The mixture was then cooled to 4 °C in an ice bath. Allyl, propargyl, or benzyl chloroformate (0.75 mmol) was added dropwise to the mixture. The mixture was stirred overnight and allowed to warm up to room temperature (r.t.). The solvents were then removed in vacuo, the crude redissolved with 25% isopropanol (IPA) in CHCl3 (20 mL), and washed with H2O (20 mL). The aqueous layer was washed with 25% IPA in CHCl3 (3 × 20 mL) and the combined organic layers dried over MgSO4, filtered, and concentrated in vacuo.
4-Amino-1-(2-deoxy-2,2-difluoro-5-O-[allyloxycarbon-yl]-β-d-erythro-pentofuranosyl)pyrimidin-2(1H)-one] (2a)
The crude was purified by column chromatography using 8% MeOH in DCM (Rf 0.29, 10% MeOH in DCM), which yielded a light green solid (90 mg, 52% yield). 1H NMR (500 MHz, DMSO) δ 7.50 (d, J = 7.5, 1H), 7.42 (d, J = 5.7, 2H), 6.44 (d, J = 6.3, 1H), 6.17 (s, 1H), 5.99–5.89 (m, 1H), 5.79 (d, J = 7.5, 1H), 5.38–5.23 (m, 2H), 4.63 (dt, J = 5.5, 1.3, 2H), 4.47–4.37 (m, 2H), 4.18 (s, 1H), 4.07–4.00 (m, 1H). 13C NMR (126 MHz, DMSO) δ 165.63 (C), 154.48 (C), 154.07 (C), 141.16 (CH), 132.02 (CH), 122.60 (t, JC–F = 264.6, C), 118.56 (CH2), 94.87 (CH), 83.89 (CH), 77.22 (CH), 70.09 (t, JC–F = 25.2, CH), 68.16 (CH2), 66.31 (CH2). LRMS (ESI) m/z 346.0 [M – H]−. HRMS (FAB) m/z [M – H]− calcd for C13H14O6N3F2, 346.0856; found, 346.0859.
4-Amino-1-(2-deoxy-2,2-difluoro-5-O-[propargyloxy-carbonyl]-β-d-erythro-pentofuranosyl)pyrimidin-2(1H)-one] (2p)
The crude was purified by column chromatography using 8% MeOH in DCM (Rf 0.23, 10% MeOH in DCM), which yielded a pale yellow solid (81 mg, 47%). 1H NMR (500 MHz, DMSO) δ 7.65 (d, J = 7.5, 1H), 7.44 (d, J = 13.0, 2H), 6.24 (t, J = 5.0, 1H), 5.81 (d, J = 7.5, 1H), 5.28–5.26 (m, 2H), 4.88 (s, 2H), 4.22–4.16 (m, 1H), 3.79–3.62 (m, 3H). 13C NMR (126 MHz, DMSO) δ 165.71 (C), 154.41 (C), 152.63 (C), 141.43 (CH), 121.48 (t, JC–F = 262.1, C), 94.83 (CH), 83.76 (CH), 79.16 (CH), 78.11 (CH), 77.25 (C), 73.39 (t, JC–F = 26.5, CH), 59.18 (CH2), 56.37 (CH2). LRMS (ESI) m/z 379.9 [M + Cl]−. HRMS (FAB) m/z [M – H]− calcd for C13H12O6N3F2, 344.0700; found, 344.0728.
4-Amino-1-(2-deoxy-2,2-difluoro-5-O-[benzyloxy-carbonyl]-β-d-erythro-pentofuranosyl)pyrimidin-2(1H)-one] (2b)
The crude was purified by column chromatography using 6% MeOH in DCM (Rf 0.05, 4% MeOH in DCM), which yielded a white solid (70 mg, 53% yield). 1H NMR (500 MHz, DMSO) δ 7.49 (d, J = 7.5, 1H), 7.44–7.33 (m, 7H), 6.44 (d, J = 6.2, 1H), 6.17 (s, 1H), 5.76 (d, J = 7.5, 1H), 5.17 (s, 2H), 4.52–4.35 (m, 2H), 4.18 (s, 1H), 4.03 (t, J = 6.1, 1H). 13C NMR (126 MHz, DMSO) δ 165.60 (C), 154.45 (C), 154.20 (C), 141.15 (CH), 135.04 (C), 128.50 (CH)2, 128.42 (CH), 128.17 (CH)2, 122.57 (t, JC–F = 258.3, C), 94.83 (CH), 83.86 (CH), 77.18 (CH), 70.10 (t, JC–F = 23.4, CH), 69.22 (CH2), 66.39 (CH2). LRMS (ESI) m/z 398.0 [M + H]+. HRMS (FAB) m/z [M – H]− calcd for C17H16O6N3F2, 396.1013; found, 396.1016.
Synthesis of Carbamate-Protected Derivatives 5a,p,b
Synthesis of Silylated Derivative 3
Gemcitabine HCl (500 mg, 1.67 mmol) was dissolved in dry DMF (5 mL) with imidazole (398 mg, 5.85 mmol) and TBS-Cl (301 mg, 2.0 mmol) stirred at r.t. overnight. The mixture was then concentrated in vacuo, redissolved in EtOAc (50 mL), and washed with H2O (50 mL). The aqueous layer was washed twice with EtOAc and, subsequently, the organic layers combined, washed with brine (150 mL), and dried over MgSO4. The crude was purified by column chromatography (eluent 8% MeOH in DCM) to yield intermediate 3 as a white solid 552 mg (88%). 1H NMR (500 MHz, DMSO) δ 7.63 (d, J = 7.5, 1H), 7.38 (s, 2H), 6.31 (d, J = 5.1, 1H), 6.14 (t, J = 7.7, 1H), 5.76 (d, J = 7.5, 1H), 4.19–4.06 (m, 1H), 3.95 (d, J = 11.8, 1H), 3.90–3.77 (m, 2H), 0.90 (s, 9H), 0.09 (d, J = 2.1, 6H). 13C NMR (126 MHz, DMSO) δ 165.58 (C), 154.56 (C), 139.91 (CH), 122.95 (t, JC–F = 258.3, C), 94.51 (CH), 83.45 (t, JC–F = 31.5, CH), 79.76 (CH), 68.23 (t, JC–F = 22.7, CH), 60.72 (CH2), 25.71 (CH3), 17.99 (C), −5.52(CH3)3, −5.63 (CH3)2. MS (ESI) m/z 378.0 [M + H]+.
Synthesis of Intermediate 4a
Compound 3 (100 mg, 0.27 mmol) was dissolved in dry THF (2.5 mL) with pyridine (61 μL, 0.75 mmol) and the mixture cooled to 4 °C in an ice bath. Allyl chloroformate (55 μL, 0.52 mmol) was added dropwise to the mixture and stirred for 1 h. The solvent was removed in vacuo and purified via flash chromatography (eluent: 3% MeOH in DCM) to yield compound 4a as a white solid.
4-(Allyloxycarbonylamino)-1-(2-deoxy-2,2-difluoro-5-O-[tert-butyl(dimethyl)silyl]-β-d-erythro-pentofuranosyl)pyrimidin-2(1H)-one] (4a)
Eighty-three milligrams, 69% yield (Rf 0.53, 10% MeOH in DCM). 1H NMR (500 MHz, MeOD) δ 8.29 (d, J = 7.7, 1H), 7.32 (d, J = 7.6, 1H), 6.25 (t, J = 6.7, 1H), 6.04–5.94 (m, 1H), 5.39 (dq, J = 17.2, 1.5, 1H), 5.27 (dq, J = 10.5, 2.6, 1.3, 1H), 4.70 (dt, J = 5.6, 1.4, 2H), 4.33–4.24 (m, J = 12.5, 8.8, 1H), 4.10 (d, J = 12.0, 1H), 4.03–3.89 (m, 2H), 0.97 (s, 9H), 0.17 (s, 6H). 13C NMR (126 MHz, MeOD) δ 165.38 (C), 157.33 (C), 154.35 (C), 144.81 (CH), 133.24 (CH), 123.87 (t, JC–F = 259.7, C), 118.88 (CH2), 96.95 (CH), 86.22 (t, JC–F = 32.8, CH), 82.46 (CH), 69.56 (t, JC–F = 23.9, CH), 67.56 (CH2), 61.68 (CH2), 26.37 (CH3)3, 19.26 (C), −5.35 (CH3), −5.45 (CH3). MS (ESI) m/z 462.0 [M + H]+.
Synthesis of Intermediates 4p,b
Compound 3 (100 mg, 0.27 mmol) was dissolved in dry DMF (2.5 mL) with pyridine (61 μL, 0.75 mmol) and the mixture cooled to 4 °C in an ice bath. Propargyl (63 μL, 0.65 mmol) or benzyl chloroformate (56 μL, 0.39 mmol) was added dropwise to the mixture. The mixture was stirred at r.t. overnight (for 4b) or for 48 h (for 4p). The solvents were removed and the mixture redissolved with EtOAc (30 mL) and washed with H2O (30 mL). The aqueous layer was washed twice more with EtOAc (30 mL each) and the combined organic layers dried over MgSO4, solids filtered off, and concentrated in vacuo. The crude was purified via flash chromatography (eluent: 3% MeOH in DCM) to yield compounds 4p,b as white solids.
4-(Propargyloxycarbonylamino)-1-(2-deoxy-2,2-difluoro-5-O-[tert-butyl(dimethyl)silyl]-β-d-erythro-pentofuranosyl)pyrimidin-2(1H)-one] (4p)
Fifty-three milligrams, 44% yield. (Rf 0.6, 10% MeOH in DCM) 1H NMR (500 MHz, MeOD) δ 8.30 (d, J = 7.6, 1H), 7.29 (d, J = 7.6, 1H), 6.25 (t, J = 6.7, 1H), 4.84 (d, J = 2.5, 2H), 4.33–4.24 (m, 1H), 4.10 (d, J = 12.0, 1H), 4.03–3.90 (m, 2H), 2.99 (t, J = 2.3, 1H), 0.98 (s, 9H), 0.17 (s, 6H). 13C NMR (126 MHz, MeOD) δ 165.28 (C), 157.29 (C), 154.57 (C), 144.97 (CH), 123.88 (t, JC–F = 259.6, C), 96.93 (CH), 86.15 (t, JC–F = 32.8, CH), 82.49 (CH), 77.70 (C), 76.92 (CH), 69.57 (t, JC–F = 22.7, CH), 61.69 (CH2), 54.34 (CH2), 26.36 (CH3)3, 19.26 (C), −5.36 (CH3), −5.46 (CH3). MS (ESI) m/z 460.2 [M + H]+.
4-(Benzyloxycarbonylamino)-1-(2-deoxy-2,2-difluoro-5-O-[tert-butyl(dimethyl)silyl]-β-d-erythro-pentofuranosyl)pyrimidin-2(1H)-one] (4b)
Fifty-six milligrams, 42% yield (Rf 0.5, 10% MeOH in DCM) which. 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 7.5, 1H), 7.40–7.32 (m, 5H), 7.25–7.20 (m, 1H), 6.40–6.32 (m, 1H), 5.22 (s, 2H), 4.41–4.31 (m, 1H), 4.09–3.85 (m, 3H), 0.93 (s, 9H), 0.12 (s, 6H). 13C NMR (126 MHz, DMSO) δ 162.90 (C), 154.93 (C), 152.35 (C), 144.42 (CH), 135.02 (C), 128.85 (CH)2, 128.43 (CH)2, 122.32 (t, JC–F = 258.3, C), 95.54 (CH), 84.56 (t, JC–F = 32.8, CH), 81.62 (CH), 69.31 (t, JC–F = 26.5, CH), 68.22 (CH2), 60.69 (CH2), 25.97 (CH3)3, 18.5 (C), −5.32 (CH3), −5.41 (CH3). MS (ESI) m/z 534.1 [M + Na]+.
Synthesis of Carbamate-Protected Derivatives 5a,p,b
Compounds 4a,p,b (48 mg, 0.1 mmol, 31 mg, 0.07 mmol, 73 mg, 0.14 mmol respectively) and TBAF solution (1.5 equiv for 4a,b and 2.5 equiv for 4p) were dissolved in anhydrous THF (3 mL) and stirred at room temperature for 3 h (for 5b) or overnight (for 5a,p). The resulting mixture was concentrated in vacuo and purified via flash chromatography (eluent: 6% MeOH in DCM) to yield compounds 5a,p,b as white solids.
4-(Allyloxycarbonylamino)-1-(2-deoxy-2,2-difluoro-β-d-erythro-pentofuranosyl)pyrimidin-2(1H)-one [5a]
Thirty-two milligrams (92% yield). Rf 0.27 (10% MeOH in DCM). 1H NMR (500 MHz, DMSO) δ 10.96 (s, 1H), 8.23 (d, J = 7.6, 1H), 7.10 (d, J = 7.6, 1H), 6.31 (d, J = 6.5, 1H), 6.16 (t, J = 7.5, 1H), 6.00–5.89 (m, 1H), 5.42–5.21 (m, 3H), 4.64 (d, J = 5.3, 2H), 4.24–4.13 (m, 1H), 3.88 (dt, J = 8.5, 3.1, 1H), 3.84–3.61 (m, 2H). 13C NMR (126 MHz, DMSO) δ 163.40 (C), 153.99 (C), 152.82 (C), 144.45 (CH), 132.39 (CH), 122.94 (t, JC–F = 258.3, C), 118.04 (CH2), 94.82 (CH), 84.07 (t, JC–F = 32.8, CH), 80.98 (CH), 68.37 (t, JC–F = 22.7, CH), 65.64 (CH2), 58.79 (CH2). LRMS (ESI) m/z 346.0 [M – H]−. HRMS (FAB) m/z [M – H]− calcd for C13H14O6N3F2, 346.0856; found, 346.0853.
4-(Propargyloxycarbonylamino)-1-(2-deoxy-2,2-difluoro-β-d-erythro-pentofuranosyl)pyrimidin-2(1H)-one (5p)
Fifteen milligrams (62% yield). Rf 0.41 (10% MeOH in DCM). 1H NMR (500 MHz, DMSO) δ 11.06 (s, 1H), 8.25 (d, J = 7.6, 1H), 7.08 (d, J = 7.6, 1H), 6.30 (d, J = 6.5, 1H), 6.16 (t, J = 7.4, 1H), 5.29 (t, J = 5.4, 1H), 4.80 (d, J = 2.3, 2H), 4.25–4.13 (m, 1H), 3.89 (dt, J = 8.5, 3.1, 1H), 3.84–3.63 (m, 2H), 3.62 (t, J = 2.3, 1H). 13C NMR (126 MHz, DMSO) δ 163.26 (C), 153.93 (C), 152.37 (C), 144.61 (CH), 122.92 (C), 94.80 (CH), 84.09 (CH), 81.01 (CH), 78.21 (CH), 78.18 (C) 68.37 (CH), 58.78 (CH2), 53.01 (CH2). LRMS (ESI) m/z 344.0 [M – H]−. HRMS (FAB) m/z [M – H]− calcd for C13H12O6N3F2, 344.0700; found, 344.0719.
4-(Benzyloxycarbonylamino)-1-(2-deoxy-2,2-difluoro-β-d-erythro-pentofuranosyl)pyrimidin-2(1H)-one (5b)
Fifty-one milligrams, 92% yield. Rf 0.42 (10% MeOH in DCM). 1H NMR (500 MHz, DMSO) δ 10.98 (s, 1H), 8.23 (d, J = 7.6, 1H), 7.45–7.32 (m, 5H), 7.11 (d, J = 7.6, 1H), 6.31 (d, J = 6.5, 1H), 6.16 (t, J = 7.4, 1H), 5.29 (t, J = 5.5, 1H), 5.20 (s, 2H), 4.25–4.13 (m, 1H), 3.88 (dt, J = 8.5, 3.1, 1H), 3.84–3.62 (m, 2H). 13C NMR (126 MHz, DMSO) δ 163.38 (C), 153.99 (C), 153.00 (C), 144.46 (CH), 135.81 (C), 128.48 (CH)2, 128.19 (CH), 127.96 (CH)2, 122.93 (t, JC–F = 259.6, C), 94.86 (CH), 84.08 (t, JC–F = 30.2, CH), 80.98 (CH), 68.37 (t, JC–F = 22.7, CH), 66.67 (CH2), 58.78 (CH2). LRMS (ESI) m/z 396.1 [M – H]−. HRMS (FAB) m/z [M – H]− calcd for C17H16O6N3F2, 396.1013; found, 396.1016.
Synthesis of Nonfluorescent Probes 8a,p
Probes 8a(14) and 8p(8) were prepared from rhodamine 110 chloride, 7, as previously described.14,8
Synthesis of Nonfluorescent Probe 8b
Under a nitrogen atmosphere, rhodamine 110 chloride (150 mg, 0.41 mmol) was dissolved in dry DMF (4 mL). Separately, benzyl chloroformate (350 μL, 2.45 mmol) and pyridine (298 μL, 3.69 mmol) were dissolved in DMF (1 mL) and added dropwise to the mixture. The reaction mixture was stirred at r.t. for 48 h, solvents removed in vacuo, and the resulting crude resuspended in 25% isopropanol in CHCl3 (30 mL) and washed with H2O. The aqueous layer was then washed 5 times with 25% isopropanol in CHCl3 (30 mL). The combined organic layers were dried over anhydrous MgSO4 and the solids filtered off and concentrated in vacuo. The crude was purified via flash chromatography (hexane/ethyl acetate 2:1) to yield bis-N,N′-benzyloxycarbonyl-rhodamine 110 (8b) as a white solid (31 mg, 13%). Rf 0.23 (hexane/ethyl acetate 2:1); 1H NMR (500 MHz, DMSO) δ 10.12 (s, 2H), 8.01 (d, J = 7.6, 1H), 7.78 (t, J = 7.1, 1H), 7.72 (t, J = 7.3, 1H), 7.58 (d, J = 1.7, 2H), 7.45–7.33 (m, 10H), 7.27 (d, J = 7.6, 1H), 7.20–7.12 (m, 2H), 6.70 (d, J = 8.7, 2H), 5.18 (s, 4H). 13C NMR (126 MHz, DMSO) δ 168.66 (C), 153.25 (C)2, 152.51 (C)2, 150.94 (C), 141.43 (C)2, 136.31 (C)2, 135.71 (CH), 130.21 (CH), 128.53 (CH)2, 128.47 (CH)4 128.19 (CH)4, 128.14 (CH)2 125.71 (C), 124.77 (CH), 123.95 (CH), 114.46 (CH)2, 112.48 (C)2, 105.10 (CH)2, 81.94 (C), 66.07 (CH2)2. MS (ESI) m/z 597.2 [M – H]−.
Biological Studies
General Methods
Cell lines were grown in culture media supplemented with serum (10% FBS) and l-glutamine (2 mM) and incubated in a tissue culture incubator at 37 °C and 5% CO2. Human pancreas adenocarcinoma BxPC-3 cells (a kind gift from Dr. Mark Duxbury) were cultured in Roswell Park Memorial Institute (RPMI) media. Human pancreatic carcinoma Mia PaCa-2 cells (a kind gift from Dr. Simon Wilkinson) were cultured in Dulbecco’s modified Eagle’s media (DMEM).
Cell Viability Studies
Cells were seeded in a 96 well plate format at the appropriate cell concentration (2,500 cells/well for BxPC-3 cells and 1,000 cells/well for MiaPaCa-2 cells) and incubated for 48 h before treatment. Each well was then replaced with fresh media containing compound 1 or 2a,p,b/5a,p,b and incubated for 4 days. Untreated cells were incubated with DMSO (0.1% v/v). PrestoBlue cell viability reagent (10% v/v) was added to each well and the plate incubated for 1 h. Fluorescence emission was detected using a PerkinElmer Victor2 multilabel reader (excitation filter at 540 nm and emissions filter at 590 nm). All conditions were normalized to the untreated cells (100%) and curves fitted using GraphPad Prism using a sigmoidal variable slope curve.
Pd0-Mediated Deprotection of 5a,p,b in Biocompatible Conditions
Compounds 5a, 5p, and 5b (100 μM) were dissolved in PBS (1 mL) with 1 mg of Pd0-resins and shaken at 1400 rpm and 37 °C in a Thermomixer. Reaction crudes were monitored at 0, 6, 24, and 48 h by analytical HPLC (Agilent) using an UV detector at 280 nm to avoid the detection of PBS salts. Eluent A, water and formic acid (0.1%); eluent B, acetonitrile and formic acid (0.1%); A/B = 95:5 to 5:95 in 3 min, isocratic 1 min, 5:95 to 95:5 in 1 min, and isocratic 1 min.
Pd0-Mediated Dealkylation of 5a,p,b in Cell Culture
BxPC-3 and MiaPaCa-2 cells were plated as described before. Each well was then replaced with fresh media containing Pd0-resins (0.67 mg/mL) with DMSO (0.1% v/v); 5a,p,b (3, 10, 30, 100, and 300 nM) with DMSO (0.1% v/v); gemcitabine (3, 10, 30, 100, and 300 nM) with DMSO (0.1% v/v); or a combination of 0.67 mg/mL of Pd0-resins + 5a,p,b (3, 10, 30, 100, and 300 nM) with DMSO (0.1% v/v). Untreated cells were incubated with DMSO (0.1% v/v).
Cell Viability Assay
Cells were incubated with drugs for 5 days. PrestoBlue cell viability reagent (10% v/v) was added to each well and the plate incubated for 60 min. Fluorescence emission was detected and results normalized as described above.
Time-Lapse Imaging Study
BxPC-3 cell growth studies were carried out at a single dose of gemcitabine (1) or 5a,p (30 nM). Each well was imaged every 3 h over 5 d under standard incubation conditions using an IncuCyte ZOOM microscope (placed inside the incubator). Image-based analysis of cell confluence and Supporting Information movie 1 were produced using the IncuCyte software.
DNA Damage Study
Mia PaCa-2 cells were seeded in an 8-well chamber slide with a density of 2,000 cells/well and incubated for 48 h prior to treatment. Each well was then replaced with fresh media containing 0.1% DMSO (v/v) and the following treatments: Pd0-resins (0.67 mg/mL); 5p (300 nM); 1 (300 nM); or a combination of Pd0-resin + 5p (300 nM). The combination of 5p and Pd0 resins were incubated for 24 h at 37 °C prior to treatment with cells. Following 24 h of treatment, cells were fixed with paraformaldehyde (4%, 20 min) and permeabilized cells (Triton X-100, 0.1%) subsequently treated with rabbit monoclonal antibody against Phospho-Histone H2AX (Ser139) (1:400, Cell Signaling Technologies, cat. no. 9718) for 1 h at room temperature. This was followed by 1 h of incubation at room temperature with secondary Alexa Fluor 488 linked antibody (1:1000, goat antirabbit, IgG, Life Technologies), Hoechst 33342 (1:8000, Life Technologies), and Alexa Fluor 594 Phalloidin (1:500, Life Technologies). Cells seeded in the chamber slide were imaged using an Olympus FV1000 microscope and merged using the ImageJ software (National Institutes of Health).
Zebrafish Studies
Wild type zebrafish embryos were collected from AB-TPL breeding pairs and reared at 28 °C in E3 embryo medium. Twenty-four hours postfertilization, embryos were treated with the anesthetic tricaine and pierced in the yolk with a fine needle. Either nonfunctionalized resin or a Pd0-resin was then rapidly inserted into the yolk. Embryos that lost significant yolk in the procedure were removed from the experiment. Embryos were then gently transferred to fresh E3 medium and returned to 28 °C to ensure the yolk wound was closed. The corresponding probe (8a,p,b) was added to the embryo medium (final concentration 5 μM) and fish incubated for additional 24 h at 31 °C. Fish were imaged using fluorescent microscopy (Olympus Scan-R). The fold change in fluorescence with the Pd0 resin + 8p in comparison to that with the inactive resin + 8p were quantified using ImageJ software. A line was drawn horizontally over the yolk sac of the embryo encompassing both the resin beads and the area surrounding them (Figure 8b,c), and the pixel intensities along the lines were calculated (Figure 8d). Experiments were repeated at least twice with n = 4/per condition. Zebrafish husbandry and experiments were performed under Home Office License in compliance with the Animals (Scientific Procedures) Act 1986 and approved by the University of Edinburgh Ethics Committee.
Acknowledgments
J.T.W. is grateful to the College of Medicine and Veterinary Medicine and the University of Edinburgh for a Darwin International Scholarship and an Edinburgh Global Research Scholarship. N.O.C. and A.U.B. thank RCUK and IGMM, respectively, for an Academic Fellowship. W.R., C.F., and E.E.P. are funded by the MRC. We are grateful to the Edinburgh Cancer Research UK Centre for funding this research through the CRUK Development Fund. This work has been partly funded by a Heriot Watt University – IGMM pilot project and the MSD Scottish Life Sciences Fund. We also thank Dr. Mark Duxbury for his helpful comments regarding the clinical applications of the prodrug approach.
Glossary
Abbreviations Used
- BOOM
bioorthogonal organometallic
- Alloc
allyloxycarbonyl
- Poc
propargyloxycarbonyl
- Cbz
carboxybenzyl
- TBS-Cl
tert-butyldimethylsilyl chloride
- TBAF
tetrabutylammonium fluoride
- dpf
days postfertilization
- TLC
thin layer chromatography
- DBU
1,8-diazabicycloundec-7-ene
- DMF
dimethylformamide
- DCM
dichloromethane
- r.t.
room temperature
- CD3OD
deuterated methanol
- RPMI
Roswell Park Memorial Institute
- DMEM
Dulbecco’s modified Eagle’s media
- DMSO
dimethyl sulfoxide
Supporting Information Available
NMR spectra of compounds 2a,p,b, 5a,p,b, and 8a,p,b and 5-day time-lapse motion picture of pancreatic BxPC-3 cell proliferation under treatment with 30 nM 5a; Pd0-resins + 30 nM 5a; 30 nM of 5p; and Pd0-resins + 30 nM of 5p. This material is available free of charge via the Internet at http://pubs.acs.org.
The opinions expressed in this publication are those of the authors and do not necessarily represent those of MSD, nor its affiliates.
The authors declare no competing financial interest.
Supplementary Material
References
- Sletten E. M.; Bertozzi C. R. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramil C. P.; Lin Q. Bioorthogonal chemistry: strategies and recent developments. Chem. Commun. 2013, 49, 11007–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertozzi C. R. A decade of bioorthogonal chemistry. Acc. Chem. Res. 2011, 44, 651–653and articles within the “Bioorthogonal Chemistry in Biology” special issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. Bioorthogonal reactions for labeling proteins. ACS Chem. Biol. 2014, 9, 16–20. [DOI] [PubMed] [Google Scholar]
- Castano A. P.; Mroz P.; Hamblin M. R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R. K.; Lin Q. Photoinducible bioorthogonal chemistry: a spatiotemporally controllable tool to visualize and perturb proteins in live cells. Acc. Chem. Res. 2011, 44, 828–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velema W. A.; Szymanski W.; Feringa B. L. Photopharmacology: beyond proof of principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. [DOI] [PubMed] [Google Scholar]
- Weiss J. T.; Dawson J. C.; Macleod K. G.; Rybski W.; Fraser C.; Torres-Sánchez C.; Patton E. E.; Bradley M.; Carragher N. O.; Unciti-Broceta A. Extracellular palladium-catalysed dealkylation of 5-fluoro-1-propargyl-uracil as a bioorthogonally activated prodrug approach. Nat. Commun. 2014, 5, 3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E.; Bertozzi C. R. Cell surface engineering by a modified Staudinger reaction. Science 2000, 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J.; Bertozzi C. R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. From mechanism to mouse: a tale of two bioorthogonal reactions. Acc. Chem. Res. 2011, 44, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Fan J.; Peng X. Colourimetric and fluorescent probes for the optical detection of palladium ions. Chem. Soc. Rev. 2013, 42, 7943–7962. [DOI] [PubMed] [Google Scholar]
- Sasmal P. K.; Streu C. N.; Meggers E. Metal complex catalysis in living biological systems. Chem. Commun. 2013, 49, 1581–1587. [DOI] [PubMed] [Google Scholar]
- Unciti-Broceta A.; Johansson E. M. V.; Yusop R. M.; Sánchez-Martín R. M.; Bradley M. Synthesis of polystyrene microspheres and functionalization with Pd(0) nanoparticles to perform bioorthogonal organometallic chemistry in living cells. Nat. Protoc. 2012, 7, 1207–1218. [DOI] [PubMed] [Google Scholar]
- Salic A.; Mitchison T. J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 2415–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streu C.; Meggers E. Ruthenium-induced allylcarbamate cleavage in living cells. Angew. Chem. Int. Ed. 2006, 45, 5645–5648. [DOI] [PubMed] [Google Scholar]
- Mascareñas J. L.; Sánchez M. I.; Penas C.; Vázquez M. E. Metal-catalyzed uncaging of DNA-binding agents in living cells. Chem. Sci. 2014, 5, 1901–1907. [PMC free article] [PubMed] [Google Scholar]
- Sasmal P. K.; Streu C. N.; Meggers E. Metal complex catalysis in living biological systems. Chem. Commun. 2013, 49, 1581–1587. [DOI] [PubMed] [Google Scholar]
- Yusop R. M.; Unciti-Broceta A.; Johansson E. M. V.; Sánchez-Martín R. M.; Bradley M. Palladium-mediated intracellular chemistry. Nat. Chem. 2011, 3, 239–243. [DOI] [PubMed] [Google Scholar]
- Li N.; Lim R. K.; Edwardraja S.; Lin Q. Copper-free Sonogashira cross-coupling for functionalization of alkyne-encoded proteins in aqueous medium and in bacterial cells. J. Am. Chem. Soc. 2011, 133, 15316–15319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer C. D.; Triemer T.; Davis B. G. Palladium-mediated cell-surface labelling. J. Am. Chem. Soc. 2012, 134, 800–803. [DOI] [PubMed] [Google Scholar]
- Michel B. W.; Lippert A. R.; Chang C. J. A reaction-based fluorescent probe for selective imaging of carbon monoxide in living cells using a palladium-mediated carbonylation. J. Am. Chem. Soc. 2012, 134, 15668–15671. [DOI] [PubMed] [Google Scholar]
- Spicer C. D.; Davis B. G. Rewriting the bacterial glycocalyx via Suzuki-Miyaura cross-coupling. Chem. Commun. 2013, 49, 2747–2749. [DOI] [PubMed] [Google Scholar]
- Eli Lilly and Company. Gemzar (Gemcitabine) Prescribing Information. http://pi.lilly.com/us/gemzar.pdf.
- Rahma O. E.; Duffy A.; Liewehr D. J.; Steinberg S. M.; Greten T. F. Second-line treatment in advanced pancreatic cancer: a comprehensive analysis of published clinical trials. Ann. Oncol. 2013, 24, 1972–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira N. M. F. S. A.; Fernández P. A.; Ramos M. J. Understanding ribonucleotide reductase inactivation by gemcitabine. Chem.—Eur. J. 2007, 13, 8507–8515. [DOI] [PubMed] [Google Scholar]
- Hazra S.; Ort S.; Konra M.; Lavie A. Structural and kinetic characterization of human deoxycytidine kinase variants able to phosphorylate 5-substituted deoxycytidine and thymidine analogues. Biochemistry 2010, 49, 6784–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccolini J.; Mercier C.; Dahan L.; André N. Integrating pharmacogenetics into gemcitabine dosing-time for a change?. Nat. Rev. Clin. Oncol. 2011, 8, 439–444. [DOI] [PubMed] [Google Scholar]
- Kocienski P. J. In Protecting Groups; Georg Thieme: Stuttgart, Germany, 1994; and references therein. [Google Scholar]
- Guibé F. Allylic protecting groups and their use in a complex environment part II: Allylic protecting groups and their removal through catalytic palladium π-allyl methodology. Tetrahedron 1998, 54, 2967–3042. [Google Scholar]
- Jarowicki K.; Kocienski P. Protecting groups. J. Chem. Soc., Perkin Trans. 1 2001, 2109–2135. [Google Scholar]
- Ramesh R.; Bhat R. G.; Chandrasekaran S. Highly selective deblocking of propargyl carbonates in the presence of propargyl carbamates with tetrathiomolybdate. J. Org. Chem. 2005, 70, 837–840. [DOI] [PubMed] [Google Scholar]
- Papageorgiou E. A.; Gaunt M. J.; Yu J.-Q.; Spencer J. B. Selective hydrogenolysis of novel benzyl carbamate protecting groups. Org. Lett. 2000, 2, 1049–1051. [DOI] [PubMed] [Google Scholar]
- Sanchez-Martin R. M.; Muzerelle M.; Chitkul N.; How S. E.; Mittoo S.; Bradley M. Bead-based cellular analysis, sorting and multiplexing. ChemBioChem 2005, 6, 1341–1345. [DOI] [PubMed] [Google Scholar]
- Unciti-Broceta A.; Díaz-Mochón J. J.; Sánchez-Martín R. M.; Bradley M. The use of solid supports to generate nucleic acid carriers. Acc. Chem. Res. 2011, 45, 1140–1152. [DOI] [PubMed] [Google Scholar]
- Dhaliwal K.; Alexander L.; Escher G.; Unciti-Broceta A.; Jansen M.; Mcdonald N.; Cardenas-Maestre J. M.; Sanchez-Martin R.; Simpson J.; Haslett C.; Bradley M. Multi-modal molecular imaging approaches to detect primary cells in preclinical models. Faraday Discuss. 2011, 149, 107–114. [DOI] [PubMed] [Google Scholar]
- Santra M.; Ko S.-K.; Shin I.; Ahnz K. H. Fluorescent detection of palladium species with an O-propargylated fluorescein. Chem. Commun. 2010, 3964–3966. [DOI] [PubMed] [Google Scholar]
- Liu B.; Wang H.; Wang T.; Bao Y.; Du F.; Tian J.; Li Q.; Bai R. A new ratiometric ESIPT sensor for detection of palladium species in aqueous solution. Chem. Commun. 2012, 48, 2867–2869. [DOI] [PubMed] [Google Scholar]
- Rambabua D.; Bhavani S.; Swamy N. K.; Rao M. V. B.; Pal M. Pd/C-mediated depropargylation of propargyl ethers/amines in water. Tetrahedron Lett. 2013, 54, 1169–1173. [Google Scholar]
- Kuo L. J.; Yang L. γ-H2AX – A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–310. [PubMed] [Google Scholar]
- Ewald B.; Sampath D.; Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol. Cancer Ther. 2007, 6, 1239–1248. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Zheng S.; Cai J.; Wang P.; Feng J.; Yang X.; Zhang L.; Ji M.; Wu F.; He N.; Wan N. Fluorescent artificial enzyme-linked immunoassay system based on Pd/C nanocatalyst and fluorescent chemodosimeter. Anal. Chem. 2013, 85, 11602–11609. [DOI] [PubMed] [Google Scholar]
- Sasmal P. K.; Carregal-Romero S.; Parak W. J.; Meggers E. Light-triggered ruthenium-catalyzed allylcarbamate cleavage in biological environments. Organometallics 2012, 31, 5968–5970. [Google Scholar]
- Wang J.; Song F.; Wang J.; Peng X. A near-infrared and ratiometric fluorescent chemosensor for palladium. Analyst 2013, 138, 3667–3672. [DOI] [PubMed] [Google Scholar]
- Bielski R.; Witczak Z. Strategies for coupling molecular units if subsequent decoupling is required. Chem. Rev. 2013, 113, 2205–2224. [DOI] [PubMed] [Google Scholar]
- Li J.; Yu J.; Zhao J.; Wang J.; Zheng S.; Lin S.; Chen L.; Yang M.; Jia S.; Zhang X.; Chen P. R. Palladium-triggered deprotection chemistry for protein activation in living cells. Nat. Chem. 2014, 6, 352–361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.