

Abstract
Much of the bone, cartilage and smooth muscle of the vertebrate face is derived from neural crest (NC) cells. During craniofacial development, the anterior neural ridge (ANR) and olfactory pit (OP) signaling centers are responsible for driving the outgrowth, survival, and differentiation of NC populated facial prominences, primarily via FGF. While much is known about the functional importance of signaling centers, relatively little is understood of how these signaling centers are made and maintained. In this report we describe a dramatic craniofacial malformation in mice mutant for the zinc finger transcription factor gene Sp8. At E14.5 they show facial prominences that are reduced in size and underdeveloped, giving an almost faceless phenotype. At later times they show severe midline defects, excencephaly, hyperterlorism, cleft palate, and a striking loss of many NC and paraxial mesoderm derived cranial bones. Sp8 expression was primarily restricted to the ANR and OP regions during craniofacial development. Analysis of an extensive series of conditional Sp8 mutants confirmed the critical role of Sp8 in signaling centers, and not directly in the NC and paraxial mesoderm cells. The NC cells of the Sp8 mutants showed increased levels of apoptosis and decreased cell proliferation, thereby explaining the reduced sizes of the facial prominences. Perturbed gene expression in the Sp8 mutants was examined by laser capture microdissection coupled with microarrays, as well as in situ hybridization and immunostaining. The most dramatic differences included striking reductions in Fgf8 and Fgf17 expression in the ANR and OP signaling centers. We were also able to achieve genetic and pharmaceutical partial rescue of the Sp8 mutant phenotype by reducing Sonic Hedgehog (SHH) signaling. These results show that Sp8 primarily functions to promote Fgf expression in the ANR and OP signaling centers that drive the survival, proliferation, and differentiation of the NC and paraxial mesoderm that make the face.
Keywords: Craniofacial development, neural crest, anterior neural ridge, SP8, FGF8, FGF17, cyclopamine, olfactory pits, signaling center
Introduction
In 1968 Wilhelm His, a Swiss embryologist, made the controversial discovery that cells could migrate from the developing neural tube and form non-neural tissues (Horstadius, 1950). These observations identified an ectodermal origin for mesenchyme and skeletal tissues traditionally believed to only arise from mesoderm. We now know that these neural crest (NC) cells contribute many components of the vertebrate face, including smooth muscle, bone, teeth and cartilage. In time two distinct models for NC development emerged. The first declared that NC cells are pre-patterned, acquiring their developmental properties prior to their exit from the neuroepithelium (Noden, 1983; Artinger and Bronner-Fraser, 1992; Hunt et al., 1998).The second argued that ongoing interactions between the migrating NC cells and their surrounding tissues drive differentiation and morphogenesis (Baker et al., 1997; Couly et al., 2002; Lee et al., 2001; Tyler and Hall, 1977). We now realize that there is a combination of initial developmental potential and continuous dynamic dialogue with flanking cells that together regulate NC developmental direction (Le Douarin et al., 2004; Noden and Schneider, 2006).
During craniofacial development there is essential crosstalk between the cephalic NC and the neuroepithelium of the developing brain. In the chick model system it has been shown that surgical extirpation of the early NC results in loss of Fgf8 expression in the anterior neural ridge (ANR) of the telencephalon, a key signaling center for both brain and NC development (Creuzet et al., 2006). FGF8 acts as a diffusible morphogen with organizer activity in the neocortex (Fukuchi-Shimogori and Grove, 2001; Toyoda et al., 2010). While the NC is required to establish this telencephalon signaling center, the resulting FGF is in turn required for proper development of the NC (Creuzet et al., 2004). FGF8 is chemotactic for NC (Sato et al., 2011), and promotes its survival and proliferation (Trumpp et al., 1999). The olfactory pit (OP) is another important signaling center during craniofacial development, and provides an additional source of FGF8 (Szabo-Rogers et al., 2009). Studies of mice with reduced FGF8 signaling further demonstrate that it is a key mediator of proper orientation and polarity of facial primordia and subsequent frontonasal skeletal morphogenesis (Griffin et al., 2013). Altogether, these studies demonstrate the essential roles of FGF8 during craniofacial development. While signaling centers are known to drive migration, survival, proliferation and differentiation of NC cells, we understand relatively little of how these signaling centers are made and maintained.
We previously reported the generation of a transgene insertional mutant mouse, named legless, that showed shortened hindlimbs, randomized laterality, and craniofacial malformations (McNeish et al., 1988). The transgene insertion resulted in a large deletion, encompassing many genes. We cloned the Lrd dynein gene from the deleted region, showed that it is expressed in the node during development, and is required for nodal cilia motility, thereby beginning to explain the randomized laterality (Supp et al., 1999; Supp et al., 1997). However, mice with a targeted mutation in Lrd did not show limb or craniofacial defects, suggesting that another altered gene was responsible (Supp et al., 1999). We subsequently showed that the transgene insertion is near, but does not delete or disrupt, the Sp8 zinc finger transcription factor gene, creating a hypomorph allele (Bell et al., 2003). Targeted null mutation of Sp8 gives a much more severe limb phenoytpe, with extreme truncation of both forelimbs and hindlimbs, as well as an absent tail (Bell et al., 2003). During limb development the apical ectodermal ridge (AER) is a signaling center that produces FGF8 and drives limb outgrowth. In the Sp8 mutants the AER fails to mature and Fgf8 expression is lost, thereby explaining the truncated limbs (Bell et al., 2003).
In addition to limb defects the Sp8 mutant mice show dramatic craniofacial malformations. In this report we describe the developmental time course of the Sp8 mutant, showing that at E14.5 the reduced facial rudiments are underdeveloped, giving an almost faceless phenotype. We also better define the expression pattern of Sp8, finding elevated levels in the anterior neural ridge (ANR) as well as the olfactory placodes and the epidermal ectoderm during craniofacial development. Further, to determine key functional domains of Sp8 expression we generated an extensive series of conditional Cre-driven compartment specific mutants. To identify possible downstream targets we globally examined altered gene expression in the Sp8 mutants in multiple facial rudiments using laser capture microdissection (LCM) and microarrays. The results indicated disrupted FGF signaling as well as altered apoptosis and cell proliferation. Perturbed pathways were further studied by in situ hybridizations, and functionally validated by genetic interaction and pharmaceutical rescue. The results show that in the developing face, as in the limb, Sp8 is required for the proper function of key signaling centers.
Materials and Methods
Mice
The floxed-Sp8 (Bell et al., 2003), FoxG1-Cre (Hebert and McConnell, 2000), Wnt1-Cre (Danielian et al., 1998), Mesp1-Cre (Saga et al., 1999), Pax3-Cre (Engleka et al., 2005) R26R-GFP, and BAT-gal (Harfe et al., 2004; Maretto et al., 2003) mice were previously described. SHH-CreGfp knock-in transgenic mice (Harfe et al., 2004) were used for genetic rescue experiments. Experiments were carried out on a mixed genetic background. All mice used in this study were housed in the Cincinnati Children’s Animal Care Facility according to NIH and institutional guidelines.
Immunofluorescence, Histology, and Skeletal Preparations
Immunofluorescence was performed as previously described (Olsson et al., 1997). Primary antibodies included goat anti-SP8 (1:8000, Santa Cruz Biotechnology), rabbit anti-GFP conjugated to Alexa488 (1:500, Invitrogen), goat anti-GFP (1:5000, Abcam), rabbit anti-SHH (1:500, Santa Cruz Biotechnology), rabbit anti-cleaved caspase 3 (1:200, Cell Signaling Technology), and rabbit anti-phosphohistone H3 (1:500, Upstate Cell Signaling Solutions). Secondary antibodies included donkey anti-rabbit or donkey anti-goat conjugated antibodies conjugated to either Alexa555 or Alexa488 (1:200, Invitrogen). The processing and histological staining of embryonic tissue was performed as previously described (Little et al., 2007). Skeletal Staining using Alizarin red and Alcian blue was performed as previously described (Kuczuk and Scott, 1984). Inner Canthal Distance was measured using Leica LAS v3.8 imaging software. The student’s two-tailed t test was used to determine differences between paired groups. A p-value of less than 0.05 was considered significant.
Quantification of Cell Death and Cell Proliferation
Cells undergoing apoptosis were labeled using rabbit anti-cleaved caspase 3 (1:200, Cell Signaling Technology) and cells undergoing proliferation were labeled using rabbit anti-phosphohistone H3 (1:500, Upstate Cell Signaling Solutions). NC cells were labeled by Wnt1-Cre driving the expression of the Rosa26-flox-stop-flox-GFP reporter. Neuroepithelium and OP epithelium were determined by cell morphology and location within the section. E8.5, E9.5, and E10.5 embryos were harvested, fixed overnight in 4% PFA at 4°C, embedded in OCT, and cryosectioned at 10 microns. Fluorescent images of the cranial region were taken at 5X, 10X, or 20X magnification. Comparable WT and Sp8 mutant sections were used during quantification, approximately 5 sections per embryo were analyzed (Table S1). The cells of interest undergoing either apoptosis or proliferation were manually scored using Image J software. The student’s two-tailed t test was used to determine differences between paired groups. A p-value of less than 0.05 was considered significant.
Whole-Mount In Situ Hybridization
Whole Mount in situ hybridization was performed as previously described (Bell et al., 2003; Little et al., 2007). Mouse cDNA probes were kindly provided by Sheila Bell (Fgf8 and Erm) (Bell et al., 2003) and Rulang Jiang and Yu Lan (Dlx5) (Baek et al., 2011). The Fgf17 riboprobe was made from pCR4-TOPO vector clone ID number 8733782 using forward primer CTGTATGAACAAGAGGGGCAA and reverse primer GAAATTAATACGACTCACTATAGGCTGCCTCTGGCCTCAAACCT (with T7 RNA Polymerase binding site). The Sprouty1 riboprobe was made from liver genomic DNA using forward primer GAGATAATACGACTCACTATAAAGCCATCAGAGGCAGCAAT (with T7 RNA Polymerase binding site) and reverse primer GAGAATTTAGGTGACACTATAGGTGGGGTCCTCTTTCAAGG (with SP6 RNA Polymerase binding site).
Western Blotting
We manually dissected the middle to anterior facial structures including the ANR from E9.5 embryos. The western blotting was performed as previously described (Chang et al., 2012). Primary Antibodies used were GLI3 (1:1000, R&D), β-Actin (1:10,000, Abcam), and α-Tubulin (1:10,000, Abcam). Secondary Antibodies used were Anti-Goat-HRP conjugated (1:5000, Santa Cruz Biotechnology) and Anti-Mouse-HRP (1:5000, Bio-Rad). The relative densities of western blots were calculated using Image J software. The student’s two-tailed t test was used to determine differences between paired groups. A p-value of less than 0.05 was considered significant.
RNA purification and Q-PCR Analysis
We manually dissected the cranial neural folds from E8.5 embryos, the middle to anterior facial structures including the ANR from E9.5 embryos, and the OP, LNP and MNP from E10.5. Total RNA was purified using the ZR RNA Microprep Kit (Zymo Research). Gene expression was determined by quantitative real-time Q-PCR using the SYBR Green PCR Master Mix (Bio-Rad) and the CFX96 Real Time thermocycler (Bio-Rad). Specific primer pairs were designed to detect target gene expression levels. Gene expression levels were normalized to β-actin or Tubbβ5. Primers used for Q-PCR:
| Gene | Forward Strand | Reverse Strand | Amplicon Size (bp) |
|---|---|---|---|
| Fgf8 | TTGGAAGCAGAGTCCGAGTT | ATACGCAGTCCTTGCCTTTG | 108 |
| Fgf17 | TCGTGGAGACAGATACATTCG | GCTTGCCCCTCTTGTTCATA | 87 |
| Gli1 | GCCTTGAAAACCTCAAGACG | ATGGCTTCTCATTGGAGTGG | 144 |
| Gli2 | ACCATGCCTACCCAACTCAG | CCTCAGCCTCAGTCTTGACC | 145 |
| Gli3 | GAGCAGAAACCGTTCAAAGC | TGGGTTTTCAGGTTTTCGAG | 131 |
| Ptch1 | CCACGGTTGCTGTAGATTGT | GCCGCAGTTCTTTTGAATGT | 122 |
| Shh | CCAATTACAACCCCGACATC | GGCCAAGGCATTTAACTTGT | 98 |
| Smo | TCAATGCGTGTTTCTTCGTG | ACAGGGTCTCACTGGAGGTG | 129 |
| Smo | TCGGGCAAGACATCCTATTT | CTCCATCTACCTGAGCCACA | 91 |
| β-Actin | AATGAGGCTGGTGATAAGTGG | AAGTTCAGTGTGCTGGGAGTC | 99 |
| Tubbβ5 | GCAGTTGGAGAAAGCTGAGG | GGAAGAGGATTTCGGAGAGG | 97 |
Laser Capture Microdissection, Microarray Hybridization, and Data Analysis
Tissues were embedded, sectioned, and microdissected as previously described (Brunskill et al., 2008). RNA purification, target amplification, and microarray data analysis was performed as previously described (Brunskill et al., 2011). Microarray data is available on the FACEBASE.ORG public data Website, listed as Sp8−/− E10 LNE, Sp8−/− E10 LNE2, Sp8−/− E10 LNE3, Sp8−/− E10 MNE, Sp8−/− E10 MNE2, Sp8−/− E10 MNE3, Sp8−/− E10 OP, Sp8−/− E10 OP2, Sp8−/− E10 OP3, and corresponding wild type samples.
Embryonic Exposure to Cyclopamine
Cyclopamine (Selleck, S1146) was dissolved in 0.1M Sodium Citrate/Phosphate pH3 containing 30% 2-hydropropyl-b-cyclodextrin (HPBCD) (Sigma 332607) as previously described (Lipinski et al., 2008; Lipinski et al., 2010). Cyclopamine was administered using Alzet Microosmotic Pump model 2001D (8 μl/hour for 1 day) (Durect Corp, Cupertino, CA). The microosmotic pumps were implanted subcutaneously under the back skin of the pregnant dam at E8.25 and were removed at E10.5. Pumps were filled with 2 mg cyclopamine/100 ul 30% HPBCD to achieve approximate 160 mg/kg/day (one pump implanted) or 260 mg/kg/day (two pumps implanted) cyclopamine release rates (Lipinski et al., 2008; Lipinski et al., 2010). In separate experiments Cyclopamine was administered by single IP injection at 25 mg/kg or 50 mg/kg dissolved in 45% HPBCD (Sigma, H5784). The injected volume was approximately 150 μl and administered every 12 hours from E7.5-E10.0 (Lipinski et al., 2008).
Results
Sp8 is required for normal craniofacial development
Sp8−/− mutant mice showed dramatic craniofacial malformations (Fig. 1). At E14.5 they displayed hypertelorism, severe midline defects and an absence of many normal facial structures (Fig. 1A,B). The tongue and mandible appeared normal, but the medial nasal prominences (MNP) remained severely underdeveloped and were not merged with other facial elements. The lateral nasal prominences (LNP) and maxillary prominences (MXP) were more normal in size, but also failed to undergo normal development, giving the face a very primitive appearance, resembling that of a much younger embryo. The MXP ordinarily gives rise to the upper jaw, sides of the face, and secondary palate, while the LNP produces the lateral walls of the nose, and the MNP gives rise to more medial structures (Helms et al., 2005). To better define the developmental progression of Sp8 mutant malformations we examined embryos from E8.5 to birth. The earliest gross evidence of craniofacial defect was seen at E9.5, with failure of anterior neuropore closure (data not shown). At E10.5, E11.5 and E12.5 all prominences were formed in mutants, but the MNP and LNP were dramatically reduced in size and underdeveloped (Fig. 1G–R).
Figure 1. Sp8 is required for proper nasal prominence morphogenesis.

(A,B) E14.5 Sp8 mutant embryos (B) display severe failure of midline fusion and a loss of many facial structures compared to WT (A).
(C–F) Wild type (C,E) and Sp8 mutant (D,F) skeletal preparations show loss of many NC derived bones in mutants, including the maxilla, palate, and frontal bone. Paraxial mesoderm-derived parietal and interparietal bones are also absent in Sp8 mutants. Dotted circle in panel F indicates a hole in the skull where the brain is exposed in the Sp8 mutant.
(G–R) Facial prominences were digitally pseudo-colored red (maxillary prominence), blue (lateral nasal prominence), green (medial nasal prominence), and purple (frontonasal region) in E10.5, E11.5, and E12.5 WT (G,I,K,M,O,Q) and Sp8 mutants
(H,J,L,N,P,R). Mutant lateral and medial nasal prominences are reduced in size and dysmorphic in shape.
Exoccipital, EO; Frontal, FR; Interparietal, IP; Lateral Nasal Prominence, LNP; Mandible, MD; Maxilla, MX; Medial Nasal Prominence, MNP; Nasal, NS; Neuroepithelium, NE; Parietal, PR; Premaxilla, PMX; Supraoccipital, SO; Tongue, T
Skeletal and cartilage preparations of Sp8 mutants at E18.5 showed loss or malformation of multiple NC-derived bones. The palate, maxilla, premaxillary and frontal bones, all NC derivatives, were absent or reduced in size and misshapen. Interestingly, there was also a reduction of the paraxial mesoderm derived parietal, interparietal, and supraoccipital bones (Fig. 1C–F). The result was a large hole in the skull between the residual frontal and parietal bones, where the excencephalic brain was exposed.
We also investigated palate development in the Sp8 mutants. The palatal shelves normally develop vertically aligned next to the tongue at E13.5, and then elevate and fuse to form the palate by E16.5 (Bush and Jiang, 2012). Histological analysis revealed that Sp8 mutant palatal shelves were reduced in size, prematurely elevated and failed to fuse along the midline even at E16.5, resulting in a cleft palate (Fig. S1).
Sp8 is expressed in craniofacial signaling centers
Although Sp8 expression has been previously examined (Bell et al., 2003; Kawakami et al., 2004; Sahara et al., 2007; Waclaw et al., 2006), it has not been well defined during early craniofacial development. Immunostain analysis at E8.5 showed that the Sp8 gene was expressed in the neuroectoderm and epidermal ectoderm, with particularly strong expression in the critical ANR signaling center (Helms et al., 2005)(Fig. 2A,C; Fig. S2). Of interest, Sp8 expression was not detected in the NC or paraxial mesoderm, which together make up the facial mesenchyme. Similar to the E8.5 time point, at E9.5 Sp8 showed robust expression in the ANR, and also in the hindbrain neuroepithelium (Fig. 2B,D). At E10.5 Sp8 was expressed in the OP epithelium, facial epidermal ectoderm, and robustly in the midline of the telencephalon (Fig. 2E). Similar to the ANR, the OP epithelium is a signaling center promoting survival and patterning of the underlying NC and mesoderm derived mesenchyme (Kawauchi et al., 2005; Szabo-Rogers et al., 2009). These results show that Sp8 is expressed in signaling centers that drive craniofacial development, and not detectably in the NC and paraxial mesoderm cells that directly contribute to the face. This is similar to what we previously observed in the developing limb, where Sp8 is expressed in the AER signaling center (Bell et al., 2003), but not in the mesenchyme that makes the muscles and bones of the limbs.
Figure 2. SP8 is made in the anterior neural ridge and olfactory pit signaling centers.

(A,C) Immunofluorescence for SP8 on transverse sections of E8.5 embryos at 9 somites showed high SP8 levels in the hindbrain, anterior neural ridge (arrows), and epidermal ectoderm.
(B,D) Sagittal sections at E9.5 revealed SP8 localization in the anterior neural ridge (arrow) and hindbrain.
(E) Coronal section at E10.5 showed robust SP8 localization in the olfactory pit epithelium signaling center (arrows) and medial telencephalon (arrowhead).
First Branchial Arch, 1BA; Foregut, FG; Forebrain, FB; Hindbrain, HB; Lateral Nasal Prominence, LNP; Medial Nasal Prominence, MNP; Midbrain, MB; Olfactory Pit, OP
Conditional deletion of Sp8 reveals critical roles in signaling centers that flank facial mesenchyme
To better define the critical domains of Sp8 expression during craniofacial development we made mice with compartment specific Sp8 deletion. We used a Cre mediated conditional knock-out strategy to inactivate Sp8 expression in the NC, paraxial mesoderm, neuroepithelium, and epidermal ectoderm. To begin, we better characterized the expression patterns of the Wnt1-Cre, Pax3-Cre, Mesp1-Cre and Foxg1-Cre strains of mice by crossing with a floxed-stop-Rosa26-GFP reporter mouse. Although each transgenic Cre line was previously described (Danielian et al., 1998; Engleka et al., 2005; Hebert and McConnell, 2000; Saga et al., 1999), we were interested in defining precise spatiotemporal expression domains during craniofacial development. The results showed Pax3-Cre expression at E8.5 in the NC, paraxial mesoderm, hindbrain neuroepithelium, and in the anterior junction of the neuroepithelium and epidermal ectoderm at the tips of the neural folds (Fig. S3A). At E9.5 Pax3-Cre expression continued in the ANR, as well as more caudal neuroepithelium (Fig. S3E). At E10.5 Pax3-Cre was expressed in both the OP epithelium and underlying facial mesenchyme, consisting of NC and paraxial mesoderm (Fig. S3I). Importantly, Pax3-Cre expression overlapped Sp8 expression in the E8.5 ANR and in the E10.5 OP epithelial signaling centers. Wnt1-Cre showed expression in the mid and hindbrain neuroepithelium, and NC, but not in the ANR or OP (Fig. S3B,F,J). Foxg1-Cre was expressed throughout the neuroepithelium and epidermal ectoderm (Fig. S3C,G,K). At E8.5 however, expression appeared weak, with mosaic reporter expression (Fig. S3C). Later, at E10.5, FoxG1-Cre drove robust reporter expression in forebrain neuroepithelium and the OP epithelium (Fig. S3K). Mesp1-Cre was expressed in the paraxial mesoderm (Fig. S3D,H,L). See Table S2A for a summary of all transgenic Cre domains of expression.
We then used each Cre strain in combination with a floxed Sp8 allele to create tissue specific conditional mutants. The Wnt1-Cre, driving Sp8 deletion in the NC, gave no detectable craniofacial phenotype (Fig. 3M,P,S,V). This was perhaps not surprising, since there was no Sp8 expression observed in NC. Nevertheless, it confirmed that there was no functionally important low-level NC expression for Sp8. For nine of ten Mesp1-Cre conditional Sp8 mutants, with paraxial mesoderm specific deletion, there were no craniofacial defects, while one mutant showed a partial facial midline cleft (Fig. 3N,Q,T,W). Consistent with the expression analysis, these results argue that Sp8 expression is not required directly in the NC and paraxial mesoderm cells that make the face.
Figure 3. Conditional Inactivation of Sp8 expression reveals important roles of Sp8 in Pax3-Cre and FoxG1-Cre expressing cells.
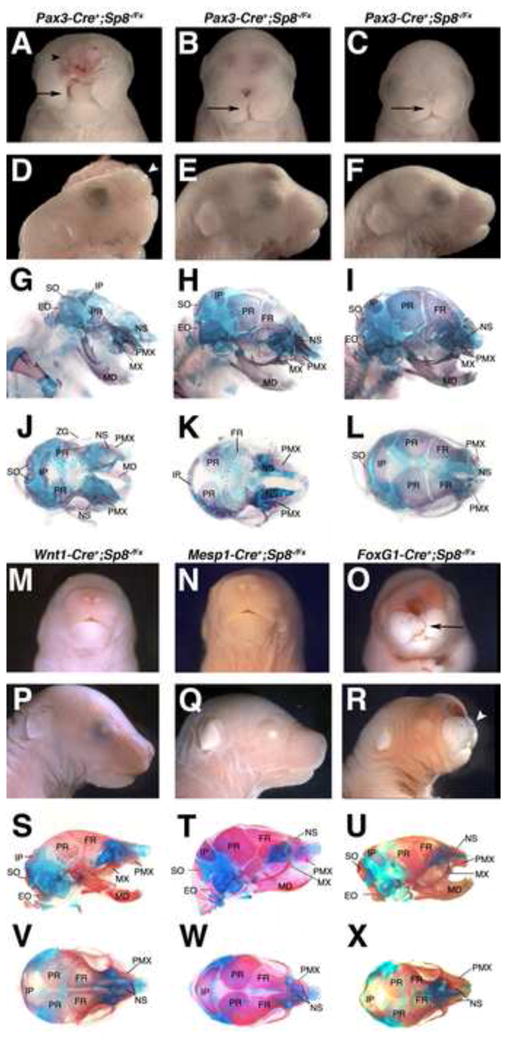
Transgenic Cre lines selectively removed Sp8 expression in specific tissue compartments when combined with one floxed and one null Sp8 allele. Whole mount and skeletal preparations were performed.
(A–L) Pax3-Cre conditional mutants displayed variable phenotypes. Embryos with severe malformations including excencephaly (black arrowhead), failure of facial prominence fusion along the midline (arrow), truncation of the anterior snout structures (white arrowhead), and an absence of many cranial bones closely resembled the Sp8 null mutant (n=4, A,D,G,J). The moderate phenotypes displayed cleft lip and palate, failure of facial prominence fusion along the midline (arrows), and a truncation of anterior structures (B,C, E,F, H,I, K,L).
(O,R,U,X) The FoxG1-Cre conditional mutants displayed malformations of the telencephalon, cleft lip (arrow), and a truncation of anterior facial structures (arrowhead).
(M,N, P,Q, S,T, V,W) Neither the Wnt1-Cre (M,P,S,V) nor the Mesp1-Cre (N,Q,T,W) conditional mutants displayed craniofacial phenotypes with the exception of one Mesp1-Cre mutant displaying a mild midline defect (not shown).
Exoccipital, EO; Frontal, FR; Interparietal, IP; Mandible, MD; Maxilla, MX; Nasal, NS; Parietal, PR; Premaxilla, PMX; Supraoccipital, SO
The Pax3-Cre conditional mutants showed the most severe phenotypes, closely resembling the Sp8 null mutants, with excencephaly, an absence of frontal and parietal bones, and a dramatic midline cleft (n=5, Fig. 3A,D,G,J). There was, however, variable expressivity. The moderate phenotype Pax3-Cre conditional mutants displayed cleft lip and palate, midline defects, and a truncation of anterior facial structures (n=7, Fig. 3B,C,E,F,H,I,K,L). In addition, five Pax3-Cre conditional mutants did not show a detectable craniofacial phenotype.
The FoxG1-Cre conditional mutants also gave variable craniofacial malformations, although none as striking as the most severe Pax3-Cre conditional mutants. The moderate FoxG1-Cre mutants displayed cleft lip and/or palate, failure of facial midline fusion (n=13), and a truncation of anterior facial structures (n=13, Fig. 3O,R,U,X). See Table S2B for a summary of transgenic Cre phenotypes observed.
Taken together these results indicate that Sp8 expression is critically important in the neuroepithelium and the epidermal ectoderm, including the OP, which flank the NC. It is interesting to note that the most severe phenotypes were observed with Pax3-Cre, which gave more robust E8.5 early expression than Foxg1-Cre, although in more restricted domains of the neuroepithelium.
Laser capture microdissection/microarray analysis of Sp8 mutants
To globally define the altered gene expression state of the Sp8−/− mutants we used LCM coupled with microarrays. We examined E10.5 wild type and mutant embryos using LCM to isolate three major compartments of the developing face, the LNP, MNP, and OP epithelium (Fig. S4G–J). Affymetrix mouse Gene 1.0 ST arrays were used for gene expression profiling. While large numbers of differentially expressed genes were found (Tables S3–5), three differences particularly stood out. The first was a significant down regulation of Fgf17 in the mutant OP epithelium. As discussed previously, FGFs are known to be key regulators of craniofacial development, and the tissue specific targeting results suggested that Sp8 expression in the olfactory epithelium is important for proper craniofacial development. The microarray data also strongly suggested increased levels of apoptosis in mutants, with both the LNP and the MNP showing about 30 genes involved in the regulation of cell death showing elevated expression in mutants. Finally, gene ontology analysis of the array data showed a dramatic cell proliferation signature. In the mutant LNE there were 43 cell cycle process genes with elevated expression, including four (Msh2, Men1, Ppm1q and Ilk) involved in cell cycle arrest. Similarly, the mutant MNP showed upregulation of 71 genes involved in the cell cycle, again with four (Msh2, Men1, Ppm1q and Sesn1) involved in cell cycle arrest.
Sp8−/− neural crest show increased apoptosis and reduced proliferation
Several results suggested an increase in apoptosis and/or decrease in cell proliferation in the Sp8 mutants, including the reduced sizes of the facial prominences during development, the striking absence of many NC and mesoderm derived cranial bones at birth, and the LCM/microarray results. We therefore monitored apoptosis by quantifying the number of Cleaved Caspase 3 positive cells in wild type and Sp8−/− mutant embryos. We identified NC cells by labeling with Wnt1-Cre activation of floxed-stop-Rosa26-GFP. At E8.5 there was a striking 3-fold increase in apoptosis in the anterior neuroepithelium of the cranial neural folds, which includes the ANR, but no elevated apoptosis in the NC (Fig. 4A and Fig. 4SA,B). This extends previous results showing elevated neuroepithelium apoptosis in Sp8−/− mutants at later developmental time points (Waclaw et al., 2006; Zembrzycki et al., 2007). Further, at E9.5 there was a 4-fold increase in apoptosis in the NC of the facial mesenchyme in Sp8−/− mutants (Fig. 4A and Fig. S4C,D). Similarly, at E10.5 apoptosis was significantly increased in both the MNP, LNP and OP (Fig. 4A and Fig. S4E,F).
Figure 4. Increased apoptosis and reduced proliferation in SP8−/− facial mesenchyme.
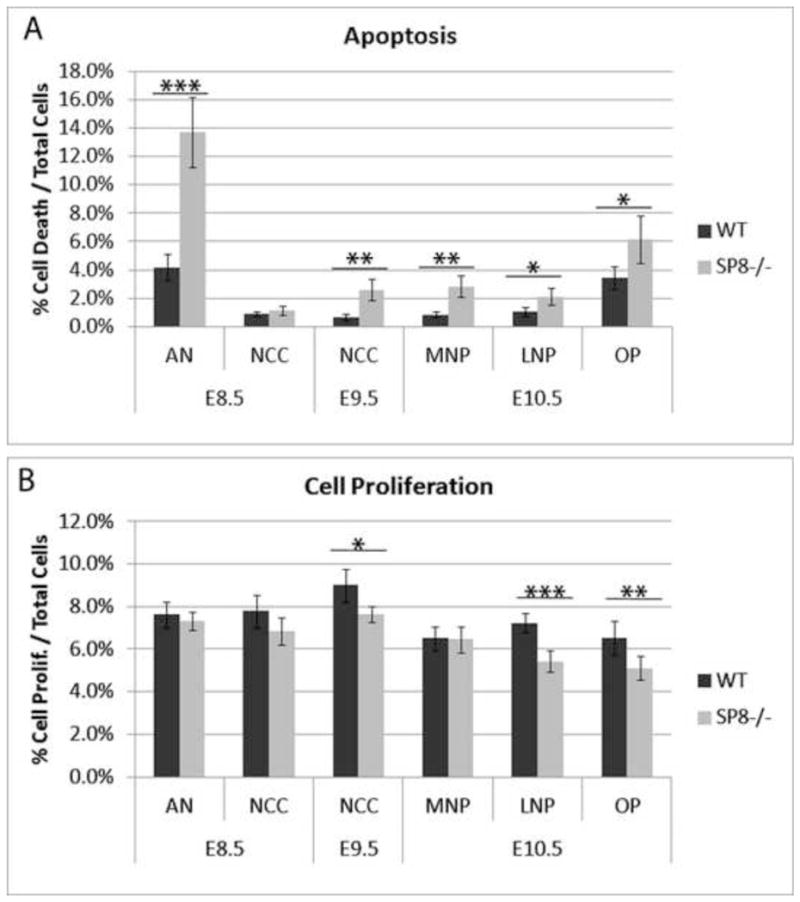
(A) Immunofluorescence for cleaved caspase 3 was used to label cells undergoing apoptosis. Wnt1-Cre activation of R26R-GFP labeled the neural crest. Apoptosis was quantified in various compartments of E8.5, E9.5, and E10.5 Sp8−/− and WT embryos. Results showed a significant increase in apoptosis in the anterior neuroepithelium at E8.5. The neural crest underwent elevated apoptosis at E9.5 as did the medial nasal prominence, lateral nasal prominence, and olfactory pit at E10.5.
(B) Immunofluorescence for phospho-histone H3 was used to label cells undergoing proliferation. The neural crest was labeled by Wnt1-Cre activation of R26R-GFP. Proliferation was quantified in various compartments of E8.5, E9.5, and E10.5 of Sp8−/− and WT embryos. Results showed a significant decrease in the neural crest at E9.5 as well as in the lateral nasal prominence and olfactory pit at E10.5.
Anterior Neuroepithelium, AN; Lateral Nasal Prominence, LNP; Medial Nasal Prominence, MNP; Neural Crest Cells, NCC; Olfactory Pit, OP
p<0.05 (*), p<0.01 (**), p<0.001 (***)
We examined cell proliferation in the Sp8−/− mutants by immunostaining for phosphohistone H3. Interestingly, there was a significant reduction of cell proliferation in the NC at E9.5, but not at E8.5 (Fig. 4B). At E10.5 there was a significantly reduced cell proliferation rate in the OP and LNP, but not the MNP. Together these results suggest that a combination of increased apoptosis and reduced cell division contribute to the dramatic craniofacial phenotype of the Sp8−/− mutants.
Reduced Fgf8 and Fgf17 expression in Sp8−/− mutant signaling centers
In situ hybridizations and Q-PCR were used to validate LCM/microarray results. Wild type embryos expressed Fgf17 in the ANR at E9.5 and along the midline and in the OP epithelium at E10.5 (Fig. 5D, E). Fgf17 was dramatically down regulated in the Sp8 mutant ANR at E9.5 and in the midline and OP epithelium at E10.5 (Fig. 5D, E). Q-PCR confirmed significant reduction of Fgf17 expression in E8.5 neural folds, E9.5 ANR, and E10.5 OP (Fig. 5G).
Figure 5. Disrupted Fgf8, Fgf17 and Gli3 expression in Sp8−/− embryos.
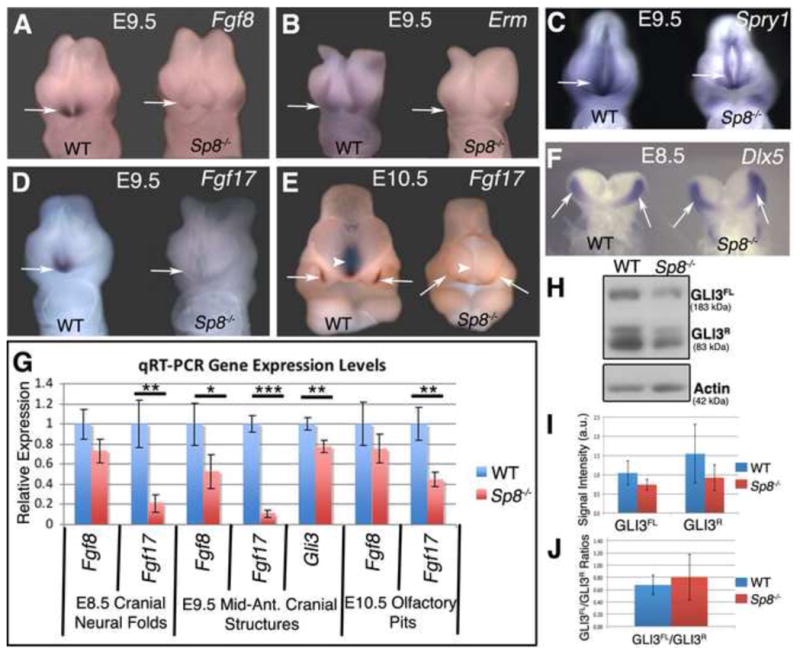
(A–E) In-situ hybridization revealed a down-regulation of Fgf17, Fgf8, and FGF8 downstream targets Erm and Spry in E9.5 Sp8 mutant anterior neural ridge (arrows) (AD). Fgf17 expression was also lost along the midline (arrowhead) and in the olfactory pit (arrows) at E10.5 (D).
(F) In-situ hybridization showed no change in expression of the E8.5 anterior neural ridge marker Dlx5 in E8.5 Sp8 mutants (arrows).
(G) Q-PCR analysis showed a down-regulation of Fgf17 at E8.5, E9.5, and E10.5. Additionally at E9.5, Gli3, and Fgf8 expression levels were significantly decreased in mutants.
(H–J) Western blot analysis (H) and relative levels (I) of GLI3 full length (transcriptional activator) and GLI3 repressor proteins showed a reduction in GLI3 full length and GLI3 repressor, although the change was not statistically significant. No significant change in the ratio of GLI3 full length to repressor levels was detected (J).
p<0.05 (*), p<0.01 (**), p<0.001 (***)
We also examined the expression pattern of Fgf8 in Sp8 mutants. As previously noted, Sp8 functions to maintain Fgf8 expression in the AER during limb bud development (Bell et al., 2003). In addition, previous studies showed that SP8 can directly bind and activate the Fgf8 promoter (Sahara et al., 2007), making it an excellent candidate downstream target. In situ hybridizations showed that Fgf8 expression was indeed down regulated in the Sp8−/− mutant ANR at E9.5 (Fig. 5A). Erm and Sprouty are downstream targets of FGF8 signaling and both Erm and Sprouty expression were also reduced in the mutant ANR (Fig. 5B,C). Quantification by Q-PCR confirmed that Fgf8 transcripts were significantly down regulated at E9.5, but not at E10.5 or E8.5, consistent with in situ hybridization and microarray results (Fig. 5G).
Due to the increased apoptosis in the Sp8 mutant NC and neuroepithelium, we analyzed the expression of an ANR marker, Dlx5, to determine whether the cells that make up the ANR are present or absent in the mutant. In-situ analysis of Dlx5 expression showed E8.5 ANR specific expression, which was not lost in the mutant (Fig. 5F). Therefore, the persistence of Dlx5 expression in the E8.5 mutant ANR together with the reduced, but not absent expression of Erm and Sprouty suggested that the cells that make up the ANR are not completely absent.
SHH and WNT signaling in Sp8−/− mutants
We also investigated SHH signaling in the Sp8 mutants, which showed a hypertelorism phenotype, with abnormally wide spacing between the eyes. A loss of SHH signaling results in cyclopia, while a gain of SHH signaling gives rise to hypertelorism (Belloni et al., 1996; Hu and Helms, 1999; Hu and Marcucio, 2009; Lipinski et al., 2010). In addition there is evidence that FGF and SHH signaling can cross regulate. In the chick system ectopic SHH in the forebrain inhibits FGF8 expression (Hu and Marcucio, 2009), while loss of Fgf8 expression in the ANR results in an expansion of Shh expression (Creuzet et al., 2006). Further, the FGF8 downstream targets Pea3 and Erm have been shown to inhibit Shh expression during limb development (Lettice et al., 2012; Mao et al., 2009; Zhang et al., 2009). Therefore both the hypertelorism phenotype and the reduced FGF expression in the Sp8 mutants were consistent with possible elevated SHH signaling.
We examined multiple components of the SHH signaling pathway using in situ hybridizations, Q-PCR, immunostain, transgenic reporter mice, and western blot techniques. No significant differences in SHH expression were detected in Sp8 mutants at E8.5, E9.5 or E10.5 by immunofluorescence or using the Ptch-lacZ SHH reporter mouse (Fig. S5B-E and data not shown). Q-PCR analysis of manually dissected E10.5 combined MNP, LNP and OP revealed no change in the SHH signaling components Shh, and Ptch1 (Fig. S5F). Likewise, Q-PCR analysis of cranial neural folds at E8.5 showed no change in Shh, Ptch1, Smo, Gli1, Gli2 or Gli3 (Fig. S5F). However, Gli3 mRNA levels were significantly down regulated in the E9.5 ANR and telencephalon (Fig. 5G). GLI3 is a zinc finger transcription factor that mediates SHH signal. Full length GLI3 functions as a transcriptional activator, while the cleaved form is a transcriptional repressor (Dai et al., 1999; Liu et al., 2005; Sasaki et al., 1999). To further define the perturbed Gli3 expression we performed western blot analysis. The results showed no significant change in the ratio of cleaved to full length forms suggesting that GLI3 processing is not affected in the mutants (Fig. 5H–J). The western blot results, although not as quantitative as Q-PCR, were nevertheless consistent with reduced Gli3 expression. It is interesting to note that Gli3 mutations in humans can cause Greig cephalopolysyndactyly syndrome (GCPS), associated with hypertelorism and polydactyly (Vortkamp et al., 1991). GCPS is the result of reduced Gli3 dosage and is found in heterozygotes with one wild type and one null allele. Nevertheless, in mice even the homozygous loss of Gli3 is not associated with hypertelorism (Veistinen et al., 2012).
In addition we used the Bat-gal reporter mice to analyze WNT signaling in mutants and no change was detected (Fig. S5A).
Inhibition of Hedgehog signaling partially rescues craniofacial development in the Sp8−/− face
Due to the hypertelorism phenotype and reduced Gli3 expression we hypothesized that the elevated SHH signaling contributes to the Sp8 mutant craniofacial phenotype. To test this we first reduced SHH signaling by deleting one wild type Shh allele in Sp8 mutants. The resulting Sp8−/−;Shh+/− mice displayed a partially rescued craniofacial phenotype (n=4). The medial facial prominences partially merged with the lateral and maxillary prominences to form a recognizable snout by E18.5 (Fig. 6A-C). Sp8−/−;Shh+/+ embryos had a significant increase in the inner canthal distance (ICD), a measure of hypertelorism, of 6.1mm ±0.45mm compared to wild type ICD of 4.4 mm ±0.09mm (p=1.56E-06). However, the mutant hypertelorism was partially and significantly rescued to 5.33mm ±0.26mm in Sp8−/−;Shh+/− embryos (p= 9.3E-02). Despite the rescue of anterior structures, the more dorsal structures of the skull and forehead were not rescued.
Figure 6. Reduction of SHH signaling partially rescued Sp8 craniofacial malformations.
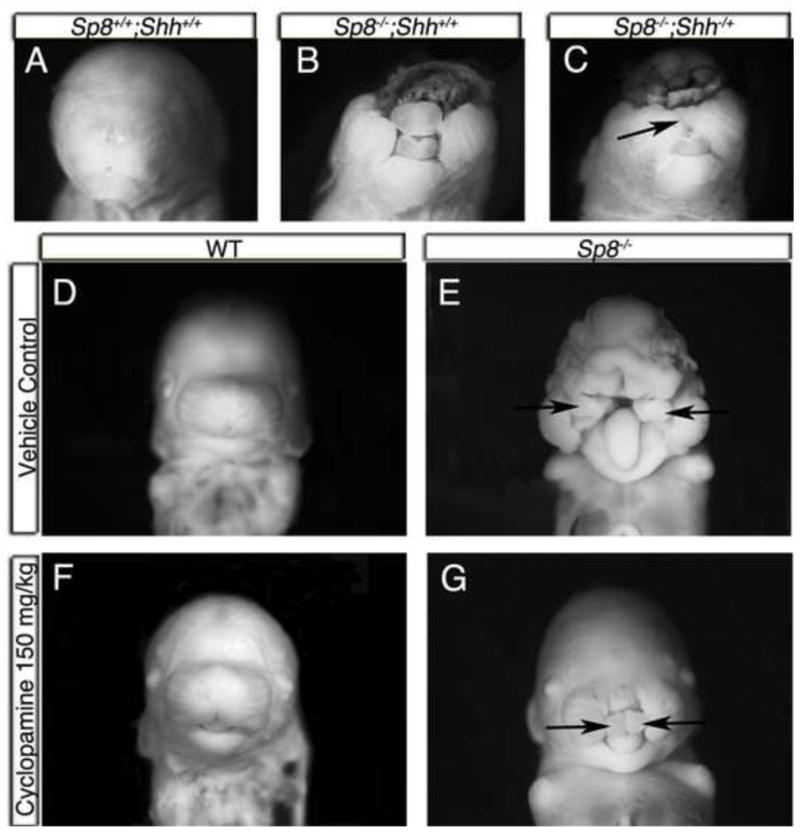
(A–C) SHH levels were reduced in Sp8 mutants by deleting one allele of Shh. Sp8−/−;Shh−/+ mutants displayed partial craniofacial rescue of anterior, but not dorsal, structures at E18.5 (n=4, arrow in C).
(D–G) SHH signaling was reduced through embryonic exposure to cyclopamine. Wild type embryos appeared normal after exposure to 150 mg/kg cyclopamine (F).
Approximately one-third (n=13) of the cyclopamine treated mutants (n=41) displayed a partial rescue of facial midline (arrows), excencephaly, and hypertelorism (G). Sp8−/− mutants exposed to vehicle alone showed the standard mutant phenotype (E).
To further interrogate SHH signaling during craniofacial development in Sp8 mutants, we used a SHH signaling inhibitor, cyclopamine, to reduce HH signaling in-utero. Cyclopamine was initially administered via intraperitoneal (IP) injection twice per day from E7.5-E10.0. The result was one mutant showing a partial rescue with the facial prominences being closer together than the unexposed mutants (Fig. S6). Due to the low penetrance of the rescue via IP injection (1 rescue/10 total exposed mutants), we used microosmotic pumps to provide a more effective method of delivering cyclopamine. Microosmotic pumps implanted subcutaneously in pregnant mice were used to release cyclopamine continuously for 24 hours from E8.0-E9.0. Mutants exposed to cyclopamine displayed a robust, but partial, rescue of the midline at E14.5 (n=13, Fig. 6D–G). The rescue resulted in a much more normal looking face (Fig. 6G versus 6E), with improved midfacial development, although the MNPs remained distinct. The degree of hypertelorism was measured by ICD in the cyclopamine exposed litters. The ICD of cyclopamine exposed embryos that showed no rescue (3.68mm ±0.05mm) was partially and significantly rescued to 3.26mm ±0.12mm in the rescued mutants (p=2.18E-04). It is important to note that the severe craniofacial phenotype of untreated Sp8 mutants is near 100% penetrant. We have not observed any mice resembling the partial rescue phenotypes in several hundred control Sp8−/− mutants. The cyclopamine doses used were relatively low (up to 260 mg/kg/day), and did not result in detectable phenotypes in any WT littermates. There was variable response of the mutant embryos to the cyclopamine, with many (n=28) still showing the typical Sp8 mutant phenotype. We suspect this variability was the result of varying cyclopamine concentrations along the length of the uterus.
Discussion
In this report we show that mutation of the Sp8 zinc finger transcription factor gene results in a striking craniofacial phenotype, with the multiple facial prominences severely reduced in size and underdeveloped, even at E14.5. Interestingly, however, the Sp8 gene did not show detectable expression in the NC and paraxial mesoderm cells that together will construct the bone, muscle and cartilage of the face. Instead, Sp8 showed elevated expression in the neuroepithelium and epidermal ectoderm, which are known to provide signaling centers that drive craniofacial development.
Cre mediated compartment specific deletion of Sp8 confirmed its function in signaling centers. Wnt1-Cre and Mesp1-Cre, used to remove Sp8 function in the NC and paraxial mesoderm respectively, gave no detectable craniofacial malformations. Of interest, Wnt1-Cre also showed strong activity in the neuroepithelium of the hindbrain, even at the earliest time point monitored, E8.5. The wild type appearance of the Wnt1-Cre;Sp8flox/− mouse indicated that the hindbrain expression of Sp8 was not required for normal craniofacial development.
Pax3-Cre was active in the neuroepithelium of the forebrain, as well as the epidermal ectoderm and OP of the developing face. In addition it was expressed in the NC cells, but the Wnt1-Cre results showed that any low level Sp8 expression present in these cells was not functionally significant. The Pax3-Cre;Sp8flox/− mice showed the most severe craniofacial phenotypes, suggesting important roles for Sp8 expression in the forebrain, OP and epidermal ectoderm. The Foxg1-Cre results confirmed this conclusion, although the resulting phenotypes were less severe, perhaps due to somewhat delayed and more mosaic expression compared to Pax3-Cre.
Expression analysis, using LCM/microarray, in situ hybridization, and Q-PCR showed reduced expression of Fgf8, Fgf17, Erm and Spry in the Sp8 mutant E9.5 ANR. In addition, Fgf17 expression in the E10.5 medial forebrain neuroepithelium and olfactory pit was dramatically reduced in mutants. Both FGF8 and FGF17 have been shown to drive proliferation and differentiation of neural cells (Xu et al., 2000). The connection between FGF8 and cell proliferation is particularly strong (Lee et al., 1997; MacArthur et al., 1995; Shamim et al., 1999; Xu et al., 2000). Indeed, it has been shown that FGF signaling is important at many stages of NC development, including emigration from the neural tube (Martinez-Morales et al., 2011), driving cartilage outgrowth (Abzhanov and Tabin, 2004), providing chemotactic attraction (Sato et al., 2011), survival (Trumpp et al., 1999) and induction (Monsoro-Burq et al., 2003). A recent report of mice with reduced FGF8 signaling exquisitely defined a number of craniofacial abnormalities approximating those seen in Sp8 mutants (Griffin et al., 2013). These included reduced MNPs, midline clefts, shallow OP, and a dramatic midfacial cleft of the skull. The results of these previous studies are, again, consistent with FGFs being key effectors of Sp8 during craniofacial development. It therefore appears likely that the reduced FGF signaling observed in the Sp8 mutants can account for the majority of the observed craniofacial phenotype.
The connection between the Sp8 mutant phenotype and perturbed SHH signaling is also of interest. The Sp8 mutants showed a hypertelorism phenotype, which is also associated with excessive SHH signaling. In addition it is known that reduced FGF levels, as observed in the Sp8 mutants, can result in elevated SHH (Lettice et al., 2012; Mao et al., 2009; Zhang et al., 2009). It therefore seemed reasonable to hypothesize that increased SHH signaling was in part responsible for the dramatic Sp8 mutant craniofacial phenotype. Indeed in utero treatment with the SHH inhibitor cyclopamine did result in a striking partial rescue of the mutant phenotype in a large fraction of mutants. Nevertheless, an extensive analysis of the Sp8 mutants looking more directly for perturbed SHH signaling levels revealed relatively little. The only consistent change observed was a moderate reduction in Gli3 expression. In situ hybridizations, immunostains, the Ptch-lacZ SHH reporter mouse and Q-PCR failed to find significant differences in a number of hedgehog signaling components, including Smo, Indian Hedgehog, Gli1, Gli2 and Ptch, looking at multiple developmental time points. It is, however, very difficult to prove a negative result. In situ hybridization and immunostain methods are not very quantitative. Q-PCR is quantitative but requires the precise dissection of equivalent wild type and mutant tissues. Further, the gross morphological differences of the Sp8 mutants can make interpretations of results challenging, since exactly corresponding structures are not always present. It is difficult to correlate expression patterns when the underlying embryologic structures are distinct. It remains possible, therefore, that perturbations in SHH signaling were missed.
The results, however, suggest an alternative possibility. Perhaps the hypertelorism of the Sp8 mutants is not the result of increased SHH signaling, but instead Sp8 and SHH are acting in independent parallel pathways. For example, the Ephrin pathway (Babbs et al., 2011), the PTPN11 gene (LEOPARD) (Limongelli et al., 2008), and certain mutations of the TWIST1 gene (Saethre-Chotzen) (Firulli et al., 2005) have also been associated with hypertelorism. That is, the Sp8 mutation could cause hypertelorism through a non-SHH related mechanism. In this case the reduction of SHH signaling through cyclopamine treatment could partially rescue the Sp8−/− phenotype through its independent ability to regulate the hypo-hyper-telorism axis.
It is also interesting to note that the frontonasal ectodermal zone (FEZ), with both organizer and growth promoting properties, is established at the SHH/FGF8 boundary (Hu et al., 2003). The FEZ regulates both proximodistal gowth and dorsoventral patterning within the frontonasal prominence. The dramatically reduced Fgf8 expression in Sp8 mutants would be expected to perturb FEZ formation and function, and could in part explain the disrupted distal extension of snout structures.
A previous study used Foxg1-Cre to conditionally delete Fgf8 in signaling centers during craniofacial development (Kawauchi et al., 2005). The resulting phenotype resembled the Sp8 mutant mice in several respects, with reduced MNP size, increased apoptosis, and severe reduction or absence of olfactory structures. It is also interesting to note that there was no detected change in Shh expression. Another study of the telencephalic signaling centers used a series of mutants to vary FGF8 doses, resulting in increased apoptosis and decreased cell proliferation in the developing forebrain (Storm et al., 2006). Of particular interest, Sp8 expression in the ANR was dramatically reduced in Fgf8null/neo mice, which have dramatically reduced Fgf8 expression. This suggests the presence of a positive feedback loop between Sp8 and Fgf8, with Sp8 activating Fgf8 expression, which in turn activates Sp8.
In this report we identify a crucial role for the Sp8 transcription factor in the function of the ANR and OP signaling centers. The OP has been shown to be capable of inducing bone and cartilage formation, as well as providing a source of olfactory neurons and sensory epithelium (Szabo-Rogers et al., 2009). Further, many key OP functions are mediated by FGF signaling (Szabo-Rogers et al., 2008). Appropriate levels of the growth factors WNT, BMP and FGF signaling are necessary for olfactory placode induction (Ahrens and Schlosser, 2005; Brugmann et al., 2004; Litsiou et al., 2005). In addition the transcription factors Pax6 (Grindley et al., 1995), Dlx5 (Bhattacharyya and Bronner-Fraser, 2008), Six1, Six4 (Chen et al., 2009), Sox2 and Pou2f1 (Donner et al., 2007) have all been shown to play critical roles in the formation of the OP. In contrast, we observed that Sp8 is not needed to establish the OP, but rather is required for proper FGF signaling function. This is quite similar to what we previously observed in the AER signaling center of the developing limb, where Sp8 is not essential for its establishment, but is needed for AER maturation and FGF signaling function (Bell et al., 2003).
Relatively little is known of the genetic regulatory program that produces the ANR. The ANR signaling center plays a key role in both prosencephalon (Paek et al., 2009) and craniofacial development. The ANR is generally described as the rostral most part of the forebrain, defined by FGF8 expression as embryos reach five somites (Shimamura and Rubenstein, 1997). Many of the same genes are expressed in the OP and ANR, including Fgf8, Dlx5, Pax6 and Six3. In this report we show that the FGF signaling function of the ANR is disrupted in Sp8 mutants.
In summary, we have shown that Sp8 is required for Fgf8 and Fgf17 expression in important craniofacial development signaling centers. Increased apoptosis coupled with reduced cell proliferation of NC cells likely account for the reduced facial prominences in Sp8 mutants. In addition we showed that genetic and pharmaceutical reduction of SHH signaling in mutants resulted in a conspicuous, but partial, craniofacial rescue. There is a striking commonality of Sp8 function in the developing limbs and face. In each case Sp8 is required for FGF production in key signaling centers that direct development.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights.
Mutation of the Sp8 transcription factor gene results in dramatic craniofacial malformations.
Sp8 mutation results in the loss of many neural crest and paraxial mesoderm derived cranial bones.
Sp8 is required in signaling centers and not directly in the neural crest or paraxial mesoderm.
Sp8 mutants show increased apoptosis and decreased proliferation of neural crest cells.
Sp8 is necessary for Fgf8 and Fgf17 expression in the anterior neural ridge.
Acknowledgments
We thank Andrew Potter for help in performing the wild type LCM. We also thank Ching-Fang Chang for helping with the western blots. This work was supported by NIH UO1 DE020049.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abzhanov A, Tabin CJ. Shh and Fgf8 act synergistically to drive cartilage outgrowth during cranial development. Dev Biol. 2004;273:134–148. doi: 10.1016/j.ydbio.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Ahrens K, Schlosser G. Tissues and signals involved in the induction of placodal Six1 expression in Xenopus laevis. Dev Biol. 2005;288:40–59. doi: 10.1016/j.ydbio.2005.07.022. [DOI] [PubMed] [Google Scholar]
- Artinger KB, Bronner-Fraser M. Partial restriction in the developmental potential of late emigrating avian neural crest cells. Dev Biol. 1992;149:149–157. doi: 10.1016/0012-1606(92)90271-h. [DOI] [PubMed] [Google Scholar]
- Baek JA, Lan Y, Liu H, Maltby KM, Mishina Y, Jiang R. Bmpr1a signaling plays critical roles in palatal shelf growth and palatal bone formation. Dev Biol. 2011;350:520–531. doi: 10.1016/j.ydbio.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CV, Bronner-Fraser M, Le Douarin NM, Teillet MA. Early- and late-migrating cranial neural crest cell populations have equivalent developmental potential in vivo. Development. 1997;124:3077–3087. doi: 10.1242/dev.124.16.3077. [DOI] [PubMed] [Google Scholar]
- Bell SM, Schreiner CM, Waclaw RR, Campbell K, Potter SS, Scott WJ. Sp8 is crucial for limb outgrowth and neuropore closure. Proc Natl Acad Sci U S A. 2003;100:12195–12200. doi: 10.1073/pnas.2134310100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell HF, Donis-Keller H, Helms C, Hing AV, Heng HH, Koop B, Martindale D, Rommens JM, Tsui LC, Scherer SW. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet. 1996;14:353–356. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Bronner-Fraser M. Competence, specification and commitment to an olfactory placode fate. Development. 2008;135:4165–4177. doi: 10.1242/dev.026633. [DOI] [PubMed] [Google Scholar]
- Brugmann SA, Pandur PD, Kenyon KL, Pignoni F, Moody SA. Six1 promotes a placodal fate within the lateral neurogenic ectoderm by functioning as both a transcriptional activator and repressor. Development. 2004;131:5871–5881. doi: 10.1242/dev.01516. [DOI] [PubMed] [Google Scholar]
- Brunskill EW, Aronow BJ, Georgas K, Rumballe B, Valerius MT, Aronow J, Kaimal V, Jegga AG, Yu J, Grimmond S, McMahon AP, Patterson LT, Little MH, Potter SS. Atlas of gene expression in the developing kidney at microanatomic resolution. Dev Cell. 2008;15:781–791. doi: 10.1016/j.devcel.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill EW, Georgas K, Rumballe B, Little MH, Potter SS. Defining the molecular character of the developing and adult kidney podocyte. PLoS One. 2011;6:e24640. doi: 10.1371/journal.pone.0024640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush JO, Jiang R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139:231–243. doi: 10.1242/dev.067082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CF, Ramaswamy G, Serra R. Depletion of primary cilia in articular chondrocytes results in reduced Gli3 repressor to activator ratio, increased Hedgehog signaling, and symptoms of early osteoarthritis. Osteoarthritis Cartilage. 2012;20:152–161. doi: 10.1016/j.joca.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Kim EH, Xu PX. Initiation of olfactory placode development and neurogenesis is blocked in mice lacking both Six1 and Six4. Dev Biol. 2009;326:75–85. doi: 10.1016/j.ydbio.2008.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couly G, Creuzet S, Bennaceur S, Vincent C, Le Douarin NM. Interactions between Hox-negative cephalic neural crest cells and the foregut endoderm in patterning the facial skeleton in the vertebrate head. Development. 2002;129:1061–1073. doi: 10.1242/dev.129.4.1061. [DOI] [PubMed] [Google Scholar]
- Creuzet S, Schuler B, Couly G, Le Douarin NM. Reciprocal relationships between Fgf8 and neural crest cells in facial and forebrain development. Proc Natl Acad Sci U S A. 2004;101:4843–4847. doi: 10.1073/pnas.0400869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creuzet SE, Martinez S, Le Douarin NM. The cephalic neural crest exerts a critical effect on forebrain and midbrain development. Proc Natl Acad Sci U S A. 2006;103:14033–14038. doi: 10.1073/pnas.0605899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Akimaru H, Tanaka Y, Maekawa T, Nakafuku M, Ishii S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J Biol Chem. 1999;274:8143–8152. doi: 10.1074/jbc.274.12.8143. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- Donner AL, Episkopou V, Maas RL. Sox2 and Pou2f1 interact to control lens and olfactory placode development. Dev Biol. 2007;303:784–799. doi: 10.1016/j.ydbio.2006.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engleka KA, Gitler AD, Zhang M, Zhou DD, High FA, Epstein JA. Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Dev Biol. 2005;280:396–406. doi: 10.1016/j.ydbio.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Firulli BA, Krawchuk D, Centonze VE, Vargesson N, Virshup DM, Conway SJ, Cserjesi P, Laufer E, Firulli AB. Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat Genet. 2005;37:373–381. doi: 10.1038/ng1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi-Shimogori T, Grove EA. Neocortex patterning by the secreted signaling molecule FGF8. Science. 2001;294:1071–1074. doi: 10.1126/science.1064252. [DOI] [PubMed] [Google Scholar]
- Griffin JN, Compagnucci C, Hu D, Fish J, Klein O, Marcucio R, Depew MJ. Fgf8 dosage determines midfacial integration and polarity within the nasal and optic capsules. Dev Biol. 2013;374:185–197. doi: 10.1016/j.ydbio.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindley JC, Davidson DR, Hill RE. The role of Pax-6 in eye and nasal development. Development. 1995;121:1433–1442. doi: 10.1242/dev.121.5.1433. [DOI] [PubMed] [Google Scholar]
- Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell. 2004;118:517–528. doi: 10.1016/j.cell.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Helms JA, Cordero D, Tapadia MD. New insights into craniofacial morphogenesis. Development. 2005;132:851–861. doi: 10.1242/dev.01705. [DOI] [PubMed] [Google Scholar]
- Horstadius S. The Neural Crest 1950 [Google Scholar]
- Hu D, Helms JA. The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development. 1999;126:4873–4884. doi: 10.1242/dev.126.21.4873. [DOI] [PubMed] [Google Scholar]
- Hu D, Marcucio RS. A SHH-responsive signaling center in the forebrain regulates craniofacial morphogenesis via the facial ectoderm. Development. 2009;136:107–116. doi: 10.1242/dev.026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Marcucio RS, Helms JA. A zone of frontonasal ectoderm regulates patterning and growth in the face. Development. 2003;130:1749–1758. doi: 10.1242/dev.00397. [DOI] [PubMed] [Google Scholar]
- Hunt P, Clarke JD, Buxton P, Ferretti P, Thorogood P. Stability and plasticity of neural crest patterning and branchial arch Hox code after extensive cephalic crest rotation. Dev Biol. 1998;198:82–104. doi: 10.1006/dbio.1998.8886. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Esteban CR, Matsui T, Rodriguez-Leon J, Kato S, Izpisua Belmonte JC. Sp8 and Sp9, two closely related buttonhead-like transcription factors, regulate Fgf8 expression and limb outgrowth in vertebrate embryos. Development. 2004;131:4763–4774. doi: 10.1242/dev.01331. [DOI] [PubMed] [Google Scholar]
- Kawauchi S, Shou J, Santos R, Hebert JM, McConnell SK, Mason I, Calof AL. Fgf8 expression defines a morphogenetic center required for olfactory neurogenesis and nasal cavity development in the mouse. Development. 2005;132:5211–5223. doi: 10.1242/dev.02143. [DOI] [PubMed] [Google Scholar]
- Kuczuk MH, Scott WJ., Jr Potentiation of acetazolamide induced ectrodactyly in SWV and C57BL/6J mice by cadmium sulfate. Teratology. 1984;29:427–435. doi: 10.1002/tera.1420290314. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM, Creuzet S, Couly G, Dupin E. Neural crest cell plasticity and its limits. Development. 2004;131:4637–4650. doi: 10.1242/dev.01350. [DOI] [PubMed] [Google Scholar]
- Lee SH, Fu KK, Hui JN, Richman JM. Noggin and retinoic acid transform the identity of avian facial prominences. Nature. 2001;414:909–912. doi: 10.1038/414909a. [DOI] [PubMed] [Google Scholar]
- Lee SM, Danielian PS, Fritzsch B, McMahon AP. Evidence that FGF8 signalling from the midbrain-hindbrain junction regulates growth and polarity in the developing midbrain. Development. 1997;124:959–969. doi: 10.1242/dev.124.5.959. [DOI] [PubMed] [Google Scholar]
- Lettice LA, Williamson I, Wiltshire JH, Peluso S, Devenney PS, Hill AE, Essafi A, Hagman J, Mort R, Grimes G, DeAngelis CL, Hill RE. Opposing functions of the ETS factor family define Shh spatial expression in limb buds and underlie polydactyly. Dev Cell. 2012;22:459–467. doi: 10.1016/j.devcel.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limongelli G, Sarkozy A, Pacileo G, Calabro P, Digilio MC, Maddaloni V, Gagliardi G, Di Salvo G, Iacomino M, Marino B, Dallapiccola B, Calabro R. Genotype-phenotype analysis and natural history of left ventricular hypertrophy in LEOPARD syndrome. Am J Med Genet A. 2008;146A:620–628. doi: 10.1002/ajmg.a.32206. [DOI] [PubMed] [Google Scholar]
- Lipinski RJ, Hutson PR, Hannam PW, Nydza RJ, Washington IM, Moore RW, Girdaukas GG, Peterson RE, Bushman W. Dose- and route-dependent teratogenicity, toxicity, and pharmacokinetic profiles of the hedgehog signaling antagonist cyclopamine in the mouse. Toxicol Sci. 2008;104:189–197. doi: 10.1093/toxsci/kfn076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski RJ, Song C, Sulik KK, Everson JL, Gipp JJ, Yan D, Bushman W, Rowland IJ. Cleft lip and palate results from Hedgehog signaling antagonism in the mouse: Phenotypic characterization and clinical implications. Birth Defects Res A Clin Mol Teratol. 2010;88:232–240. doi: 10.1002/bdra.20656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litsiou A, Hanson S, Streit A. A balance of FGF, BMP and WNT signalling positions the future placode territory in the head. Development. 2005;132:4051–4062. doi: 10.1242/dev.01964. [DOI] [PubMed] [Google Scholar]
- Little MH, Brennan J, Georgas K, Davies JA, Davidson DR, Baldock RA, Beverdam A, Bertram JF, Capel B, Chiu HS, Clements D, Cullen-McEwen L, Fleming J, Gilbert T, Herzlinger D, Houghton D, Kaufman MH, Kleymenova E, Koopman PA, Lewis AG, McMahon AP, Mendelsohn CL, Mitchell EK, Rumballe BA, Sweeney DE, Valerius MT, Yamada G, Yang Y, Yu J. A high-resolution anatomical ontology of the developing murine genitourinary tract. Gene Expr Patterns. 2007;7:680–699. doi: 10.1016/j.modgep.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- MacArthur CA, Lawshe A, Shankar DB, Heikinheimo M, Shackleford GM. FGF-8 isoforms differ in NIH3T3 cell transforming potential. Cell Growth Differ. 1995;6:817–825. [PubMed] [Google Scholar]
- Mao J, McGlinn E, Huang P, Tabin CJ, McMahon AP. Fgf-dependent Etv4/5 activity is required for posterior restriction of Sonic Hedgehog and promoting outgrowth of the vertebrate limb. Dev Cell. 2009;16:600–606. doi: 10.1016/j.devcel.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci U S A. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Morales PL, Diez del Corral R, Olivera-Martinez I, Quiroga AC, Das RM, Barbas JA, Storey KG, Morales AV. FGF and retinoic acid activity gradients control the timing of neural crest cell emigration in the trunk. J Cell Biol. 2011;194:489–503. doi: 10.1083/jcb.201011077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeish JD, Scott WJ, Jr, Potter SS. Legless, a novel mutation found in PHT1-1 transgenic mice. Science. 1988;241:837–839. doi: 10.1126/science.3406741. [DOI] [PubMed] [Google Scholar]
- Monsoro-Burq AH, Fletcher RB, Harland RM. Neural crest induction by paraxial mesoderm in Xenopus embryos requires FGF signals. Development. 2003;130:3111–3124. doi: 10.1242/dev.00531. [DOI] [PubMed] [Google Scholar]
- Noden DM. The role of the neural crest in patterning of avian cranial skeletal, connective, and muscle tissues. Dev Biol. 1983;96:144–165. doi: 10.1016/0012-1606(83)90318-4. [DOI] [PubMed] [Google Scholar]
- Noden DM, Schneider RA. Neural crest cells and the community of plan for craniofacial development: historical debates and current perspectives. Adv Exp Med Biol. 2006;589:1–23. doi: 10.1007/978-0-387-46954-6_1. [DOI] [PubMed] [Google Scholar]
- Olsson M, Campbell K, Turnbull DH. Specification of mouse telencephalic and mid-hindbrain progenitors following heterotopic ultrasound-guided embryonic transplantation. Neuron. 1997;19:761–772. doi: 10.1016/s0896-6273(00)80959-9. [DOI] [PubMed] [Google Scholar]
- Paek H, Gutin G, Hebert JM. FGF signaling is strictly required to maintain early telencephalic precursor cell survival. Development. 2009;136:2457–2465. doi: 10.1242/dev.032656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki J, Inoue T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development. 1999;126:3437–3447. doi: 10.1242/dev.126.15.3437. [DOI] [PubMed] [Google Scholar]
- Sahara S, Kawakami Y, Izpisua Belmonte JC, O’Leary DD. Sp8 exhibits reciprocal induction with Fgf8 but has an opposing effect on anterior-posterior cortical area patterning. Neural Dev. 2007;2:10. doi: 10.1186/1749-8104-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915–3924. doi: 10.1242/dev.126.17.3915. [DOI] [PubMed] [Google Scholar]
- Sato A, Scholl AM, Kuhn EN, Stadt HA, Decker JR, Pegram K, Hutson MR, Kirby ML. FGF8 signaling is chemotactic for cardiac neural crest cells. Dev Biol. 2011;354:18–30. doi: 10.1016/j.ydbio.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamim H, Mahmood R, Logan C, Doherty P, Lumsden A, Mason I. Sequential roles for Fgf4, En1 and Fgf8 in specification and regionalisation of the midbrain. Development. 1999;126:945–959. doi: 10.1242/dev.126.5.945. [DOI] [PubMed] [Google Scholar]
- Shimamura K, Rubenstein JL. Inductive interactions direct early regionalization of the mouse forebrain. Development. 1997;124:2709–2718. doi: 10.1242/dev.124.14.2709. [DOI] [PubMed] [Google Scholar]
- Storm EE, Garel S, Borello U, Hebert JM, Martinez S, McConnell SK, Martin GR, Rubenstein JL. Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development. 2006;133:1831–1844. doi: 10.1242/dev.02324. [DOI] [PubMed] [Google Scholar]
- Supp DM, Brueckner M, Kuehn MR, Witte DP, Lowe LA, McGrath J, Corrales J, Potter SS. Targeted deletion of the ATP binding domain of left-right dynein confirms its role in specifying development of left-right asymmetries. Development. 1999;126:5495–5504. doi: 10.1242/dev.126.23.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supp DM, Witte DP, Potter SS, Brueckner M. Mutation of an axonemal dynein affects left-right asymmetry in inversus viscerum mice. Nature. 1997;389:963–966. doi: 10.1038/40140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo-Rogers HL, Geetha-Loganathan P, Nimmagadda S, Fu KK, Richman JM. FGF signals from the nasal pit are necessary for normal facial morphogenesis. Dev Biol. 2008;318:289–302. doi: 10.1016/j.ydbio.2008.03.027. [DOI] [PubMed] [Google Scholar]
- Szabo-Rogers HL, Geetha-Loganathan P, Whiting CJ, Nimmagadda S, Fu K, Richman JM. Novel skeletogenic patterning roles for the olfactory pit. Development. 2009;136:219–229. doi: 10.1242/dev.023978. [DOI] [PubMed] [Google Scholar]
- Toyoda R, Assimacopoulos S, Wilcoxon J, Taylor A, Feldman P, Suzuki-Hirano A, Shimogori T, Grove EA. FGF8 acts as a classic diffusible morphogen to pattern the neocortex. Development. 2010;137:3439–3448. doi: 10.1242/dev.055392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev. 1999;13:3136–3148. doi: 10.1101/gad.13.23.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler MS, Hall BK. Epithelial influences on skeletogenesis in the mandible of the embryonic chick. Anat Rec. 1977;188:229–239. doi: 10.1002/ar.1091880208. [DOI] [PubMed] [Google Scholar]
- Veistinen L, Takatalo M, Tanimoto Y, Kesper DA, Vortkamp A, Rice DP. Loss-of-Function of Gli3 in Mice Causes Abnormal Frontal Bone Morphology and Premature Synostosis of the Interfrontal Suture. Front Physiol. 2012;3:121. doi: 10.3389/fphys.2012.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waclaw RR, Allen ZJ, 2nd, Bell SM, Erdelyi F, Szabo G, Potter SS, Campbell K. The zinc finger transcription factor Sp8 regulates the generation and diversity of olfactory bulb interneurons. Neuron. 2006;49:503–516. doi: 10.1016/j.neuron.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Xu J, Liu Z, Ornitz DM. Temporal and spatial gradients of Fgf8 and Fgf17 regulate proliferation and differentiation of midline cerebellar structures. Development. 2000;127:1833–1843. doi: 10.1242/dev.127.9.1833. [DOI] [PubMed] [Google Scholar]
- Zembrzycki A, Griesel G, Stoykova A, Mansouri A. Genetic interplay between the transcription factors Sp8 and Emx2 in the patterning of the forebrain. Neural Dev. 2007;2:8. doi: 10.1186/1749-8104-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Verheyden JM, Hassell JA, Sun X. FGF-regulated Etv genes are essential for repressing Shh expression in mouse limb buds. Dev Cell. 2009;16:607–613. doi: 10.1016/j.devcel.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.