

Abstract
Adenosine (ADO) is an endogenous vasodilatory purine widely recognized to be a significant contributor to functional hyperaemia. Despite this, many aspects of the mechanisms by which ADO induces dilation in small resistance arterioles are not established, or appear contradictory. These include: identification of the primary receptor subtype; its location on endothelial (EC) or vascular smooth muscle cells; whether ADO acts on KATP channels in these resistance vessels; and the contribution of cAMP/protein kinase A (PKA) signalling to the response. In intravital microscopy studies of intact or EC-denuded skeletal muscle arterioles, we show that ADO acts via A2A receptors located on ECs to produce vasodilation via activation of KATP channels located on vascular smooth muscle cells. Importantly, we found that the signalling pathway involves cAMP as expected, but that a requirement for PKA activation is demonstrable only if the vessel is not pre-exposed to ADO. That is, PKA-dependent signalling varies with pre-exposure to ADO. Further, we show that PKA activation alone is not sufficient to dilate these arterioles; an additional EC calcium-dependent signalling mechanism is required for vasodilation to ADO. The ability of arterioles in situ to respond to occupancy of a specific receptor by utilizing different cell signalling pathways under different conditions to produce the same response allows the arteriole to respond to key homeostatic requirements using more than a single signalling mechanism. Clearly, this is likely to be physiologically advantageous, but the role for this signalling flexibility in the integrated arteriolar response that underlies functional hyperaemia will require further exploration.
Key points
The adenosine-dependent component of functional dilation of small resistance arterioles acts via A2A receptors located on endothelium to activate KATP channels on associated vascular smooth muscle.
A2A receptors are Gs-coupled, hence receptor occupancy should activate cAMP/protein kinase A (PKA) cell signalling pathways However, pre-exposure to adenosine alters the PKA dependence of the response and renders the vessel insensitive to PKA inhibition.
The adenosine pre-exposure effect is mimicked by pre-activation of PKA and is specific to adenosine, as the PKA dependence of dilation to isoproterenol (another Gs-coupled agonist) is not affected by pre-exposure.
Activation of PKA alone does not induce dilation. An additional signalling mechanism, dependent on increased EC Ca2+ via activation of cyclic nucleotide gated channels, is required together with PKA activation to produce A2A-dependent vasodilation.
This novel identification of variability in signalling downstream from a single receptor to produce the same response may reflect a mechanism for integration of key homeostatic responses.
Introduction
The close matching between blood flow and tissue metabolic rate (functional hyperaemia) is known to be impaired in many pathological conditions such as hypertension, obesity and diabetes. However, despite the considerable amount known in this field and its potential therapeutic value, there is still much that is not established concerning the mechanisms that normally mediate functional hyperaemia. The local response of resistance arterioles, which is critical for delivery of nutrients and oxygen supply to metabolically active tissue, is an important component of functional hyperaemia; many mediators of this response have been identified (Sarelius & Pohl, 2010). The majority of evidence supports the nucleoside adenosine (ADO), an endogenous vasodilatory agent, as a significant contributor to both functional and exercise hyperaemia (Bockman et al. 1976; Kille & Klabunde, 1984; Proctor, 1984; Poucher et al. 1990; Hellsten et al. 1998; Radegran & Calbet, 2001; Murrant & Sarelius, 2002; Marshall, 2007; Ross et al. 2013) with its contribution usually being of the order of 25–30% of the total dilatory response (Poucher et al. 1990; Radegran & Calbet, 2001; Murrant & Sarelius, 2002; Duza & Sarelius, 2004a). The involvement of ADO in functional hyperaemia has led to extensive research aimed at shedding light on the cellular location of ADO receptors, the receptor subtype and the signal transduction mechanisms that mediate ADO-induced vasodilation.
Many aspects of the cellular signalling pathway(s) by which ADO induces dilation in resistance arterioles are not clear. For example, despite being oppositely coupled to adenylate cyclase, evidence from several different arteriolar models indicates that both A1 and A2 (A2A and A2B) receptors can mediate dilation to ADO (Danialou et al. 1997; Hein et al. 1999, 2001; Ngai et al. 2001; Nicholls et al. 2002; Ray & Marshall, 2009; Feng & Navar, 2010). Furthermore, they appear to act via similar downstream signalling that includes KATP channel activation, but how this could occur is not established. Importantly, it is not known which of the two principal cell types in the arteriolar wall, endothelial cells (EC) or vascular smooth muscle cells (VSMC), predominates in the ADO-dependent component of the functional dilation that occurs in small resistance vessels. This question cannot be answered solely by looking at receptor expression, as A1, A2A and A2B receptors have all been identified in both EC and VSMC (Lynge & Hellsten, 2000; Wang et al. 2005). In a previous study, we found that vasodilation to ADO involved increased EC calcium (Duza & Sarelius, 2003), and, in a separate study, showed that an intact endothelium is required for dilation of small resistance arterioles in response to muscle contraction (Duza & Sarelius, 2004a). Taken together, these studies support the hypothesis that the primary action of ADO to dilate resistance arterioles is via endothelial expression of the relevant ADO receptor. Thus, a primary goal of the present study was to test the hypothesis that ADO acts primarily via receptors located on EC to produce functional dilation in small resistance arterioles.
While the principal ADO receptor that mediates the response of skeletal muscle resistance arterioles to ADO is not yet clear, most studies indicate that ADO most often induces arteriolar dilation by acting on A2A receptors (Hein et al. 1999, 2001; Ngai et al. 2001; Nicholls et al. 2002; Ray & Marshall, 2009); these studies are consistent with reports that identify an important role for A2A receptors in exercise hyperaemia (Poucher, 1996; Ray & Marshall, 2009). Although it is established that A2A receptors can induce vasodilation, the signalling downstream from A2A receptor stimulation that mediates this dilation appears to be ambiguous. A2A receptors are coupled to Gs protein signalling, thus it is remarkable that the involvement of cAMP and its downstream effector protein kinase A (PKA) have not been consistently demonstrated. For example, ADO-induced dilation in both coronary and cerebral arterioles is mediated by A2A receptors (Hein et al. 2001; Ngai et al. 2001), but PKA inhibition in these arterioles, while significantly attenuating cAMP-mediated dilation, does not affect dilation induced by ADO (Hein & Kuo, 1999; Hein et al. 2001; West et al. 2003). On the other hand, A2A receptor-mediated vasodilation of rat preglomerular microvessels is significantly dependent on both cAMP production and PKA activation (Carroll et al. 2006).
Additional evidence that contributes to the apparent inconsistency concerning PKA involvement in dilation to ADO stems from the involvement of KATP channels in this pathway. Like ADO, KATP channels have also been implicated in functional hyperaemia (Banitt et al. 1996; Saito et al. 1996; Murrant & Sarelius, 2002). Most, although not all (Murrant & Sarelius, 2002), evidence supports a role for these channels in ADO-induced arteriolar dilation (Jackson, 1993; Hein & Kuo, 1999; Hein et al. 2001). Interestingly, in arterioles where ADO acts via both A2A receptors (Hein et al. 2001; Nicholls et al. 2002) and KATP channels (Jackson, 1993; Hein & Kuo, 1999; Hein et al. 2001) to induce dilation, blocking KATP channels does not invariably affect dilation to the adenylate cyclase activator forskolin or to cAMP analogues (Jackson, 1993; Hein & Kuo, 1999; Hein et al. 2001), suggesting that at least a portion of the ADO-induced response is not mediated via cAMP signalling. In contrast, patch clamp studies of isolated VSMC from rat mesenteric arteries supply direct evidence that both ADO and A2A agonists activate KATP channels, and, further, show that this activation is sensitive to PKA inhibitors (Kleppisch & Nelson, 1995). Reports demonstrating direct cAMP/PKA activation of KATP channels are supported by the identification of multiple PKA phosphorylation sites on vascular KATP channels and their involvement in channel activation (Quinn et al. 2004; Shi et al. 2007). Thus, a second important goal of the present study was to establish the contribution of PKA and its downstream cell signalling pathways to the vasodilation induced in small resistance arterioles in skeletal muscle by A2A receptor occupancy.
To achieve these goals, we used in situ blood perfused mouse cremaster arterioles to examine the signalling by which ADO dilates these small resistance vessels. In particular, our study aimed to establish whether the relevant ADO receptor(s) acted via EC or VSMC pathways, and was designed to better understand the signalling that determines whether PKA participates in ADO-induced dilation in these vessels.
Methods
Animals and preparation
All procedures were approved by the Institutional Review Board of the University of Rochester. Male C57BL/6 mice aged 12–15 weeks were anaesthetized with pentobarbital sodium (65 mg kg−1 i.p.) and tracheotomized to ensure a patent airway. A right jugular vein catheter was placed for delivery of supplemental anaesthetic as needed. The body temperature of the mouse was maintained throughout the experiment via convective heat. The right cremaster muscle was exteriorized and prepared for intravital microscopy as previously described (Lau et al. 2000; Duza & Sarelius, 2004a,b). Briefly, the cremaster was exteriorized through a longitudinal skin incision, separated from the testis and epididymis, and gently spread over an optical quartz pillar. During surgery and experimental protocols, the tissue was continuously superfused with warmed (36°C) physiological solution composed of (mm): 131.9 NaCl, 4.7 KCl, 2.0 CaCl2, 1.2 MgSO4 and 18 NaHCO3, and equilibrated with gas containing 5% CO2–95% N2 to maintain pH at 7.4. In experiments where the A2A receptor agonist CGS21680 was used, the superfusion was equilibrated with gas containing 21% O2–5% CO2–74% N2 to facilitate maintenance of resting arteriolar tone. The cremaster microvasculature was visualized by transillumination using an Olympus BX50WI microscope with tungsten lamp, a ×25 objective (Leitz Wetzlar, Germany; numerical aperture 0.35), and a ×1.6 magnification changer. Final magnification of the site was ×400. The microscope image was displayed via a video camera (Dage MTI, Michigan City, IN, USA; CCD72) on a monitor and recorded using a DVD recorder (Sony, San Diego, CA, USA; DVO-1000MD). Before data acquisition, all preparations were allowed to stabilize for approximately 30 min. Small arterioles of the terminal microvasculature (average maximum diameter approximately 30 μm) were chosen for observation based on vessel size, presence of vascular tone and clarity. On completion of the protocols, animals were killed by anaesthetic overdose followed by pneumothorax.
Endothelial denudation
To identify whether ECs or VSMCs are the principal mediators of the response to ADO, selected regions (approximately 200 μm long) of blood-perfused arterioles were denuded of ECs using a locally introduced air embolism as described previously (Duza & Sarelius, 2003). Briefly, a sharp, triple-bevelled micropipette containing air was introduced into an arteriole that fed a targeted microvascular region. Brief, rapid pressurization was used to drive an air bubble into the microvasculature; the pipette was immediately depressurized and removed, enabling restoration of blood flow. The air bubble dispersed into the microvasculature within 5–10 min. Lack of dilation to locally applied acetylcholine (ACh; 10−4 m, 2 min) with continuing ability to dilate to locally applied sodium nitroprusside (SNP; 10−4 m, 2 min) was used to verify selective endothelial disruption and continuing vascular smooth muscle function in the target arteriole.
General protocols and agonist/antagonist application (concentrations and sources)
Drugs were either globally or locally applied as previously described (Lau et al. 2000; Murrant & Sarelius, 2002; Duza & Sarelius, 2003, 2004a,b). Global application of a drug was achieved by diluting it to the required concentration in the bicarbonate-buffered superfusion solution; the exposure was ≥15 min unless specified otherwise. For local application, drug was delivered using glass micropipettes (10–15 μm tip diameter) placed in close proximity to the arteriolar wall; the micropipette was pressurized via a manometer system (approximately 30 cmH2O ejection pressure) to generate flow out of the pipette. To confirm free flow from the micropipette and to verify that the micropipette contents flowed across the arteriole, FITC-dextran (4000 MW; 75 mm) was added to the contents of the micropipette, allowing flow detection using brief epi-illumination. Arteriolar responses are unaffected by the tracer (Frame & Sarelius, 1995). Unless noted otherwise, all agonists/activators were applied locally and all antagonists/inhibitors were applied globally. For diameter measurement protocols, each vessel was recorded during a 1 min baseline period, and a 2–3 min drug application period. In EC denuded vessels, local drug application was for 5 min to ensure that the peak local response was obtained. Recovery was monitored for at least 5 min but was not used in the analysis. Unless otherwise specified, both control and test data were obtained from the same site on the same arteriole. Maximum vessel diameter for each arteriole was obtained at the end of the protocol after restoration of control superfusate, and following at least 10 min superfusion of the entire preparation with either ADO (10−4 m) or SNP (10−4 m) or a combination of both.
Concentrations of ADO, the KATP channel activator pinacidil and the KATP channel blocker glibenclamide (60 μm, 10 μm and 10 μm respectively; Sigma, St Louis, MO, USA) were all chosen based on concentration–response curves that we completed in preliminary protocols. We chose the EC65 (60 μm) as the test concentration for locally applied ADO to ensure detection of responses to ADO antagonists. Other agonist/antagonists used to explore the cell signalling pathways activated by ADO receptors were used at effective concentrations (identified in parentheses) that were identified either from previous literature or in our preparation. We tested a series of ADO receptor subtypes (all were from Tocris Biosciences, Bristol, UK) as follows: A1 antagonist, DPCPX (0.5 μm); A3 antagonist, MRS-1334 (1 μm); A2B antagonist, MRS-1754 (1 μm); A2B antagonist, PSB-603 (1 μm); A2A antagonist, SCH-442416 (1 μm); A2A agonist, CGS-21680 (0.4 mm); and A2B agonist, BAY 60–6583 (10 μm). To explore the role of cAMP in the ADO response, we used the cAMP activator forskolin (10 μm; Sigma), and the adenylate cyclase inhibitor SQ-22536 (160 μm; Biomol International, Farmingdale, NY, USA). We also used ACh (100 μm; Sigma) and isoproterenol (0.1 μm; Sigma) as controls in selected protocols (see Results).
Endothelial cell calcium measurements
EC Ca2+ levels in confocally imaged blood perfused arterioles in situ were measured using Fluo-4 as previously described (Duza & Sarelius, 2004a,b). Briefly, the arteriole feeding a targeted region of the network was cannulated with a triple-bevelled micropipette containing 5 μm Fluo-4 AM solution (Duza & Sarelius, 2004a). Dye was perfused for approximately 15 min, after which the pipette was withdrawn and blood flow was allowed to resume. An interval of 10–15 min was allowed for the intracellular dye to de-esterify, and for vascular tone to be re-established. Adequate EC loading of Fluo-4 was confirmed by monitoring the EC Ca2+ response to locally applied ACh (10−4 m). EC Ca2+ changes were measured off-line in individual ECs by defining a region of interest that encompassed individual ECs selected because they were in focus in the confocal plane as described previously (Duza & Sarelius, 2004a). Fluorescence intensity was background subtracted and normalized to baseline as described elsewhere (Duza & Sarelius, 2004a). To explore the source of increased EC Ca2+ (see Results) we used the cyclic nucleotide gated channel blocker l-cis-diltiazem (0.3 mm, Enzo Life Sciences, Farmingdale, NY, USA).
Contribution of protein kinase A-dependent signalling to the response to adenosine
PKA involvement in the ADO-induced dilation was tested using the myristoylated PKA inhibitory peptide PKI[14–22] (PKI 25 μm; Invitrogen Grand Island, NY, USA). Initially, we explored the involvement of PKA by recording the response to ADO after PKI had been applied locally for ≥15 min. In this protocol, the vessel response to local ADO was tested before and after exposure to PKI (pre-exposure protocol). Based on the outcome of these experiments (see Results) we also used a second protocol in which the vessel was not tested with ADO before application of PKI (no pre-exposure protocol). To confirm that PKI could indeed inhibit PKA in our system, we used another G-protein coupled receptor (GPCR) agonist, isoproterenol (0.1 μm), which also acts via PKA (Morgan & Baker, 1991), to verify that in these intact arterioles, a dilation mediated by PKA could be inhibited by PKI.
To explore further the role of PKA, we also used the selective PKA activator N6-benzoyl-cAMP (N6-BNZ, 0.2–0.3 mm; Calbiochem, La Jolla, CA, USA) in three different protocols. In the first protocol, the effect of PKA activation on vasodilation was tested using 2 min local application of 0.3 mm N6-BNZ. In the second protocol, N6-BNZ was used to explore the role of PKA activation in the ADO pre-exposure effect described in Results. For this, N6-BNZ (0.2 mm) was locally applied for ≥10 min after which PKI (25 μm) was locally applied for ≥25 min and then followed by 2 min local application of ADO (60 μm). Lastly, we used N6-BNZ to examine the effect of PKA on KATP channel activation. In this protocol, we measured the response to the KATP channel activator pinacidil (10 μm) alone or following ≥20 min local application of N6-BNZ (0.3 mm).
Diameter measurements and statistical analyses
The diameter of each vessel was recorded during baseline (1 min), 2–3 min agonist application, and 3–5 min recovery, unless stated otherwise. Diameter was measured offline using video calipers (Colorado Video; model 308A) and expressed as change in diameter from baseline, or as fraction of maximal capacity to dilate, calculated as (Dtest – Dbaseline)/(Dmax – Dbaseline). Group means are reported ± s.e.m. Numbers of observations (n) are given in figure legends or text; n refers to the number of arteriolar sites, with not more than three sites per tissue and at least three animals per group, unless stated otherwise. Data were compared using paired and unpaired Student's t tests, or ANOVA, as appropriate. When the ANOVA identified significant differences, Bonferroni's post hoc analysis was used to compare means between conditions. Differences were considered significant if P ≤ 0.05.
Results
Mean baseline and maximal arteriolar diameters are shown in Table 1.
Table 1.
Baseline and maximal diameters (μm ± s.e.m.) for grouped data shown in the figures*
| Baseline diameter | Maximum diameter | |
|---|---|---|
| Fig. 1A | 5.9 ± 0.3 | 33.4 ± 0.6 |
| 1B | 5.1 ± 0.3 | 36.1 ± 1.0 |
| 1C | 5.6 ± 0.3 | 36.9 ± 1.1 |
| 1D | 5.8 ± 0.5 | 35.7 ± 1.1 |
| Fig. 2 (+EC) | 8.1 ± 0.7 | 38.1 ± 1.1 |
| (–EC) | 9.7 ± 0.7 | 28.4 ± 1.3† |
| Fig. 3A | 5.1 ± 0.2, 4.9 ± 0.2† | 34.3 ± 0.6 |
| 3B (–EC) | 9.3 ± 0.8 | 26.0 ± 2.4† |
| Fig. 4 | 6.9 ± 1.1 | 31.0 ± 1.0 |
| Fig. 5 | 7.7 ± 1.2 | 29.1 ± 1.1 |
| Fig. 6 | 7.6 ± 2.7 | 28.8 ± 2.3 |
| Fig. 8 | 6.9 ± 1.5 | 29.2 ± 3.1 |
Abbreviations: EC, endothelial cell.
Diameter measurements were not obtained for the fluorescence measurements shown in Fig. 7. Unless indicated, the table shows group means of data from all panels in each figure. †With glibenclamide.
Maximum in the EC denuded region.
Arteriolar dilation resulting from adenosine exposure or muscle contraction is mediated by A2A receptors
ADO concentrations used in this study are based on the concentration–response curve for ADO in blood-perfused arterioles (Fig. 1A). Figure 1B shows that the A2A receptor antagonist SCH-442416, but not antagonists of A1, A2B or A3 receptors, significantly attenuated the ADO-induced dilation (from 15.3 ± 2.7 to 5.6 ± 1.8 μm), indicating that ADO acts on A2A receptors to dilate skeletal muscle arterioles. Furthermore, the combination of both A2A and A2B antagonists had no further effect over A2A inhibition alone (data not shown), ruling out any significant contribution of A2B receptors in these intact microvessels. Consistent with this finding, the A2A receptor agonist CGS-21680 induced a dilation (15.2 ± 1.7 μm) that was significantly inhibited by the A2A antagonist (3.4 ± 1.0 μm), thus confirming both the presence of A2A receptors in these arterioles, and the specificity of the A2A receptor antagonist (Fig. 1C). Figure 1C also shows that the A2B agonist BAY 60–6583 can dilate these vessels, and that this dilation can be specifically blocked by the A2B antagonist MRS-1754. Together with the finding that A2B antagonists do not block the response to ADO (Fig 1B), this indicates that although these receptors are present on the arteriole, they are not utilized by ADO to induce dilation.
Figure 1. Dilation to ADO and to muscle contraction is mediated by ADO A2A receptors.
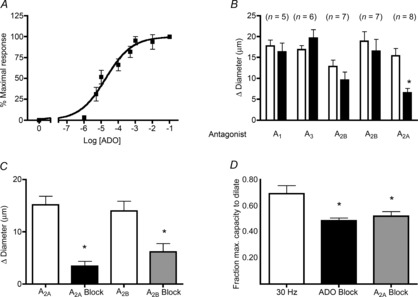
A, concentration–response curve for ADO (n = 5–7). B, A2A antagonist SCH442416 significantly attenuates the dilation to ADO, unlike the A1 antagonist DPCPX, the A3 antagonist MRS1334, and the A2B antagonists MRS1754 and PSB603, which do not. White bars, ADO; black bars, ADO+antagonist. C, dilation to the A2A agonist CGS21860 is significantly decreased in the presence of the A2A antagonist SCH442416 (black bar, n = 5), and dilation to the A2B agonist BAY 60–6583 is significantly decreased by the A2B antagonist MRS1754 (grey bar, n = 11). D, dilation induced by muscle contraction (white bar, field stimulation, 30 Hz, 15 s, n = 7) is significantly attenuated by the non-specific ADO inhibitor, xanthine amine congener (black bar, n = 5) and is equally attenuated by the A2A antagonist SCH442416 (grey bar, n = 7). Each column represents 2 min average change in diameter (A–C) or fraction of the maximum capacity to dilate (D). Bars are means ± s.e.m. *Significantly different from control response (P ≤ 0.05). ADO, adenosine.
We used electrical field stimulation (15 s at 30 Hz, 0.2 ms duration, 5–10 V) as described previously (Duza & Sarelius, 2004a) to ask whether the ADO-dependent component of muscle contraction-induced dilation was also mediated by A2A receptors. Figure 1D shows that the dilation induced by muscle contraction is inhibited by approximately 30% by the non-specific ADO inhibitor xanthine amine congener (10 μm). Importantly, we show in Fig. 1D that the xanthine amine congener-dependent inhibition of the functional arteriolar response is not significantly different from the inhibition produced by the A2A antagonist SCH442416, indicating that the ADO-dependent component of functional dilation in these arterioles is indeed mediated by A2A receptors.
cAMP is a required signalling intermediate for the adenosine-mediated arteriolar dilation
The A2A receptor is a GPCR that acts via Gαs-mediated adenylate cyclase activation and generation of cAMP. Thus, it is important to establish whether ADO-induced vasodilation is mediated by cAMP signalling in our system. For this, we used the non-specific adenylate cyclase inhibitor SQ-22536. This significantly attenuated the dilation to ADO from 15.2 ± 1.2 to 2.5 ± 1.3μm (n = 8), indicating that cAMP production is essential for the A2A-mediated vasodilation in these arterioles.
Endothelium is required for dilation to adenosine A2A receptors
We hypothesized that the A2A-dependent dilation is endothelium-dependent. To test this, we denuded ECs from regions of target arterioles as described in Methods, and tested the local responses of the resultant blood perfused, endothelium-denuded arterioles. Observations were made in vessel regions where responses to ACh and SNP confirmed EC disruption with ongoing VSMC ability to dilate, as described in Methods. Figure 2 shows that vasodilation to the A2A agonist CGS-21680 was abolished in the absence of endothelium, despite the continuing ability of the denuded region to dilate to SNP, indicating that A2A-mediated vasodilation indeed requires intact endothelium. Interestingly, the EC-denuded region remained able to dilate to ADO (Fig. 2), indicating that in the absence of endothelium, a different ADO receptor(s) can mediate vasodilation. From the data shown in Fig. 1B and C, and from the literature, we hypothesized that this VSMC-dependent response was likely to be A2B-mediated: Fig. 2 shows that in EC-denuded arterioles, the dilation to ADO is indeed abolished by the A2B antagonist PSB-603. Given that the response to ADO was insensitive to this antagonist in intact vessels (Fig 1B), we conclude, as suggested earlier, that A2B receptors are present on VSMC but that A2A receptor activation on ECs is the dominant signalling pathway in these arterioles.
Figure 2. A2A-dependent dilation requires intact endothelium.
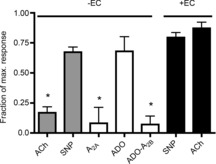
Intact blood-perfused arterioles that were denuded of their endothelium did not dilate significantly to the A2A agonist CGS21860 (white bar, n = 5), compared to the response in intact arterioles that is shown in Fig. 1. In the absence of endothelium, dilation to ADO (white bar, n = 8) was not attenuated, but was significantly attenuated by the A2B antagonist PSB603 (white bar, n = 7), which was ineffective in blocking dilation to ADO in intact arterioles (Fig. 1). Effective disruption of endothelium was confirmed by the significantly decreased dilation to ACh (grey bar, n = 8), with continuing dilation to SNP (grey bar, n = 8). Bars are means of maximal capacity to dilate, ± s.e.m. *Significantly different from mean responses to ACh and SNP in intact vessels (black bars), P ≤ 0.05. ACh, acetylcholine; ADO, adenosine; EC, endothelial cell; SNP, sodium nitroprusside.
Dilation produced by A2A receptors and by activation of adenylate cyclase requires KATP channel activation
To test whether or not KATP channels are required for dilation to ADO in the present model, we used glibenclamide (10 μm) to block KATP channel activity. Figure 3A shows that glibenclamide significantly attenuated both ADO and A2A agonist induced dilation by 63.5 ± 11.9% and 77.6 ± 8.1%, respectively, indicating a central role for KATP channels in A2A-mediated dilation to ADO in these arterioles. The specificity of this response was confirmed by the inability of glibenclamide to inhibit dilation to ACh (Fig. 3A), which acts independently of KATP channels to induce dilation (Banitt et al. 1996; de Wit, 2010).
Figure 3. Dilation mediated by ADO, A2A receptor activation, and by activation of adenylate cyclase, is dependent on KATP channels located on vascular smooth muscle.
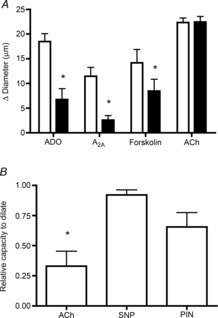
A, KATP channel blocker glibenclamide (black bars) significantly inhibits the dilation to ADO (n = 9), the A2A agonist CGS21860 (n = 6), and the adenylate cyclase activator forskolin (n = 7) but does not affect the dilation to ACh (n = 7), which acts via a different cell signalling pathway. *Significantly different from response in absence of glibenclamide (white bars). B, in endothelial denuded arterioles (confirmed by the significant decrease in dilation to ACh), the response to the KATP channel activator pinacidil is not significantly different from that produced by SNP (n = 9 for each condition). *Significantly different from dilation to SNP. All bars are group means ± s.e.m., P ≤ 0.05. ACh, acetylcholine; ADO, adenosine; PIN, pinacidil; SNP, sodium nitroprusside.
An important remaining question in these intact resistance arterioles is ‘Where are these KATP channels located – on ECs or VSMCs?’. To address this, we locally applied the KATP channel opener pinacidil to EC-denuded arterioles. As shown in Fig. 3B, pinacidil induces a significant dilation in EC-denuded vessels, indicating that KATP channels are indeed located on VSMCs.
Pre-exposing arterioles to adenosine alters the signalling that mediates arteriolar dilation
We were surprised to find that the ADO induced response (18 ± 1.3 μm) was not significantly attenuated in the presence of the specific PKA inhibitor, PKI (16.1 ± 1.8 μm, Fig. 4A), implying that PKA does not mediate the dilation to ADO in these vessels. To confirm that application of PKI is capable of blocking PKA signalling in the intact arteriolar wall, we tested the effect of PKI on the β-adrenergic receptor agonist isoproterenol (Fig. 4B), which acts via PKA (Shi et al. 2007). Dilation to isoproterenol (19.7 ± 1.8 μm) was significantly inhibited by PKI (6.1 ± 1.5 μm), verifying that indeed, application of PKI has the capacity to block PKA in the cells of the arteriolar wall.
Figure 4. Pre-exposure to ADO alters the role of protein kinase A in the dilation produced by ADO.
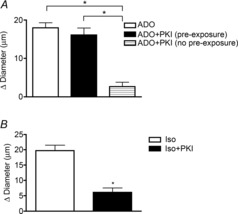
A, PKA inhibitor, PKI does not alter the dilation to ADO when the vessel was pre-exposed to ADO (black bar, n = 8) but significantly attenuates the dilation to ADO when the vessel was not pre-exposed to ADO (hatched bar, n = 6). White bar, control response to ADO (n = 8). B, after pre-exposure to isoproterenol, PKI significantly attenuates the dilation to isoproterenol (n = 6). All bars are group mean ± s.e.m. *Significantly different from control response, P ≤ 0.05. ADO, adenosine; Iso, isoproterenol; PKI, myristoylated PKI[12-22]; PKA, protein kinase A.
Given that our data show that cAMP, but not PKA, is involved in the cell signalling pathway downstream from A2A receptor activation, we asked whether cAMP-dependent, but PKA independent, signalling pathways were responsible for the ADO-dependent dilation. Another signalling molecule that is activated directly via cAMP and mediates cAMP-dependent intracellular signalling that is not via PKA, is Exchange Protein Activated by cAMP (Epac) (de Rooij et al. 1998; Oestreich et al. 2007; Cazorla et al. 2009). We therefore sought to determine whether Epac activation might account for the PKA independent response to ADO. As Epac inhibitors are not available, we instead used the cell permeable Epac activator cpTOME (Oestreich et al. 2007) to ask whether activation of Epac would induce vasodilation. However, local application of cpTOME (100 μM, 15 min) did not produce a response (6.3 ± 0.6 with cpTOME vs. 6.5 ± 1.5 μm in controls, n = 9), indicating that although A2A receptor occupancy activates cAMP, Epac is unlikely to contribute to any vasodilation, including that initiated by ADO.
Because ADO is implicated in ischaemia pre-conditioning effects (Mubagwa & Flameng, 2001), we wondered whether the surprising failure to implicate PKA signalling in the ADO-dependent response could be related to the pre-exposure to ADO that was part of our protocol. For the dilations to ADO and isoproterenol reported in Fig. 4, we used a protocol in which we first tested the response to the agonist, then exposed that site to PKI, and then retested the response to agonist at the same site. In contrast, the hatched bar in Fig. 4A shows results obtained using a modified protocol (no pre-exposure protocol) in which individual vessels were first exposed to PKI without being previously exposed to ADO, and then, following the PKI application, were tested with ADO. The figure shows that in contrast to arterioles that were pre-exposed to ADO, in arterioles that were not pre-exposed to ADO, inhibition of PKA significantly reduced the dilation to ADO (2.6 ± 1.2 μm) compared to the control ADO response (17.0 ± 1.1 μm). That is, in these arterioles we were indeed able to demonstrate involvement of PKA in the downstream cell signalling pathway, whereas, as we showed initially, when vessels had been previously exposed to ADO, inhibition of PKA was without effect on the subsequent dilation. Thus, our data indicate that prior exposure to ADO induces a ‘pre-exposure’ effect that alters how ADO produces dilation, despite that the dilation itself is not different in the two conditions (17.4 ± 2.6 μm, pre-exposure vs. 16.5 ± 1.9 μm, no pre-exposure, n = 7).
Protein kinase A activation alone is not sufficient to induce arteriolar vasodilation, but modulates the arteriolar dilation produced by KATP channel activation
As a first step toward defining this novel pre-exposure effect of ADO, we hypothesized that pre-exposure to ADO and the subsequent activation of PKA would lead to the phosphorylation of downstream effectors, likely including KATP channels. We further postulated that this phosphorylation must have been sustained throughout the time frame of our experiment, and hence the second time the arteriole was exposed to ADO, PKA inhibition would have no effect on the response. Because we routinely observe that the recovery to basal tone following ADO-induced dilation occurs within 5 min (often within 2 min) of cessation of agonist application, we infer that KATP channel phosphorylation by PKA cannot directly induce channel activation in these vessels. To address this hypothesis, we first asked whether direct activation of PKA (using the specific PKA activator N6-BNZ) could produce vasodilation. However, arteriolar diameter in response to 2 min application of N6-BNZ (6.9 ± 1.1 μm, Fig. 5A) was not significantly different from baseline (7.7 ± 1 μm), indicating that PKA activation alone does not suffice to induce dilation. Higher concentrations of N6-BNZ (up to 1 mm) were also ineffective (data not shown).
Figure 5. PKA activation alone is not sufficient to induce dilation, but can potentiate the arteriolar dilation to pinacidil. The contribution of KATP channels to the ADO dilation is not changed with ADO pre-exposure.
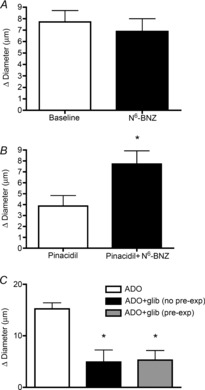
A, PKA activator N6-BNZ (black bar) does not produce arteriolar dilation above baseline (n = 7). B, arteriolar response to pinacidil (white bar) was enhanced in the presence of the PKA activator N6-BNZ (black bar). *Significantly different from pinacidil alone (n = 7, P ≤ 0.05). C, inhibition of dilation to ADO by the KATP channel blocker glibenclamide is not different after ADO pre-exposure (grey bar) compared to no pre-exposure (black bar). n = 8. *Significantly different from ADO alone (white bar), P ≤ 0.05. All bars are group means ± s.e.m. ADO, adenosine; N6-BNZ, N6-benzoyl-cAMP; glib, glibenclamide; PKA, protein kinase A.
To explore this further, we sought to provide evidence that, in intact arterioles in situ, PKA has the ability to directly modulate KATP channel activity. To do this, we asked whether PKA activation could modify the dilation induced by the KATP channel activator pinacidil. We found that dilation to pinacidil (3.9 ± 1 μm) was significantly increased (to 7.7 ± 1 μm) in the presence of the PKA activator N6-BNZ (Fig. 5B). This shows that PKA can potentiate KATP channel activation in arterioles in situ, indicating that, indeed, PKA can modulate KATP channel activity, and thus supporting our hypothesis that the ADO pre-exposure effect is mediated by PKA-dependent modulation of KATP channels.
The finding that PKA activation, although involved in the ADO response, does not by itself result in vasodilation, when taken together with our data showing that the KATP channel activator pinacidil induces dilation in these arterioles (white bar, Fig. 5B), indicates that for the response to ADO, PKA interaction with the KATP channel is not by itself sufficient for the activation of the channel and subsequent vasodilation. Furthermore, the contribution of KATP channels to ADO-dependent dilation does not change according to whether the vessel was pre-exposed to ADO (Fig. 5C).
The adenosine pre-exposure effect can be mimicked by pre-exposing arterioles to a protein kinase A activator
Our hypothesis (above) predicts that pre-exposure to a PKA activator should have the same effect as pre-exposure to ADO. To test this, we first activated PKA by pre-exposing the arteriole to the PKA activator N6-BNZ, and then examined the effect of PKI (the PKA inhibitor) on the ADO mediated dilation (Fig. 6). This response was compared with the response to ADO in the previously completed pre-exposure and no pre-exposure protocols (Fig. 4A). When the PKA inhibitor (PKI) was applied after pre-exposure of the vessel to N6-BNZ, ADO induced a significant dilation of 76 ± 2.5% of the maximal response capacity, which is not significantly different from that seen after ADO pre-exposure (76.8 ± 9.5%). In vessels that were not pre-exposed either to ADO or to the PKA activator, the dilation to ADO after PKA inhibition was significantly less (10.3 ± 5.3% of the maximal capacity to dilate). Thus, the pre-exposure effect of ADO, in which the downstream signalling pathway is no longer sensitive to PKA inhibition, can be mimicked by pre-exposure to a PKA activator. These results support the hypothesis that the pre-exposure effect of ADO is mediated either by PKA activation itself or by one of the signalling steps distal to PKA activation.
Figure 6. PKA activation is able to mimic the pre-exposure effect seen with ADO.

Pre-exposure to the PKA activator N6-BNZ prevented inhibition of dilation by the PKA inhibitor PKI (hatched bar, n = 5) in the same way that pre-exposure to ADO prevented inhibition of dilation by PKI (white bar, n = 8); in contrast, with no pre-exposure to the PKA activator, PKI inhibited the dilation to ADO as expected (black bar, n = 6). *Significantly different from control response, P ≤ 0.05. All bars are means ± s.e.m. ADO, adenosine; N6-BNZ, N6-benzoyl-cAMP; PKA, protein kinase A; PKI, myristoylated PKI[12-22].
An adenosine-induced increase in endothelial cell Ca2+ that is mediated by cyclic nucleotide gated channels is necessary for dilation
Our results so far clearly indicate that the arteriolar dilation to ADO is not mediated exclusively by the cAMP/PKA signalling pathway; thus, an additional signalling pathway must be involved. We therefore asked whether ADO can directly alter EC Ca2+ levels in these skeletal muscle arterioles as expected from earlier findings (Duza & Sarelius, 2003, 2004a). Fluo-4-loaded ECs were used to measure changes in Ca2+ in individual ECs in response to ADO. Changes in fluorescence intensity were recorded during 1 min (normalized to the averaged value during 1 min baseline) in the absence or presence of ADO (Fig. 7A). Globally applied ADO induced a significant increase in the averaged EC Ca2+ compared to control (F/F0 = 1.06 and 0.97 respectively; P < 0.05) confirming that ADO does indeed induce an increase in EC Ca2+ in these arterioles (Fig. 7B).
Figure 7. Adenosine induces an increase in EC calcium via cyclic nucleotide gated channels.
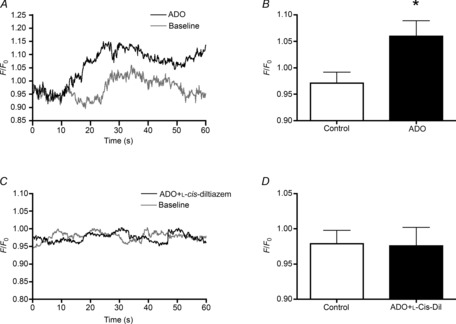
A, averaged time course of EC Ca2+ (44 cells) in baseline (control) conditions (grey line) and in response to ADO (black line) in Fluo-4 loaded intact blood-perfused arterioles. EC Ca2+ changes in (A) and (C) are expressed as relative change in fluorescence intensity. B, averaged representation of the data presented in (A). C, averaged time course of EC Ca2+ (43 cells) in baseline conditions (grey line) and in response to ADO in the presence of the cyclic nucleotide gated channel blocker l-cis-diltiazem (black line). D, averaged representation of the data presented in (C). *Significantly different from control (P ≤ 0.05). Bars in (B) and (D) are means ± s.e.m. ADO, adenosine; EC, endothelial cell.
To test if the A2A-dependent dilation we describe here also activates CNG channels in arteriolar ECs, we used the selective CNG channel blocker l-cis-diltiazem (Frings et al. 1992; Haynes, 1992; Cheng et al. 2008). Figure 7 shows that the increase in EC Ca2+ produced by ADO was abolished in the presence of l-cis-diltiazem (to F/F0 = 0.979, Fig. 7C and D). Furthermore, blocking CNG channels profoundly attenuated the ADO-mediated dilation, from 15.6 ± 1.9 to 3.6 ± 1.7 μm (Fig. 8). As expected from our hypothesis, this effect of l-cis-diltiazem was unaffected by whether or not the vessel had been pre-exposed to ADO (data not shown). The data in Figs 7 and 8 thus support a role for CNG channels in the regulation of resistance arteriolar responses, and identify the involvement of these channels in ADO-mediated vasodilation. Importantly, the nitric oxide donor SNP produced a significant dilation in the presence of l-cis-diltiazem (Fig. 8), confirming the specificity of the CNG channel blocker. Furthermore, dilation to forskolin was reduced by l-cis-diltiazem (62.6 ± 3.7 vs. 6.6 ± 3.1% of maximal capacity to dilate, n = 5), further confirming a role for CNG channels in cAMP-dependent vasodilation in these vessels.
Figure 8. Inhibition of cyclic nucleotide gated channels attenuates ADO dilation.

Dilation to ADO in the absence (white bar) or presence (black bar) of the cyclic nucleotide gated channel blocker l-cis-diltiazem. Arteriolar functionality and l-cis-diltiazem specificity is demonstrated by the ability to dilate in response to SNP in the presence of l-cis-diltiazem (striped bar). *Significantly different from ADO (P ≤ 0.05). Bars are mean of n = 6 responses, ± s.e.m. ADO, adenosine; SNP, sodium nitroprusside.
Discussion
In this study, we show that in small blood-perfused resistance arterioles in situ, ADO acts via A2A receptors and, further, we show that this A2A-dependent dilation requires endothelium. Using muscle contraction as a model of exercise hyperaemia, we show that indeed, the ADO-dependent component of this response is accounted for by endothelial A2A-mediated dilation. Our data support a model in which this A2A receptor-dependent response is coupled through cAMP to signalling pathways that result in activation of KATP channels; this pathway includes an as yet unidentified signalling intermediate, which together with increased EC Ca2+ modulates PKA-dependent enhancement of KATP channel activity. Importantly, we further show, in these small resistance arterioles, that the downstream signalling by which ADO induces vasodilation is switched from PKA-dependent to PKA independent when the vessel is pre-exposed to ADO. This pre-exposure dependent alteration in downstream signalling is specific to ADO, as we show that dilation to isoproterenol (also a Gs-coupled GPCR) remains PKA-dependent without regard for whether the vessel was pre-exposed. The physiological context of this pre-exposure effect remains to be explored, but it clearly exemplifies one way in which multiple signalling pathways can converge to support a key physiological response – in this case functional hyperaemia.
Our finding that in these small resistance arterioles, ADO acts via A2A receptors to mediate vasodilation is consistent with earlier studies that indicate a requisite role for A2A receptors both in the exercise-induced increase in muscle blood flow and in ADO-induced dilation of non-skeletal muscle arterioles (Poucher, 1996; Hein et al. 1999; Carroll et al. 2006; Marshall, 2007; Ray & Marshall, 2009). We show here that ADO-induced dilation is initiated via A2A receptors located on ECs, while A2B receptors, despite their expression in the microvasculature (Lynge & Hellsten, 2000; Feng & Navar, 2010) remain ‘silent’ and mediate dilation only when the endothelium is removed (Fig. 2). Presumably, this represents either or both of possible differences in receptor expression density, and the higher affinity of A2A vs. A2B receptors for ADO. Because the interstitial pool of ADO is critical in hyperaemic responses (Hellsten et al. 2012b), it is often inferred that the most likely target of the ADO pool generated by purine release from skeletal muscle myocytes would be receptors located on VSMCs. However, recent work has clarified that in small resistance arterioles, such as the ones studied here, there is a unique morphological arrangement of VSMCs and ECs such that EC processes project through the internal elastic lamina (IEL) and are thus intimately apposed to the overlying VSMCs (Mather et al. 2005; Sandow et al. 2009, 2012). Further, it has been shown that these projections represent regions of local signalling compartmentalization that are critical to communication between the two cell types, whether directly via gap junctions or via release of local paracrine agents (Isakson, 2008; Figueroa & Duling, 2009; Garland et al. 2011). It has also been shown that there are ‘holes’ in the IEL that are not occupied by EC projections (Mather et al. 2005; Sandow et al. 2009) and that we speculate could thus act as a low-resistance pathway for passage of material across the IEL. These observations suggest that, in these small resistance vessels, interstitial ADO could occupy A2A receptors located on the abluminal side of the ECs due to this morphological arrangement in which EC processes project through the IEL, and, in addition, ADO could diffuse through the holes present in the IEL to stimulate EC A2A receptors. Thus, interstitial ADO production need not be confined to the luminal side of the arteriole to act on ECs. Whether endothelial ADO receptors are compartmentalized to the myoendothelial projections, or are located on the EC abluminal plasma membrane adjacent to the IEL, is an important question that is beyond the scope of the present work.
Our study highlights the complexity of ADO-dependent signalling, particularly in intact blood vessels where ADO receptors and their downstream cellular signalling molecules are distributed between two cell types (VSMC and EC) that, as discussed above, directly communicate with each other using a variety of compartmentalized signalling complexes (Isakson et al. 2007; Dora, 2010; Garland et al. 2011; Sandow et al. 2012). We confirmed in our system that as expected, cAMP plays a significant role in the signalling pathway for dilation to ADO. We note that other studies have suggested that in A2A receptor-induced dilation, ADO does not act via cAMP (Hein & Kuo, 1999; Hein et al. 2001; West et al. 2003); however, the basis for that conclusion arises primarily from experiments using PKA inhibitors. Importantly, not only do these inhibitors not address other cAMP-dependent signalling such as via Epac or CNG channel activation but we now provide evidence that the contribution of PKA activation to ADO-induced dilation is dependent on whether the arteriole was pre-exposed to ADO. Thus, an important conclusion from our study is that PKA inhibition alone cannot be used to probe involvement of ADO in vascular responses, as this experimental approach is clearly open to misinterpretation.
Using the KATP channel inhibitor glibenclamide we showed that both ADO and A2A-induced dilations are mediated via activation of KATP channels (Fig. 3). While most reports support a role for KATP channels in arteriolar dilation to ADO (Kleppisch & Nelson, 1995; Danialou et al. 1997; Hein et al. 1999), the involvement of KATP channels in the skeletal muscle arteriolar response to ADO may vary; for example, we previously found that glibenclamide did not attenuate ADO-dependent dilation in hamster cremaster arterioles (Murrant & Sarelius, 2002). This could reflect a difference in the targeted ADO receptor subtype; however, it seems more likely that our previous inability to demonstrate KATP channel involvement is a reflection of the differential ability of various signalling pathways to be activated by the same receptor, as we describe in the present work. We note with interest that the cell signalling pathway that leads to KATP channel activation in response to ADO is still not fully defined. Our current data are consistent with the considerable body of work showing that KATP channel activation in response to ADO and A2A agonists is mediated via activation of cAMP/PKA (Kleppisch & Nelson, 1995). Thus, we show that ADO induced dilation is significantly attenuated in the presence of an adenylate cyclase inhibitor and, further, that the adenylate cyclase activator forskolin induces dilation via KATP channels (Fig. 3). Together, these findings support the conclusion that ADO acts via cAMP to activate KATP channels. Overall, these observations support our earlier conclusion that the involvement of cAMP in A2A receptor-mediated dilation can vary, perhaps among different tissues or in the same tissue when it is in different tissue states; moreover, they suggest the existence of multiple signalling pathways for KATP channel activation downstream from occupancy of A2A receptors. Our findings thus give strong credence to the concept that more than a single downstream signalling pathway is activated by A2A receptor occupancy.
We report the novel finding that pre-exposure to ADO changes the downstream cell signalling requirements by which dilation is achieved, translating the response from PKA-dependent to PKA independent after ADO pre-exposure. This is a most compelling piece of evidence in support of the ability of these small arterioles to produce the same response outcome via different signalling requirements consequent to stimulation of the same receptor. Importantly, we showed that activation of β-adrenergic receptors (which like A2A receptors are Gs-coupled GPCRs) does not result in a pre-exposure effect on downstream PKA activation (Fig. 4). This indicates that the pre-exposure effect is not a general feature of all Gs-coupled GPCR-mediated dilation; we speculate that the effect is specific to ADO, but addressing this possibility is beyond the scope of the present study. Similarly and importantly, further studies will be necessary to define the timeline over which the pre-exposure effect is manifest.
Considering the involvement of both PKA and KATP channels in the response to ADO reported here, and the numerous reports that demonstrate that PKA can mediate phosphorylation and activation of KATP channels (Quinn et al. 2004; Shi et al. 2007), including by ADO and A2A receptor agonists (Kleppisch & Nelson, 1995), we have hypothesized that the ADO pre-exposure phenomenon could be a reflection of the kinetics of PKA-mediated phosphorylation of KATP channels. In this scenario, PKA activation in the first exposure to ADO would cause phosphorylation of KATP channels. The channels then remain phosphorylated throughout the time course of the protocol and therefore the second ADO-induced dilation is not affected by PKA inhibition. As we observe that arterioles rapidly recover from ADO-induced dilation, we also hypothesized that this PKA-dependent phosphorylation by itself does not lead to the activation of these KATP channels, because their activation would result in vasodilation, as can be shown using pinacidil. In support of our hypothesis, we show three different results. First, we show that the ADO pre-exposure effect can be mimicked by pre-exposing the arterioles to a PKA activator instead of ADO (Fig. 6) indicating that the ADO pre-exposure effect is mediated by PKA or its downstream signalling. Second, we show that PKA activation alone cannot induce vasodilation (Fig. 5A) while the KATP channel opener pinacidil does (Fig. 5B), indicating that PKA by itself is not sufficient to activate KATP channels. This conclusion is supported by Mauerer et al. (1998) who showed in renal epithelia that PKA phosphorylation is necessary but not sufficient for KATP channel activation. Lastly, we show that PKA activation potentiates the dilation to pinacidil (Fig. 5B), demonstrating that PKA has the capacity to modulate KATP channel activity in these arterioles.
Our findings also extend current understanding of arteriolar KATP channel activation in response to ADO. While KATP channels are established as mediators of arteriolar dilation to ADO (Jackson, 1993; Hein et al. 1999, 2001), some earlier studies have concluded that PKA is not required for activation of these channels (Jackson, 1993; Hein & Kuo, 1999; West et al. 2003), and the signalling that leads to their activation has remained elusive. However, most data that are used to exclude PKA as a mediator of ADO-dependent dilation are based on PKA inhibition protocols. Given the pre-exposure effect that we describe in the current study, it appears likely that failure to demonstrate a role for PKA in previous work might reflect pre-exposure to ADO in the protocols of those studies (Hein & Kuo, 1999; West et al. 2003). Furthermore, while we demonstrated the involvement of PKA in dilation to ADO in arterioles that were not pre-exposed to ADO, we also showed that PKA activation, although capable of modulating KATP channels, is by itself not sufficient to induce dilation. This indicates that an additional signal is required to act together with the cAMP/PKA pathway to mediate dilation in response to ADO; our study points towards EC Ca2+ changes as a likely candidate for this additional signal. We have previously shown that EC Ca2+ changes are required for muscle contraction-induced arteriolar dilation (Duza & Sarelius, 2004a), and have shown in non-skeletal muscle arterioles that EC Ca2+ is increased by ADO (Duza & Sarelius, 2003). In the current study, we show that EC Ca2+ is indeed increased by ADO in these small resistance vessels. Ca2+ was measured in all in focus ECs (Fig. 7); while changes in individual ECs varied considerably (from 0 to 67%), as expected from earlier work (Duza & Sarelius, 2004a,b), the averaged response that we report is of the same order of magnitude as we have reported previously in response to purines in other small arterioles (Duza & Sarelius, 2003). Our data further show that this Ca2+ increase is most likely via activation of CNG channels. Furthermore, we show that blocking CNG channels significantly attenuated ADO-mediated dilation, indicating that this increase in EC Ca2+ is necessary for the response to ADO. This strongly supports the suggestion that the additional signal needed along with PKA to activate KATP channels is EC Ca2+, or is EC Ca2+ dependent. Potential candidates include Ca2+/calmodulin-dependent protein kinase II, protein kinase C and protein kinase G, all of which are able to phosphorylate KATP channels (Light et al. 2000; Han et al. 2002; Kawano et al. 2009). Figure 9 shows a schematic integrating our findings, illustrating our current understanding of the mechanisms activated by ADO to cause dilation of skeletal muscle resistance arterioles.
Figure 9. Schematic illustration of proposed model for cell signalling mechanisms underlying the dilation to ADO in small resistance arterioles in situ in skeletal muscle.
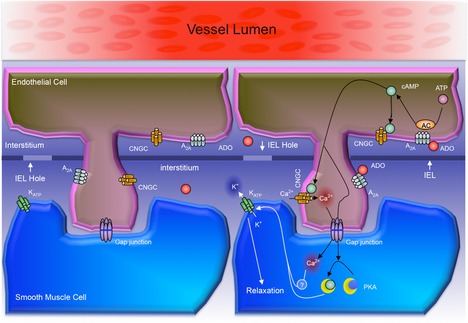
The schematic shows an EC with a process projecting through one of several openings in the IEL and thus closely apposed to the underlying VSMC. The left-hand version shows the location on the plasma membranes of the receptors and channels that we studied directly (or, for the myoendothelial gap junction, inferred from our data). Clearly, other transmembrane receptors, channels and transporters are likely to contribute to the integrated functional response. The right-hand side summarizes the cell signalling mechanisms that from our data we propose link A2A receptor activation on ECs to KATP channel activation on VSMCs. Thus, ADO binds to the A2A receptor on ECs, resulting in cAMP production. cAMP then activates both CNGCs (leading to increased EC Ca2+) and via the myoendothelial gap junction, activates PKA in the VSMC. Ca2+ (or a Ca2+ activated signalling intermediate) also then passes through a myoendothelial gap junction to the VSMC. cAMP activates PKA and, separately, Ca2+ activates the as yet unidentified intermediate: both are required to activate the KATP channel and so cause VSMC relaxation. Prolonged phosphorylation of KATP channels by PKA results in the exclusion of PKA activation from subsequent responses to ADO via A2A receptors (the pre-exposure effect): A2A receptors still activate the CNGC pathway, enabling the Ca2+-dependent component of KATP channel activation to act on the phosphorylated channel to cause VSMC relaxation. AC, adenylate cyclase, ADO, adenosine; EC, endothelial cell; IEL, internal elastic lamina; CNGC, cyclic nucleotide gated channel; PKA, protein kinase A; VSMC, vascular smooth muscle cell.
In summary, our study shows that ADO acts via A2A receptors on endothelium to produce vasodilation using downstream signalling that varies according to whether the vessel was pre-exposed to ADO. While we show that PKA is involved in this response, most likely by acting on KATP channels, we also show that PKA activation by itself is not sufficient to activate these channels, indicating that a second signal is required for KATP channel activity. Our data suggest that this signal is EC Ca2+ or is EC Ca2+ dependent. The ability of arterioles in situ to respond to occupancy of a specific receptor by utilizing multiple cell signalling pathways under different conditions to produce the same response clearly has the capacity to be physiologically advantageous. It is now widely recognized that regulation of key homeostatic responses (such as functional hyperaemia) depends on the integrated outcome of a spectrum of signalling events and pathways (Sarelius & Pohl, 2010; Hellsten et al. 2012a). In this context, the ability to respond to the same signalling molecule (ADO) by utilizing cell signalling pathways in different ways confers an additional, and previously undescribed, degree of flexibility on the ability of the system to respond to key homeostatic requirements. Clearly, the physiological role of this mechanism will need to be defined in future studies, particularly in relation to other signalling contributions to the integrated arteriolar response that subserves functional hyperaemia.
Acknowledgments
None.
Glossary
- ADO
adenosine
- CNG
cyclic nucleotide gated
- EC
endothelial cell
- Epac
exchange protein activated by cAMP
- GPCR
G-protein coupled receptor
- N6-BNZ
N6-benzoyl-cAMP
- PKA
protein kinase A
- PKI
myristoylated PKI[12–22] amide
- SNP
sodium nitroprusside
- VSMC
vascular smooth muscle cell
Additional information
Competing interests
The authors have no conflict of interest to disclose.
Author contributions
N.M. designed and completed experiments, analysed and interpreted data and contributed to writing the manuscript. P.A.T. designed and completed experiments, analysed data and critiqued the manuscript. I.H.S. designed experiments, analysed and interpreted data, and contributed to writing and revision of the manuscript.
Funding
NIH RO1 HL 76414 and NIH RO1 HL105909
References
- Banitt PF, Smits P, Williams SB, Ganz P, Creager MA. Activation of ATP-sensitive potassium channels contributes to reactive hyperemia in humans. Am J Physiol Heart Circ Physiol. 1996;271:H1594–H1598. doi: 10.1152/ajpheart.1996.271.4.H1594. [DOI] [PubMed] [Google Scholar]
- Bockman EL, Berne RM, Rubio R. Adenosine and active hyperemia in dog skeletal muscle. Am J Physiol. 1976;230:1531–1537. doi: 10.1152/ajplegacy.1976.230.6.1531. [DOI] [PubMed] [Google Scholar]
- Carroll MA, Doumad AB, Li J, Cheng MK, Falck JR, McGiff JC. Adenosine2A receptor vasodilation of rat preglomerular microvessels is mediated by EETs that activate the cAMP/PKA pathway. Am J Physiol Renal Physiol. 2006;291:F155–F161. doi: 10.1152/ajprenal.00231.2005. [DOI] [PubMed] [Google Scholar]
- Cazorla O, Lucas A, Poirier F, Lacampagne A, Lezoualc'h F. The cAMP binding protein Epac regulates cardiac myofilament function. Proc Natl Acad Sci U S A. 2009;106:14144–14149. doi: 10.1073/pnas.0812536106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Leung YK, Shen B, Kwok YC, Wong CO, Kwan HY, Man YB, Ma X, Huang Y, Yao X. CNGA2 channels mediate adenosine-induced Ca2+ influx in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:913–918. doi: 10.1161/ATVBAHA.107.148338. [DOI] [PubMed] [Google Scholar]
- Danialou G, Vicaut E, Sambe A, Aubier M, Boczkowski J. Predominant role of A1 adenosine receptors in mediating adenosine induced vasodilatation of rat diaphragmatic arterioles: involvement of nitric oxide and the ATP-dependent K+ channels. Br J Pharmacol. 1997;121:1355–1363. doi: 10.1038/sj.bjp.0701247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- de Wit C. Different pathways with distinct properties conduct dilations in the microcirculation in vivo. Cardiovasc Res. 2010;85:604–613. doi: 10.1093/cvr/cvp340. [DOI] [PubMed] [Google Scholar]
- Dora KA. Coordination of vasomotor responses by the endothelium. Circ J. 2010;74:226–232. doi: 10.1253/circj.cj-09-0879. [DOI] [PubMed] [Google Scholar]
- Duza T, Sarelius IH. Conducted dilations initiated by purines in arterioles are endothelium dependent and require endothelial Ca2+ Am J Physiol Heart Circ Physiol. 2003;285:H26–H37. doi: 10.1152/ajpheart.00788.2002. [DOI] [PubMed] [Google Scholar]
- Duza T, Sarelius IH. Increase in endothelial cell Ca2+ in response to mouse cremaster muscle contraction. J Physiol. 2004a;555:459–469. doi: 10.1113/jphysiol.2003.051029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duza T, Sarelius IH. Localized transient increases in endothelial cell Ca2+ in arterioles in situ: implications for coordination of vascular function. Am J Physiol Heart Circ Physiol. 2004b;286:H2322–H2331. doi: 10.1152/ajpheart.00006.2004. [DOI] [PubMed] [Google Scholar]
- Feng MG, Navar LG. Afferent arteriolar vasodilator effect of adenosine predominantly involves adenosine A2B receptor activation. Am J Physiol Renal Physiol. 2010;299:F310–F315. doi: 10.1152/ajprenal.00149.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa XF, Duling BR. Gap junctions in the control of vascular function. Antioxid Redox Signal. 2009;11:251–266. doi: 10.1089/ars.2008.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame MD, Sarelius IH. L-arginine-induced conducted signals alter upstream arteriolar responsivity to L-arginine. Circ Res. 1995;77:695–701. doi: 10.1161/01.res.77.4.695. [DOI] [PubMed] [Google Scholar]
- Frings S, Lynch JW, Lindemann B. Properties of cyclic nucleotide-gated channels mediating olfactory transduction. Activation, selectivity, and blockage. J Gen Physiol. 1992;100:45–67. doi: 10.1085/jgp.100.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland CJ, Hiley CR, Dora KA. EDHF: spreading the influence of the endothelium. Br J Pharmacol. 2011;164:839–852. doi: 10.1111/j.1476-5381.2010.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Kim N, Joo H, Kim E, Earm YE. ATP-sensitive K+ channel activation by nitric oxide and protein kinase G in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H1545–1554. doi: 10.1152/ajpheart.01052.2001. [DOI] [PubMed] [Google Scholar]
- Haynes LW. Block of the cyclic GMP-gated channel of vertebrate rod and cone photoreceptors by l-cis-diltiazem. J Gen Physiol. 1992;100:783–801. doi: 10.1085/jgp.100.5.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein TW, Kuo L. cAMP-independent dilation of coronary arterioles to adenosine: role of nitric oxide, G proteins, and K(ATP) channels. Circ Res. 1999;85:634–642. doi: 10.1161/01.res.85.7.634. [DOI] [PubMed] [Google Scholar]
- Hein TW, Belardinelli L, Kuo L. Adenosine A(2A) receptors mediate coronary microvascular dilation to adenosine: role of nitric oxide and ATP-sensitive potassium channels. J Pharmacol Exp Ther. 1999;291:655–664. [PubMed] [Google Scholar]
- Hein TW, Wang W, Zoghi B, Muthuchamy M, Kuo L. Functional and molecular characterization of receptor subtypes mediating coronary microvascular dilation to adenosine. J Mol Cell Cardiol. 2001;33:271–282. doi: 10.1006/jmcc.2000.1298. [DOI] [PubMed] [Google Scholar]
- Hellsten Y, Maclean D, Radegran G, Saltin B, Bangsbo J. Adenosine concentrations in the interstitium of resting and contracting human skeletal muscle. Circulation. 1998;98:6–8. doi: 10.1161/01.cir.98.1.6. [DOI] [PubMed] [Google Scholar]
- Hellsten Y, Nyberg M, Jensen LG, Mortensen SP. Vasodilator interactions in skeletal muscle blood flow regulation. J Physiol. 2012a;590:6297–6305. doi: 10.1113/jphysiol.2012.240762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten Y, Nyberg M, Mortensen SP. Contribution of intravascular versus interstitial purines and nitric oxide in the regulation of exercise hyperaemia in humans. J Physiol. 2012b;590:5015–5023. doi: 10.1113/jphysiol.2012.234963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakson BE. Localized expression of an Ins(1,4,5)P3 receptor at the myoendothelial junction selectively regulates heterocellular Ca2+ communication. J Cell Sci. 2008;121:3664–3673. doi: 10.1242/jcs.037481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakson BE, Ramos SI, Duling BR. Ca2+ and inositol 1,4,5-trisphosphate-mediated signalling across the myoendothelial junction. Circ Res. 2007;100:246–254. doi: 10.1161/01.RES.0000257744.23795.93. [DOI] [PubMed] [Google Scholar]
- Jackson WF. Arteriolar tone is determined by activity of ATP-sensitive potassium channels. Am J Physiol Heart Circ Physiol. 1993;265:H1797–H1803. doi: 10.1152/ajpheart.1993.265.5.H1797. [DOI] [PubMed] [Google Scholar]
- Kawano T, Zoga V, Gemes G, McCallum JB, Wu HE, Pravdic D, Liang MY, Kwok WM, Hogan Q, Sarantopoulos C. Suppressed Ca2+/CaM/CaMKII-dependent K(ATP) channel activity in primary afferent neurons mediates hyperalgesia after axotomy. Proc Natl Acad Sci U S A. 2009;106:8725–8730. doi: 10.1073/pnas.0901815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kille JM, Klabunde RE. Adenosine as a mediator of postcontraction hyperemia in dog gracilis muscle. Am J Physiol Heart Circ Physiol. 1984;246:H274–H282. doi: 10.1152/ajpheart.1984.246.2.H274. [DOI] [PubMed] [Google Scholar]
- Kleppisch T, Nelson MT. Adenosine activates ATP-sensitive potassium channels in arterial myocytes via A2 receptors and cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1995;92:12441–12445. doi: 10.1073/pnas.92.26.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KS, Grange RW, Isotani E, Sarelius IH, Kamm KE, Huang PL, Stull JT. nNOS and eNOS modulate cGMP formation and vascular response in contracting fast-twitch skeletal muscle. Physiol Genomics. 2000;2:21–27. doi: 10.1152/physiolgenomics.2000.2.1.21. [DOI] [PubMed] [Google Scholar]
- Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ. Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc Natl Acad Sci U S A. 2000;97:9058–9063. doi: 10.1073/pnas.160068997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynge J, Hellsten Y. Distribution of adenosine A1, A2A and A2B receptors in human skeletal muscle. Acta Physiol Scand. 2000;169:283–290. doi: 10.1046/j.1365-201x.2000.00742.x. [DOI] [PubMed] [Google Scholar]
- Marshall JM. The roles of adenosine and related substances in exercise hyperaemia. J Physiol. 2007;583:835–845. doi: 10.1113/jphysiol.2007.136416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather S, Dora KA, Sandow SL, Winter P, Garland CJ. Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res. 2005;97:399–407. doi: 10.1161/01.RES.0000178008.46759.d0. [DOI] [PubMed] [Google Scholar]
- Mauerer UR, Boulpaep EL, Segal AS. Regulation of an inwardly rectifying ATP-sensitive K+ channel in the basolateral membrane of renal proximal tubule. J Gen Physiol. 1998;111:161–180. doi: 10.1085/jgp.111.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan HE, Baker KM. Cardiac hypertrophy. Mechanical, neural, and endocrine dependence. Circulation. 1991;83:13–25. doi: 10.1161/01.cir.83.1.13. [DOI] [PubMed] [Google Scholar]
- Mubagwa K, Flameng W. Adenosine, adenosine receptors and myocardial protection: an updated overview. Cardiovasc Res. 2001;52:25–39. doi: 10.1016/s0008-6363(01)00358-3. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Sarelius IH. Multiple dilator pathways in skeletal muscle contraction-induced arteriolar dilations. Am J Physiol Regul Integr Comp Physiol. 2002;282:R969–R978. doi: 10.1152/ajpregu.00405.2001. [DOI] [PubMed] [Google Scholar]
- Ngai AC, Coyne EF, Meno JR, West GA, Winn HR. Receptor subtypes mediating adenosine-induced dilation of cerebral arterioles. Am J Physiol Heart Circ Physiol. 2001;280:H2329–H2335. doi: 10.1152/ajpheart.2001.280.5.H2329. [DOI] [PubMed] [Google Scholar]
- Nicholls J, Hourani SM, Hall JM. Characterization of adenosine receptors mediating the vasodilator effects of adenosine receptor agonists in the microvasculature of the hamster cheek pouch in vivo. Auton Autacoid Pharmacol. 2002;22:209–214. doi: 10.1046/j.1474-8673.2002.00259.x. [DOI] [PubMed] [Google Scholar]
- Oestreich EA, Wang H, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac-mediated activation of phospholipase Ce plays a critical role in b-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J Biol Chem. 2007;282:5488–5495. doi: 10.1074/jbc.M608495200. [DOI] [PubMed] [Google Scholar]
- Poucher SM. The role of the A(2A) adenosine receptor subtype in functional hyperaemia in the hindlimb of anaesthetized cats. J Physiol. 1996;492(Pt 2):495–503. doi: 10.1113/jphysiol.1996.sp021324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poucher SM, Nowell CG, Collis MG. The role of adenosine in exercise hyperaemia of the gracilis muscle in anaesthetized cats. J Physiol. 1990;427:19–29. doi: 10.1113/jphysiol.1990.sp018158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor KG. Reduction of contraction-induced arteriolar vasodilation by adenosine deaminase or theophylline. Am J Physiol Heart Circ Physiol. 1984;247:H195–H205. doi: 10.1152/ajpheart.1984.247.2.H195. [DOI] [PubMed] [Google Scholar]
- Quinn KV, Giblin JP, Tinker A. Multisite phosphorylation mechanism for protein kinase A activation of the smooth muscle ATP-sensitive K+ channel. Circ Res. 2004;94:1359–1366. doi: 10.1161/01.RES.0000128513.34817.c4. [DOI] [PubMed] [Google Scholar]
- Radegran G, Calbet JA. Role of adenosine in exercise-induced human skeletal muscle vasodilatation. Acta Physiol Scand. 2001;171:177–185. doi: 10.1046/j.1365-201x.2001.00796.x. [DOI] [PubMed] [Google Scholar]
- Ray CJ, Marshall JM. Elucidation in the rat of the role of adenosine and A2A-receptors in the hyperaemia of twitch and tetanic contractions. J Physiol. 2009;587:1565–1578. doi: 10.1113/jphysiol.2008.163683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GA, Mihok ML, Murrant CL. Extracellular adenosine initiates rapid arteriolar vasodilation induced by a single skeletal muscle contraction in hamster cremaster muscle. Acta Physiol (Oxf) 2013;208:74–87. doi: 10.1111/apha.12060. [DOI] [PubMed] [Google Scholar]
- Saito Y, McKay M, Eraslan A, Hester RL. Functional hyperemia in striated muscle is reduced following blockade of ATP-sensitive potassium channels. Am J Physiol Heart Circ Physiol. 1996;270:H1649–H1654. doi: 10.1152/ajpheart.1996.270.5.H1649. [DOI] [PubMed] [Google Scholar]
- Sandow SL, Gzik DJ, Lee RM. Arterial internal elastic lamina holes: relationship to function. J Anat. 2009;214:258–266. doi: 10.1111/j.1469-7580.2008.01020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow SL, Senadheera S, Bertrand PP, Murphy TV, Tare M. Myoendothelial contacts, gap junctions, and microdomains: anatomical links to function. Microcirculation. 2012;19:403–415. doi: 10.1111/j.1549-8719.2011.00146.x. [DOI] [PubMed] [Google Scholar]
- Sarelius I, Pohl U. Control of muscle blood flow during exercise: local factors and integrative mechanisms. Acta Physiol (Oxf) 2010;199:349–365. doi: 10.1111/j.1748-1716.2010.02129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Wu Z, Cui N, Shi W, Yang Y, Zhang X, Rojas A, Ha BT, Jiang C. PKA phosphorylation of SUR2B subunit underscores vascular KATP channel activation by beta-adrenergic receptors. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1205–R1214. doi: 10.1152/ajpregu.00337.2007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Whitt SP, Rubin LJ, Huxley VH. Differential coronary microvascular exchange responses to adenosine: roles of receptor and microvessel subtypes. Microcirculation. 2005;12:313–326. doi: 10.1080/10739680590934736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West GA, Meno JR, Nguyen TS, Ngai AC, Simard JM, Winn HR. cGMP-dependent and not cAMP-dependent kinase is required for adenosine-induced dilation of intracerebral arterioles. J Cardiovasc Pharmacol. 2003;41:444–451. doi: 10.1097/00005344-200303000-00013. [DOI] [PubMed] [Google Scholar]