

Abstract
Host specialization by pathogens requires a repertoire of virulence factors as well as fine-tuned regulation of gene expression. The fungal wheat pathogen Zymoseptoria tritici (synonym Mycosphaerella graminicola) is a powerful model system for the discovery of genetic elements that underlie virulence and host specialization. We transcriptionally profiled the early stages of Z. tritici infection of a compatible host (wheat) and a noncompatible host (Brachypodium distachyon). The results revealed infection regulatory programs common to both hosts and genes with striking wheat-specific expression, with many of the latter showing sequence signatures of positive selection along the Z. tritici lineage. Genes specifically regulated during infection of wheat populated two large clusters of coregulated genes that may represent candidate pathogenicity islands. On evolutionarily labile, repeat-rich accessory chromosomes (ACs), we identified hundreds of highly expressed genes with signatures of evolutionary constraint and putative biological function. Phylogenetic analyses suggested that gene duplication events on these ACs were rare and largely preceded the diversification of Zymoseptoria species. Together, our data highlight the likely relevance for fungal growth and virulence of hundreds of Z. tritici genes, deepening the annotation and functional inference of the genes of this model pathogen.
Keywords: RNA-seq, supernumerary chromosomes, gene duplication, plant pathogenic fungi, host adaptation, Mycosphaerella graminicola
Introduction
Many parasites have evolved strict specificities to particular hosts. Specialization is mediated by an ability to suppress host defenses and by adaptation to host substrates, within-host proliferation, dispersal, or reproduction. How pathogens acquire such attributes and achieve specialization is one of the central questions of modern ecological genetics. In plant pathogens, a handful of landmark studies have mapped genes that drive host specificity (Hacquard et al. 2013), some of which are characterized by positioning in distinct, rapidly evolving regions of the genome (Ma et al. 2010; Rouxel et al. 2011; De Jonge et al. 2012). For the majority of pathogen–host interactions, however, the genomic and molecular basis of specialization remains unknown.
The wheat pathogen Zymoseptoria tritici (synonym Mycosphaerella graminicola) is a powerful model system for the study of the evolution of host specificity. The genome of one isolate has been fully sequenced from telomere to telomere revealing 13 core chromosomes (CCs) and a set of eight repeat-rich accessory chromosomes (ACs; Goodwin et al. 2011). ACs resemble B-chromosomes of plants, and, in comparison to the core genome, they appear to evolve under less selective constraint (Stukenbrock et al. 2011) and undergo more frequent intrachromosomal recombination, translocations, and nondisjunctions during meiosis (Covert 1998; Wittenberg et al. 2009; Ma et al. 2010; Raffaele and Kamoun 2012; Croll et al. 2013). Compared with other fungal species with ACs, Z. tritici has an unusual high number of these small chromosomes. As many as eight ACs have been documented in one isolate (Goodwin et al. 2011), yet the functional relevance of these chromosomes in Z. tritici remains poorly understood.
Zymoseptoria tritici is globally distributed, has a hemibiotrophic lifestyle, and is specialized to domesticated bread wheat, Triticum aestivum, and durum wheat, T. durum (Brokenshire 1975; Eyal et al. 1985; Banke and McDonald 2005). This pathogen uses no special structures such as appressoria or haustoria for penetration of host tissues (Kema et al. 1996). Instead, Z. tritici enters its host through stomatal openings and establishes an intercellular hyphal network that resembles endophytic rather than intimate biotrophic growth (Kema et al. 1996; Brunner et al. 2013). After a long latent period, the fungus switches to necrotrophic growth. Genome and transcriptome sequencing, and comparative analysis with other Dothideomycete genomes, have provided insight into the gene content of Z. tritici but have not revealed how the pathogen can proliferate in tissue of susceptible wheat lines without inducing resistance responses (Goodwin et al. 2011; Ohm et al. 2012; Yang et al. 2013). When infecting other grass hosts such as Brachypodium distachyon or Agropyron repens, Z. tritici also penetrates the leaf surface through stomata but infection is stopped in the substomatal cavity (Haueisen J and Stukenbrock E unpublished data). This suggests an early recognition and interaction between host and pathogen at which Z. tritici can manipulate host defenses in susceptible wheat lines.
In this work, we set out to identify candidate determinants of host specificity in Z. tritici using transcriptional profiles of early-stage infections of two grasses: wheat, the compatible host, and B. distachyon, a noncompatible host. We developed a novel pipeline for analysis of RNA-seq from Z. tritici collected in planta. With the data, we characterized distinct regulatory programs activated during infection of wheat, and the evolutionary and functional signatures of genes with host-specific expression patterns. To gain insight into the functional relevance of AC-encoded genes in Z. tritici, we furthermore explored both genome and transcriptome data with respect to differences between essential CCs and ACs.
Materials and Methods
Strains and Growth Conditions
All experiments used Z. tritici isolate IPO323 (Kema and van Silfhout 1997). Cultures were inoculated onto solid YMS agar (4 g yeast extract, 4 g malt extract, 4 g sucrose, 20 g bacto agar, 1 l H2O) at 18 °C. For transcriptional profiling of axenic culture, yeast-like cells were isolated from these plates after 72 h. For transcriptional profiling of plant infections, 15-day-old wheat seedlings of the cultivar Obelisk and the B. distachyon accession Bd21 were inoculated as follows. A distinct area of the second leaf (10–15 cm) was marked, and a spore solution of 1 × 107 yeast cells/ml containing 0.1% Tween 20 was brushed onto these areas. After an initial incubation for 48 h at 100% humidity and 22 °C, inoculated plants were incubated with a 16 h light period at 75% humidity and 22 °C for 4 days.
RNA-Seq
To extract total RNA from fungal axenic culture or freeze-dried leaf tissue infected with Z. tritici, 100 mg of sample was crushed in liquid nitrogen, and total RNA was extracted using the TRIZOL reagent (Invitrogen) following the manufacturer’s protocol. RNA samples were purified twice with Dynal oligo(dT) magnetic beads (Invitrogen), following the manufacturer's instructions. Illumina RNA-seq libraries for axenic culture replicates were prepared from an input of 10 µg total purified polyA RNA according to Palma-Guerrero et al. (2013). Axenic culture libraries were sequenced on an Illumina Genome Analyzer II in the Functional Genomics Laboratory at the University of California, Berkeley, applying standard Illumina operating procedures. RNA-seq libraries for plant infection replicates were prepared from an input of 4 µg total purified polyA RNA according to recommendations of the supplier (TruSeq RNA sample preparation v2 guide, Illumina). Libraries were quantified by fluorometry, immobilized, and processed onto a flow cell with a cBot (Illumina) followed by sequencing-by-synthesis with TruSeq v3 chemistry on a HiSeq2000 at the Max Planck Genome Center, Cologne. RNA-seq data for all samples has been deposited in National Center for Biotechnology Information's (NCBI) Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE54874 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE54874, last accessed May 23, 2014).
RNA-seq Read Filtering and Mapping
Single-end RNA-seq reads were filtered for quality and for homology to the genomes of T. aestivum (Brenchley et al. 2012) and B. distachyon (accession Bd21, International Brachypodium Initiative 2010). The workflow for analyses of the RNA-seq data is given in supplementary figure S2, Supplementary Material online. Briefly, RNA-seq reads were analyzed and filtered using the grooming, trimming, filtering, and masking tools from the Galaxy server (www.galaxyproject.org, last accessed May 23, 2014). Trimming parameters for each data set were specified after quality control using the NGS QC toolkit (Patel and Jain 2012, supplementary table S1, Supplementary Material online). Fungal RNA-seq reads from infected plants were trimmed 12 bp from the left end and 15 bp from the right end, resulting in a final read length of 80 bp (supplementary table S1, Supplementary Material online). Reads from axenic culture were only trimmed from the left end, resulting in a final read length of 38 bp. Reads with an overall quality score below 20 and reads having less than 25% of nucleotides with a quality score above 20 were discarded. For the remaining reads, nucleotides with quality scores below 20 were masked with Ns.
To account for incorrect mapping of plant reads with high similarity to fungal sequences, reads from infected plant tissue were mapped to both the plant genomes and the genome of Z. tritici IPO323. All plant reads were removed from further downstream analyses. To this end, reads with 100% identity to the plant genomes and a maximum of 25% N positions were filtered out in fastq_screen v0.4.1 (www.bioinformatics.babraham.ac.uk/projects/fastq_screen, last accessed May 23, 2014) applying a sensitive local Bowtie2 alignment (supplementary fig. S1, Supplementary Material online).
Mapping of single end RNA-seq reads to the genome of the Z. tritici isolate IPO323 (Goodwin et al. 2011) was carried out with tophat v2.0.9 (Kim et al. 2013). Mapping was disabled for novel splice discovery and used the JGI transcript annotation of IPO323 as reference (Goodwin et al. 2011; http://genome.jgi.doe.gov/Mycgr3/Mycgr3.home.html, last accessed May 23, 2014). The maximum number of read mismatches and read gap length were set to 2. Multiple mappings of single reads was set to 10 (default: 20) to decrease the bias for genes with more multimapping reads. In total, 2.3% and 3.8% of fungal reads of infected T. aestivum and B. distachyon, and 5.7% reads of axenic culture were mapped to multiple positions in the genome of IPO323 (supplementary table S1, Supplementary Material online). Relative abundances for predicted transcripts of IPO323 were estimated in Cufflinks v2.1.1 (Trapnell et al. 2012). Total read counts per transcript were estimated in htseq-count (http://www-huber.embl.de/users/anders/HTSeq/doc/count.html, last accessed May 23, 2014) applying the intersection-strict mode.
Coding and protein sequences of candidate genes were surveyed for orthology and homologous domains using NCBI BLAST (Altschul et al. 1990, 1997) and SMART (Schultz et al. 1998; Letunic et al. 2009) searches.
Transcriptional Profiling
For detection of expressed genes, we set a minimal reads per kilobase per million reads (RPKM) threshold of >2 according to a comparison with the coverage of putative noncoding loci as follows. We first defined a representative set of noncoding loci by masking the genome for 1) transcripts, 2) 500 bp upstream and downstream flanking regions of each transcript, and 3) transposable elements. Transposable element predictions relied on modified parameters of Goodwin et al. (2011) (Grandaubert J and Stukenbrock E unpublished data). The remaining loci were filtered for sequences ≥300 bp length, resulting in a set of 5,394 noncoding loci of 300–11,833 bp length, covering 7.81 Mb of the genome. The median RPKM values for such noncoding loci in libraries from axenic culture, 4 dpi on T. aestivum, and 4 dpi on B. distachyon were 1.1, 2.3, and 2.1, respectively. The distribution of RPKM values of these nontranscribed loci clearly differed from the RPKM distribution of transcripts (supplementary fig. S10, Supplementary Material online).
For sliding window analyses of transcription, a gtf file with respective window sizes for the genome of IPO323 was created and applied for read mapping. Genome-wide transcription was visualized with Circos (Krzywinski et al. 2009). RNA-seq sample relations were evaluated based on multidimensional scaling using the plotMDS package of DESeq (Anders and Huber 2010) in R (www.R-project.org, last accessed May 23, 2014). Significantly differentially transcribed genes were identified using the bioconductor packages DESeq (Anders and Huber 2010) and EdgeR (McCarthy et al. 2012) in R (www.R-project.org, last accessed May 23, 2014) applying the Trimmed Mean Method for total tag count normalization in EdgeR and a false discovery rate P value of 0.01. Inference of secreted gene products was based on the identification of secretion signals by Morais do Amaral et al. (2012). Differential expression patterns in RNA-seq analyses were verified and confirmed by qRT-polymerase chain reaction (PCR) for six genes of Z. tritici, namely Mg32609, Mg41093, Mg42222, Mg49575, Mg70993, and Mg107320 (supplementary fig. S9, Supplementary Material online) using methods as follows. cDNAs from RNA-seq samples were used in a qRT-PCR experiment using the iQ SYBR Green Supermix Kit (Bio-Rad, Munich, Germany) with gene-specific primers and an annealing temperature of 59 °C. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (gapdh, Mg99044) was used as a constitutively expressed control gene. The PCR was conducted on a CFX96 Real-Time PCR Detection System (Bio-Rad).
To identify clusters of genes enriched for upregulation during plant infection, we carried out a sliding window analysis as follows. We binned the fungal genome into 10-gene bins with a step size of 5 genes, and for each window, we tabulated the number of genes called significant in the comparison between expression levels during infection of each plant host and those in axenic culture, for which the former were of larger magnitude. For each bin, we then evaluated the likelihood of observing this count of upregulated genes under a Poisson null (www.R-project.org, last accessed May 23, 2014) applying the ppois function. An analogous calculation was carried out for genes downregulated during infection.
Identification of Gene Families and Phylogenetics
To define gene families in IPO323, sequence data sets of transcripts and proteins from the Z. tritici reference genome version 2.0 from the Joint Genome Institute (http://genome.jgi.doe.gov/Mycgr3/Mycgr3.home.html, last accessed May 23, 2014) were formatted for BLAST using formatdb (NCBI). A preliminary set of paralog pairs was identified as those genes whose homology achieved e < 0.1 in an all-by-all BLAST of amino acid sequences (Altschul et al. 1997). This preliminary set was then filtered in two ways: Either applying the criteria from (Gu et al. 2002, filter scheme 1), or within the program SiliX (Miele et al. 2011), filtering for sequence identity ≥35%, alignment length ≥30 amino acids, and overlaps ≥30% (filter scheme 2). Genome-wide visualization of paralogs was done with Circos (Krzywinski et al. 2009). Gene families were grouped from paralog pairs using SiliX (Miele et al. 2011), allowing for grouping of sequences that did not overlap with all sequences of the gene family.
To screen for homologs in other Z. tritici isolates and Zymoseptoria species, each transcript from the Z. tritici IPO323 reference genome was subjected to BLAST against the 12 Zymoseptoria genomes reported in Stukenbrock et al. (2011). Homology was considered for sequences with alignment lengths >150 bp and sequence identities > 60% (with respect to the alignment length).
Ratios of Ka/Ks (nonsynonymous to synonymous substitutions) between species and Pn/Ps (nonsynonymous to synonymous polymorphisms) within species were assessed in Stukenbrock et al. (2010). The method of Nei and Gojobori was used to compute synonymous and nonsynonymous rates of substitutions and polymorphisms (Nei and Kumar 2000).
Gene ontology categories of IPO323 transcripts were taken from Goodwin et al. (2011) and analyzed for enrichments with the clusterProfiler package (Yu et al. 2012) in R (www.R-project.org, last accessed May 23, 2014).
For the phylogenetic trees in figure 7 and supplementary figure S8, Supplementary Material online, amino acid sequences were aligned using Clustal Omega applying default settings (Sievers et al. 2011). Leading and trailing gaps as well as inner gaps of the alignment were removed with gblocks (Dereeper et al. 2008). To account for larger genetic distances among the aligned sequences down to 40% sequence identity at the amino acid level, we applied in gblocks smaller final blocks, gap positions within final blocks, and less strict flanking positions (Castresana 2000). Maximum likelihood (ML) analyses (Felsenstein 1981) were performed with RAxML 7.0.4 (Stamatakis et al. 2005). RAxML 7.0.4 conducted 1000 bootstrap replicates using a rapid bootstrap algorithm (Stamatakis et al. 2008) applying the PROTGAMMAWAG approximation. In the subsequent ML search for the best scoring ML tree starting from each fifth bootstrap tree, the more accurate PROTCAT approximation was applied. Bootstrap support values were mapped on the most likely tree that was visualized and edited in FigTree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/, last accessed May 23, 2014). Filter settings of gblocks had negligible effects on inferred tree topologies (data not shown).
Fig. 7.—

Positions on core and accessory chromosomes of members of a gene family have been maintained during the evolution of Dothideomycete fungi. Shown is an unrooted maximum likelihood phylogeny of an amino acid alignment of the representative gene family J_FAM234 (see supplementary table S5, Supplementary Material online). Bootstrap support values above 50 are given next to branches. Branch lengths correspond to substitutions per site.
Results
A Survey of Expressed Genes in Z. tritici
To survey the expression patterns of Z. tritici during infection of plant hosts, we inoculated three biological replicates of the susceptible wheat host T. aestivum Obelisk with the Z. tritici strain IPO323 and generated analogous infected cultures of the nonsusceptible grass species B. distachyon (accession Bd21). We transcriptionally profiled each sample by RNA-seq after 4 days postinfection (dpi), along with axenic Z. tritici culture controls (supplementary table S1, Supplementary Material online). We developed a read-mapping pipeline to distinguish fungal and host transcripts in the in planta samples (supplementary fig. S1, Supplementary Material online); our final data sets comprised 45.1–50.2 million mapped reads from samples of Z. tritici axenic culture and 4.2–5.3 million Z. tritici reads from samples of infected plant tissue (supplementary table S1, Supplementary Material online). Of the 10,952 predicted Z. tritici genes (http://genome.jgi.doe.gov/Mycgr3/Mycgr3.home.html, last accessed May 23, 2014), 8,053 were expressed at >2 read per million in any of the three growth conditions (supplementary table S2, Supplementary Material online), and we considered this set of genes to represent the core active transcriptional program of Z. tritici in our experiments. Expressed genes were uniformly distributed across the genome except for a region of the CC 7 likely subject to transcriptional silencing (fig. 1). A small population of ribosomal DNA fragments cloned in our RNA-seq libraries mapped to a region of chromosome 7 likely to function as the rDNA cluster of Z. tritici (fig. 1, supplementary fig. S2, Supplementary Material online). Chromosome 18, an AC, was distinguished by a complete absence of expression signal (fig. 1), and multiplex PCR analyses confirmed that chromosome 18 has been lost in the Z. tritici strain IPO323 (supplementary fig. S3, Supplementary Material online).
Fig. 1.—

Genomic overview of transcription of Z. tritici isolate IPO323. Each vertical line on the outermost thick track reports the location of a Z. tritici gene on the indicated core (blue) or accessory (yellow) chromosome, with positions on each chromosome scaled at 1 Mb to 100 units. Small black circles below this first track indicate genes whose proteins are predicted to be secreted (Morais do Amaral et al. 2012). On the next three tracks, each vertical line reports expression of the corresponding gene in axenic culture (yeast malt sucrose medium, YMS) or during infection of Triticum aestivum (Ta) or Brachypodium distachyon (Bd) as indicated, with brightness reporting the expression level scaled separately for each condition. Absolute expression, as an average over the three conditions, is shown as a line plot above the outermost track, with the y axis reporting reads mapped to the indicated position per kilobase per million mapped reads of the library, RPKM. The remaining (innermost) tracks are a blowup of the accessory chromosome data with symbols as in the main figure. Positions of particularly highly expressed genes are noted as text.
Previous analyses indicated that the Z. tritici transcriptome is extensively reprogrammed during infection (Yang et al. 2013). Consistent with this picture, our in planta genome-wide transcriptional profiles of Z. tritici were more similar to one another than to profiles from axenic culture (supplementary fig. S4, Supplementary Material online). Likewise, our data agreed well with published sources, with complete overlap between the top quartile of expressed genes during infection of wheat at 4 dpi in our data set and that reported by Yang et al. (2013). To further analyze infection regulatory programs in detail in our data set, we first identified genes differentially expressed between the axenic condition and cultures on each plant host in turn, and we also tabulated genes significantly differentially expressed between hosts (fig. 2). We took the intersection of the former two plant-induction regulons to encompass the most robust set of Z. tritici transcriptional changes during infection (fig. 2, supplementary table S3, Supplementary Material online). In this infection regulatory program, we noted several genes with putative roles in small molecule detoxification, including homologs of multicopper oxidases, cytochrome P450s, and oxygenases involved in tryptophan and quinone metabolism (supplementary table S3, Supplementary Material online). Furthermore, this set of genes was enriched for Gene Ontology groups involved in peroxidase activity (GO:0004601), oxidoreductase activity (GO:0016684), and antioxidant activity (GO:0016209; supplementary table S4, Supplementary Material online), as well as an excess of genes encoding proteins with putative secretion signals (13.7% with secretion signals compared with 8.9% genome wide; supplementary table S3, Supplementary Material online), suggestive of a role in host interactions for many genes differentially regulated during infection.
Fig. 2.—
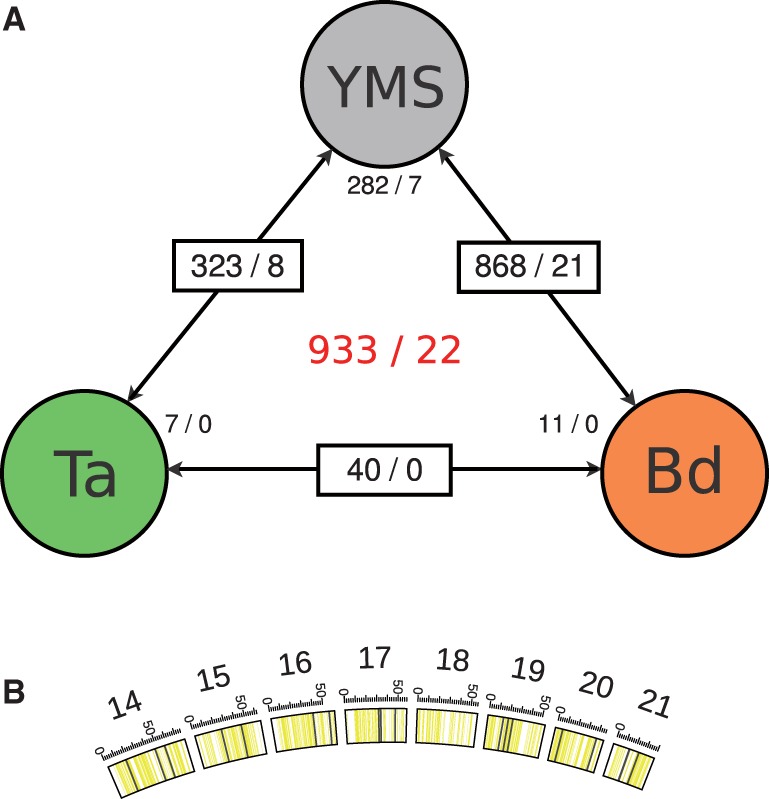
Zymoseptoria tritici genes differentially expressed between axenic culture and growth in planta. (A) Each box reports results of a comparison between expression profiles from the indicated two cultures: axenic growth in yeast malt sucrose medium (YMS), infection of T. aestivum (Ta), and infection of B. distachyon (Bd). In a given box, the value before the slash reports the number of genes on core chromosomes differentially expressed at a false discovery rate of 0.01 or less between the indicated conditions, and the value after the slash reports the analogous quantity from accessory chromosomes. Small text next to a given circle reports the number of genes differentially expressed between the indicated condition and both of the other two, on core chromosomes (before the slash) and accessory chromosomes (after the slash). Red text at center reports the total numbers of genes differentially expressed in any comparison, on core chromosomes (before the slash) and accessory chromosomes (after the slash). (B) Distribution of differentially transcribed genes (black solid lines) along ACs. Boxes depict single chromosomes with regions containing transcripts drawn in yellow. Scaling: 50 = 0.5 Mb.
In many fungal pathogens, infection-related genes cluster into “pathogenicity islands” that can sweep rapidly through populations (Kämper et al. 2006; Fedorova et al. 2008; Stergiopoulus and de Wit, 2009; Rouxel et al. 2011). We hypothesized that Z. tritici genes regulated during infection could likewise be distributed nonrandomly in the genome. To test this, we investigated the genomic positions of genes activated or repressed during infection. The results revealed a cluster of genes on Z. tritici chromosome 2 upregulated during infection, including several small molecule detoxification factors (fig. 3A); strikingly, nine of the ten genes of this locus were regulated in a host-specific manner, with 1.6-fold to 4-fold higher expression during infection of the preferred host, T. aestivum, relative to infection of B. distachyon (fig. 3B). Another locus, on Z. tritici chromosome 9, harbored a cluster of genes repressed during infection (supplementary fig. S5, Supplementary Material online). We reasoned that, if these clusters were of particular importance in the evolutionary history of Z. tritici, they could have assembled by genomic rearrangements during the divergence of this species from others in the clade. Consistent with this notion, both clusters were absent or rearranged in other Zymoseptoria genomes (fig. 3C and supplementary fig. S5, Supplementary Material online). Thus, these results provide a first compelling line of evidence for these gene clusters as putative pathogenicity islands in Z. tritici.
Fig. 3.—

Plant-induced upregulation of a gene cluster on chromosome 2. (A) Each bar denotes the number of Z. tritici genes significantly upregulated during infection of both wheat and Brachypodium (y) in sliding windows of 10 genes along the genome (x). The red circle indicates a window containing five upregulated genes, a degree of clustering unlikely under a genomic null (Poisson p = 3.9e−07). (B) Each row reports characteristics of one gene in the cluster of upregulated genes on Z. tritici chromosome 2 in (A). RPKM, reads mapped to the indicated gene per kilobase per million mapped reads in the library, in axenic culture (yeast malt sucrose medium, YMS) or during infection of T. aestivum (Ta) or B. distachyon (Bd). Secreted, inference that the encoded protein is secreted according to Morais do Amaral et al. (2012). (C) Presence–absence variation in the coregulated cluster genes in Z. tritici and other Zymoseptoria species. Boxes depict cluster genes (orange) and cluster-flanking genes (gray). Lines connect genes of the same genome contig sequence. Orthologous genes are vertically aligned.
Genomic Features of Z. tritici Genes Differentially Expressed between Hosts
We next carried out a broader-scale analysis of the genes induced in Z. tritici during infection of one plant host and not the other, which we considered as prime candidates for a potential role in host specialization in this fungus. qRT-PCR validation of six genes confirmed the RNA-seq-based prediction of differential gene expression between the two plant species (supplementary fig. S9, Supplementary Material online). Given the wheat-specific induction of small molecule metabolism genes clustering on chromosome 2 (fig. 3), we hypothesized that Z. tritici infection of wheat could involve a broader program of upregulation of such genes. As an unbiased test of this notion, we evaluated the patterns of host-specific expression among genes of the Eukaryotic Orthologs Group (KOG) annotated in Secondary metabolites, biosynthesis, transport, and catabolism (genome.jgi.doe.gov/Tutorial/tutorial/kog.html). The results bore out our prediction, with a significant signal for upregulation of the genes of this group during infection of T. aestivum relative to that of B. distachyon (fig. 4). Interestingly, genes encoding proteins with putative secretion signals were also enriched for those upregulated during infection of wheat and genes with plant-specific expression patterns (fig. 4), further attesting to the plausible role of these genes in host invasion.
Fig. 4.—
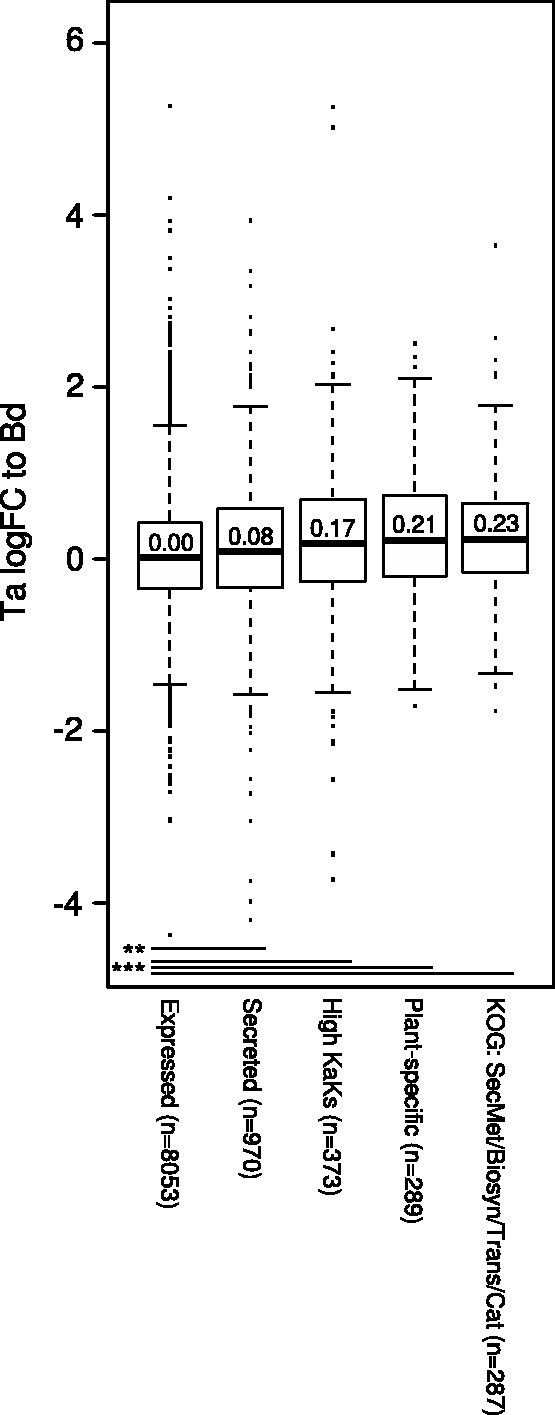
Patterns of functional and evolutionary relevance among Z. tritici genes expressed differently during infection of distinct plant hosts. Each column reports the distribution of expression fold-changes between infection of T. aestivum (Ta) and B. distachyon (Bd) for one group of genes. In a given distribution, the median is denoted as a thick horizontal bar, 25% quartiles are shown as a box, thin horizontal bars denote 1.5 times the interquartile range, and values outside the latter range are shown as points. Expressed, the set of all genes expressed at greater than 2 reads per kilobase per million mapped reads in the library in axenic culture or during either infection. Secreted, all genes encoding proteins predicted to be secreted by Morais do Amaral et al. 2012. High Ka/Ks, genes with rates of protein evolution between Z. tritici and other Zymoseptoria species reflecting positive selection, from Stukenbrock et al. 2011. Plant-specific, genes called significantly differentially expressed between axenic growth and the two plant infection conditions in FIgure 2B. The last column reports analysis of the eukaryotic orthologous group category Secondary metabolites, biosynthesis, transport, and catabolism (genome.jgi.doe.gov/Tutorial/tutorial/kog.html). At bottom, each horizontal line reports results from a comparison of significant differential expression between the indicated gene sets: **< 0.01; ***< 0.001.
We next reasoned that, if Z. tritici genes induced during infection of wheat were of evolutionary relevance as this fungus specialized to the wheat host niche, genes subject to host-specific regulation would exhibit sequence signatures of natural selection in Z. tritici. To test this, we used our previously characterized set of 373 genes with evidence for positive selection during the divergence of Z. tritici, from within- and between-species comparisons of Z. tritici with its sister species Z. pseudotritici and Z. ardabiliae, which have different host preferences (Stukenbrock et al. 2010). As predicted, genes with signatures of positive selection in Z. tritici were significantly more highly expressed during infection of the preferred host, T. aestivum, than during infection of B. distachyon (fig. 4). Taken together, our data attest to the unique inferred functions, sequence signatures of positive selection, and genomic clustering of Z. tritici genes with wheat-specific expression patterns, all lending support to a model in which these genes have contributed to host specialization of the pathogen.
Expression and Evolutionary History of Genes on Z. tritici ACs
We next sought to investigate the regulation and function of genes on ACs of Z. tritici. On average, the 654 AC-encoded genes were expressed at 13-fold lower levels than those on CCs (fig. 1), but 174 AC-encoded genes were expressed at >2 read per million in at least one sample of our data set (supplementary table S2, Supplementary Material online). We thus reasoned that highly expressed AC-encoded genes, though relatively few in number, could carry out important biological roles and be subject to evolutionary constraint. Analyses of conservation bore out this notion: Relative to the rest of the genes on ACs, expressed genes were 2.1-fold enriched for the presence of orthologs in other Zymoseptoria strains and species (supplementary fig. S6, Supplementary Material online), and 1.4-fold enriched for signatures of selective constraint on amino acid sequence (fig. 5). We also noted 22 AC-encoded genes whose expression was significantly regulated during infection of both wheat and Brachypodium (fig. 2 and table 1). Thus, genes encoded on the Z. tritici ACs exhibit signatures of activity, host-specific regulation, and selective constraint, reflecting a likely functional role in many cases.
Fig. 5.—
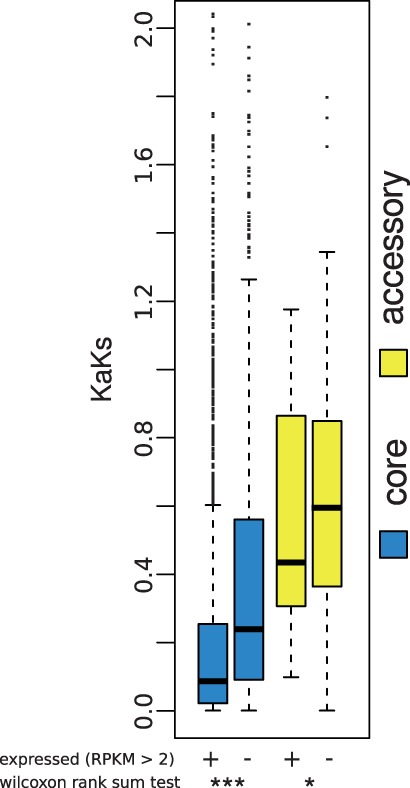
Expressed Z. tritici genes are enriched for sequence signatures of purifying selection. Each column reports the distribution of rates of protein evolution between Z. tritici and other Zymoseptoria species, from Stukenbrock et al. 2011, in one set of genes. In a given distribution, the median is denoted as a thick horizontal bar, 25% quartiles are shown as a box, thin horizontal bars denote 1.5 times the interquartile range, and values outside the latter range are shown as points. Expressed, the set of all genes expressed at greater than 2 reads per kilobase per million mapped reads in a library from axenic culture or during infection of wheat or B. distachyon.
Table 1.
Genes on Z. tritici Accessory Chromosomes Differentially Expressed between Growth Conditions
| Gene ID | Chromosome | Length (bp) | KOG ID | Sequence Homology | Mean RPKM of Replicates |
Paralogs | ||
|---|---|---|---|---|---|---|---|---|
| Ta | Bd | YMS | ||||||
| 97552 | 14 | 966 | KOG4207 | Predicted splicing factor SR protein superfamily | 3.20 | 4.01 | 0.38 | None |
| 18496 | 14 | 306 | NA | No homology | 11.73 | 9.60 | 0.67 | None |
| 78038 | 14 | 1514 | NA | C4-decarboxylate transporter | 318.21 | 364.45 | 16.34 | Mg40431 |
| 51580 | 14 | 3154 | KOG0243 | Kinesin like protein | 0.31 | 0.66 | 4.08 | Mg65848 |
| 51599 | 14 | 558 | NA | No homology | 24.29 | 21.97 | 3.63 | None |
| 106608 | 15 | 658 | NA | No homology | 0.75 | 0.50 | 3.57 | None |
| 106610 | 15 | 702 | NA | No homology | 2.73 | 2.71 | 13.12 | None |
| 111745 | 15 | 1619 | NA | HnH endonuclease | 3.56 | 4.33 | 26.82 | None |
| 97676 | 15 | 1014 | NA | No homology | 1.14 | 2.23 | 11.04 | Mg97711, Mg97810, Mg98061 |
| 106633 | 16 | 1273 | NA | No homology | 1.82 | 2.08 | 0.00 | None |
| 97764 | 16 | 3679 | KOG2992 | Nucleolar GTPase ATPase p130 (Nop14) | 2.57 | 2.06 | 7.50 | None |
| 97825 | 17 | 1380 | NA | No homology | 1.45 | 1.18 | 29.50 | Mg97877 |
| 97829 | 17 | 1164 | NA | Secretion signal | 0.38 | 0.00 | 2.02 | None |
| 51731 | 17 | 204 | NA | No homology | 0.00 | 0.00 | 117.05 | None |
| 97935 | 19 | 3901 | KOG0663 | Nucleolar GTPase ATPase p130 (Nop14) | 4.45 | 3.02 | 0.00 | None |
| 97941 | 19 | 1290 | KOG4297 | C-type lectin (chromosome segregation) | 0.68 | 2.14 | 0.22 | Mg111803 |
| 111789 | 19 | 499 | NA | No homology | 10.75 | 21.47 | 2.89 | None |
| 97959 | 19 | 489 | NA | No homology | 12.99 | 31.23 | 4.00 | None |
| 111795 | 20 | 1435 | NA | No homology | 3.63 | 0.94 | 0.68 | None |
| 98067 | 20 | 2035 | NA | No homology | 5.11 | 0.00 | 1.13 | None |
| 98089 | 21 | 519 | NA | No homology | 4.59 | 6.42 | 0.00 | None |
| 98123 | 21 | 465 | NA | No homology | 3.53 | 0.47 | 0.00 | None |
Note.—Gene ID, based on JGI genome annotation (http://genome.jgi.doe.gov/Mycgr3/Mycgr3.home.html, last accessed May 23, 2014); KOG, eukaryotic orthology group (genome.jgi.doe.gov/Tutorial/tutorial/kog.html); NA, not available. RPKM, the number of reads per kilobase per mapped reads in libraries from axenic culture (YMS, yeast malt sucrose medium) or from infections of T. aestivum (Ta) or B. distachyon (Bd).
We expected that, if a given gene on an AC carried out biological roles essential to Z. tritici biology, it would often be a unique representative of its functional class as opposed to a member of a large gene family. To test this, we catalogued the complete set of gene families in the Z. tritici genome using two homology-detection schemes, described in Materials and Methods, which define a given family on the basis of the length and proportion of genes showing high sequence identity. These strategies defined 362 and 604 gene families in the Z. tritici genome, respectively, comprising 1,020 and 1,610 genes in total (table 2, supplementary table S5, Supplementary Material online). These annotations provided no evidence for a model of rampant gene duplication on ACs in Z. tritici. Instead, only a few dozen members of gene families were encoded on ACs (27 and 44 genes, representing 4.1% and 6.7% of all AC genes, respectively; fig. 6A and table 3). Likewise, genes on CCs were more likely to be members of gene families than were genes on ACs (fig. 6B and table 3). The gene families that did include AC-encoded genes were often not uniquely comprised of such loci: Among AC-encoded gene family members, half were paralogous to CC genes (48.1–56.8%; fig. 6C). Expression patterns of unique genes and members of gene families were indistinguishable between AC-encoded and CC genes (supplementary fig. S7, Supplementary Material online). Figure 7 and supplementary figure S8, Supplementary Material online, show representative gene families in which each member in turn formed a monophyletic clade with homologs in other species, including the AC-encoded genes of the family, arguing against recent duplication on the ACs in Z. tritici. We conclude that relatively few AC genes have close paralogs in the Z. tritici genome, a signature of the potential function of many such genes that echoes our observations of expression and evolutionary constraint at these loci.
Table 2.
Composition of Paralogous Gene Families in Z. tritici
| Filter Scheme |
||
|---|---|---|
| Family size | 1 | 2 |
| Total | 362 | 604 |
| 2 | 255 | 430 |
| 3 | 54 | 94 |
| 4 | 15 | 30 |
| 5 | 12 | 20 |
| 6 | 5 | 5 |
| 7 | 6 | 8 |
| 8 | 6 | 7 |
| 9 | 4 | 3 |
| 10 | 0 | 3 |
| 11 | 0 | 1 |
| 12 | 2 | 2 |
| 14 | 0 | 1 |
| 17 | 1 | 0 |
| 20 | 1 | 0 |
| 34 | 1 | 0 |
| Max. | 34 | 14 |
Note.—Filter Schemes 1 and 2 denote protocols for evaluating paralogy; see Materials and Methods.
Fig. 6.—

Paralogous and unique genes of the Zymoseptoria tritici isolate IPO323. (A) Each set of bars reports the distribution of genes on one class of Z. tritici chromosomes that fall into paralogous gene families. The height of each colored bar reports the proportion of genes falling into gene families under one scheme for definition of the latter (schemes 1 and 2; see Methods for details). (B) Data are as in (A), except that a given set of bars reports the proportion of genes falling into paralogous gene families on each chromosome in turn, with bars from left to right reporting results from the lowest to highest numbered chromosomes of the indicated class. (C) Each panel reports analysis of paralogous gene families that include genes on accessory chromosomes, from one paralogy filter scheme. In a given panel, each curved line reports the paralogy relationship between a gene on an accessory chromosome (yellow) and the most closely related gene of its family on a core (blue) chromosome; the value at center reports the total proportion of genes on accessory chromosomes that fall into families also containing genes on core chromosomes.
Table 3.
Unique Genes and Members of Gene Families on the Core and Accessory Chromosomes of Z. tritici
| CC | AC | CC | AC | |
|---|---|---|---|---|
| Filter scheme | 1 | 1 | 2 | 2 |
| Unique | 9,305 | 627 | 8,732 | 610 |
| Paralogous | 993 | 27 | 1,566 | 44 |
| Total | 10,298 | 654 | 10,298 | 654 |
| Fisher's exact test P value | <10−6 | <10−9 | ||
Note.—CC, core chromosomes; AC, accessory chromosomes; Unique, number of genes on the indicated class of chromosomes not falling into any gene families; Paralogous, number of genes on the indicated class of chromosomes falling into a paralogous gene family; Fisher's exact test P value, result of a test for enrichment of gene family membership among genes on core chromosomes relative to those on accessory chromosomes; Filter schemes 1 and 2 denote protocols for evaluating paralogy; see Materials and Methods.
Discussion
For fungal plant pathogens, although the repertoire of virulence genes present in the genome is the fundamental basis of infectivity (Giraud et al. 2010), quantitative tuning of gene expression is also essential for establishment of infectious hyphae (Skibbe et al. 2010, Hacquard et al. 2013). As such, expression profiling serves as a powerful strategy to identify genes that underlie virulence and host specialization. In this work, we have developed methods for expression profiling of Z. tritici in planta, and we have used the results to highlight candidate determinants of host specialization in this system.
During early stages of host infection, fungal biomass is present at levels considerably lower than host biomass, and yet the two cannot be partitioned before RNA isolation or analysis. In establishing RNA-seq analysis procedures for this complex mixture of plant and fungal material, we ensured that plant reads would not contribute to estimates of fungal gene expression, mapping each complete data set’s worth of RNA-seq reads to both plant and fungal genomes. The 3–5% of high-quality reads of fungal origin emerging from our protocol is comparable to previous RNA-seq of leaf tissue at early stages of infection by the fungal pathogen Blumeria graminis (1.8% fungal reads; Hacquard et al. 2013).
Our analysis of infection regulatory programs in Z. tritici highlighted small-molecule detoxification genes, which were largely upregulated during plant infection and particularly so during growth of Z. tritici on wheat. Given the well-documented cases in which detoxification factors determine host range in other fungal plant pathogens (Bowyer et al. 1995; Coleman et al. 2009; Srivastava et al. 2013), it is tempting to speculate that such genes participate in the response of Z. tritici to reactive oxygen species produced by the host as a defense mechanism. On a genomic scale, the coincidence between signatures of positive selection and host-specific expression that we noted in Z. tritici strongly supports a role for regulatory programs in the evolution of virulence of this fungus. Qualitatively, the modest number of genes at which we detected significant differential expression between hosts (40 genes; fig. 2) dovetails with a previous report of only a few dozen genes with host-specific expression in Bl. graminis (Hacquard et al. 2013). The emerging picture is one in which expression regulation may determine a small but critical set of genes in each of these pathogen species.
In focused analyses of genes on Z. tritici ACs, we observed no evidence for elevated rates of paralogy with other elements of the Z. tritici genome, dovetailing with the high proportions of unique genes on ACs in Fusarium oxysporum (Ma et al. 2010) and Haematonectria haematococca (Coleman et al. 2009). Most of the gene composition of ACs in these species thus is unlikely to originate from the core genome. Likewise, our identification of >150 expressed genes on Z. tritici ACs, often tightly conserved within and between species, also reflect the importance of many AC genes for the fitness of the organism. Our data leave open the question of the relevance of ACs for infectivity in particular. Determinants of host specificity and virulence have been identified on ACs of other fungal plant pathogens, including the AC-encoded enzymes for detoxification of a plant phytoalexin in Nectria haematococca (Miao et al. 1991; Coleman et al. 2011) and the virulence-associated host-specific effector genes on lineage-specific chromosomes of Fusarium oxysporum (Ma et al. 2010). Though we detected no host-specific gene expression of AC genes at the very early stages of infection, 25 AC-encoded genes were upregulated during infection of wheat at 13 dpi (Yang et al. 2013). As such, future studies of expressed AC genes will continue to shed light on the biological role of these highly dynamic genomic elements.
In summary, we have constructed a pipeline for whole-transcriptome analyses of plant tissue at early stages of pathogen infection, which identified Z. tritici genes with expression-based and sequence signatures of a role in fungal growth and infectivity. Our findings will serve as a rich source of testable candidate virulence determinants in Z. tritici and underscore the power of expression profiling as a complement to genome-scale analyses in the search for the genetic basis of host–pathogen interactions.
Supplementary Material
Supplementary tables S1–S5 and figures S1–S10 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
Julien Y. Dutheil and the Fungal Biodiversity Group are acknowledged for their overall support. Plant seeds were kindly provided by Gert Kema. Franziska Scheidemantel is acknowledged for performing qRT-PCRs. This work was supported by intramural funds of the Max Planck Society to E.H.S.
Literature Cited
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke S, McDonald B. Migration patterns among global populations of the pathogenic fungus Mycosphaerella graminicola. Mol Ecol. 2005;14:1881–1896. doi: 10.1111/j.1365-294X.2005.02536.x. [DOI] [PubMed] [Google Scholar]
- Bowyer P, Clarke BR, Lunness P, Daniels MJ, Osbourn AE. Host range of a plant pathogenic fungus determined by a saponin detoxifying enzyme. Science. 1995;267:371–374. doi: 10.1126/science.7824933. [DOI] [PubMed] [Google Scholar]
- Brenchley R, et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature. 2012;491:705–710. doi: 10.1038/nature11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokenshire T. The role of graminaceous species in the epidemiology of Septoria tritici on wheat. Plant Pathol. 1975;24:33–38. [Google Scholar]
- Brunner PC, Torriani SFF, Croll D, Stukenbrock EH, McDonald BA. Coevolution and life cycle specialization of plant cell wall degrading enzymes in a hemibiotrophic pathogen. Mol Biol Evol. 2013;30:1337–1347. doi: 10.1093/molbev/mst041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Coleman JJ, White GJ, Rodriguez-Carres M, VanEtten HD. An ABC transporter and a cytochrome P450 of Nectria haematococca MPVI are virulence factors on pea and are the major tolerance mechanisms to the phytoalexin pisatin. Mol Plant Microbe Interact. 2011;24(3):368–376. doi: 10.1094/MPMI-09-10-0198. [DOI] [PubMed] [Google Scholar]
- Coleman JJ, et al. The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 2009;5:e1000618. doi: 10.1371/journal.pgen.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert SF. Supernumerary chromosomes in filamentous fungi. Curr Genet. 1998;33:311–319. doi: 10.1007/s002940050342. [DOI] [PubMed] [Google Scholar]
- Croll D, Zala M, McDonald BA. Breakage-fusion-bridge cycles and large insertions contribute to the rapid evolution of accessory chromosomes in a fungal pathogen. PLoS Genet. 2013;9:e1003567. doi: 10.1371/journal.pgen.1003567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jonge R, et al. Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc Natl Acad Sci U S A. 2012;109(13):5110–5115. doi: 10.1073/pnas.1119623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyal Z, Scharen A, Huffman M, Prescott J. Global insights into virulence and frequencies of Mycosphaerella graminicola. Ecol Epidemiol. 1985;75:1456–1462. [Google Scholar]
- Fedorova ND, et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008;4(4):e1000046. doi: 10.1371/journal.pgen.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Giraud T, Gladieux P, Gavrilets S. Linking the emergence of fungal plant diseases with ecological speciation. Trends Ecol Evol. 2010;25(7):387–395. doi: 10.1016/j.tree.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin SB, et al. Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 2011;7:e1002070. doi: 10.1371/journal.pgen.1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Cavalcanti A, Chen F-C, Bouman P, Li W-H. Extent of gene duplication in the genomes of Drosophila, nematode, and yeast. Mol Biol Evol. 2002;19:256–262. doi: 10.1093/oxfordjournals.molbev.a004079. [DOI] [PubMed] [Google Scholar]
- Hacquard S, et al. Mosaic genome structure of the barley powdery mildew pathogen and conservation of transcriptional programs in divergent hosts. Proc Natl Acad Sci U S A. 2013;110:E2219–E2228. doi: 10.1073/pnas.1306807110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- Kämper J, et al. Insights from the genome of the biotrophic plant pathogen Ustilago maydis. Nature. 2006;444:97–101. doi: 10.1038/nature05248. [DOI] [PubMed] [Google Scholar]
- Kema G, Yu D, Rijkenberg F, Shaw M, Baayen R. Histology of the pathogenesis of Mycosphaerella graminicola in wheat. Phytopathology. 1996;86:777–786. [Google Scholar]
- Kema GH, van Silfhout CH. Genetic variation for virulence and resistance in the wheat-Mycosphaerella graminicola pathosystem III. Comparative seedling and adult plant experiments. Phytopathology. 1997;87:266–272. doi: 10.1094/PHYTO.1997.87.3.266. [DOI] [PubMed] [Google Scholar]
- Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. SMART 6: recent updates and new developments. Nucleic Acids Res. 2009;37:D229–D232. doi: 10.1093/nar/gkn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L-J, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao V, Covert S, VanEtten H. A fungal gene for antibiotic resistance on a dispensable (“B”) chromosome. Science. 1991;254:1773–1776. doi: 10.1126/science.1763326. [DOI] [PubMed] [Google Scholar]
- Miele V, Penel S, Duret L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinformatics. 2011;12:116. doi: 10.1186/1471-2105-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais do Amaral A, Antoniw J, Rudd JJ, Hammond-Kosack KE. Defining the predicted protein secretome of the fungal wheat leaf pathogen Mycosphaerella graminicola. PLoS One. 2012;7:e49904. doi: 10.1371/journal.pone.0049904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Kumar S. Oxford: Oxford University Press; 2000. Molecular evolution and phylogenetics. [Google Scholar]
- Ohm RA, et al. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen dothideomycetes fungi. PLoS Pathog. 2012;8:e1003037. doi: 10.1371/journal.ppat.1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma-Guerrero J, et al. Genome wide association identifies novel Loci involved in fungal communication. PLoS Genet. 2013;9:e1003669. doi: 10.1371/journal.pgen.1003669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaele S, Kamoun S. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol. 2012;10:417–430. doi: 10.1038/nrmicro2790. [DOI] [PubMed] [Google Scholar]
- Rouxel T, et al. Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat-induced point mutations. Nat Commun. 2011;2:202. doi: 10.1038/ncomms1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibbe DS, Doehlemann G, Fernandes J, Walbot V. Maize tumors caused by Ustilago maydis require organ-specific genes in host and pathogen. Science. 2010;328:89–92. doi: 10.1126/science.1185775. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Cho IK, Cho Y. The Bdtf1 gene in Alternaria brassicicola is important in detoxifying brassinin and maintaining virulence on Brassica species. Mol Plant Microbe Interact. 2013;26(12):1429–1440. doi: 10.1094/MPMI-07-13-0186-R. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- Stergiopoulus I, de Wit P. Fungal effector proteins. Annu Rev Phytopathol. 2009;47:233–263. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- Stukenbrock EH, et al. Whole-genome and chromosome evolution associated with host adaptation and speciation of the wheat pathogen Mycosphaerella graminicola. PLoS Genet. 2010;6:e1001189. doi: 10.1371/journal.pgen.1001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenbrock EH, et al. The making of a new pathogen: insights from comparative population genomics of the domesticated wheat pathogen Mycosphaerella graminicola and its wild sister species. Genome Res. 2011;21:2157–2166. doi: 10.1101/gr.118851.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberg AHJ, et al. Meiosis drives extraordinary genome plasticity in the haploid fungal plant pathogen Mycosphaerella graminicola. PLoS One. 2009;4:e5863. doi: 10.1371/journal.pone.0005863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Li W, Jørgensen HJL. Transcriptional reprogramming of wheat and the hemibiotrophic pathogen Septoria tritici during two phases of the compatible interaction. PLoS One. 2013:e81606. doi: 10.1371/journal.pone.0081606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.