

Abstract
The non-obese diabetic (NOD) mouse is a prevalent disease model of type 1 diabetes. Immune aberrations that cause and propagate autoimmune insulitis in these mice are being continually debated, with evidence supporting both dominance of effector cells and insufficiency of suppressor mechanisms. In this study we assessed the behaviour of NOD lymphocytes under extreme expansion conditions using adoptive transfer into immunocompromised NOD.SCID (severe combined immunodeficiency) mice. CD4+ CD25+ T cells do not cause islet inflammation, whereas splenocytes and CD4+ CD25− T cells induce pancreatic inflammation and hyperglycaemia in 80–100% of the NOD.SCID recipients. Adoptively transferred effector T cells migrate to the lymphoid organs and pancreas, proliferate, are activated in the target organ in situ and initiate inflammatory insulitis. Reconstitution of all components of the CD4+ subset emphasizes the plastic capacity of different cell types to adopt effector and suppressor phenotypes. Furthermore, similar immune profiles of diabetic and euglycaemic NOD.SCID recipients demonstrate dissociation between fractional expression of CD25 and FoxP3 and the severity of insulitis. There were no evident and consistent differences in diabetogenic activity and immune reconstituting activity of T cells from pre-diabetic (11 weeks) and new onset diabetic NOD females. Similarities in immune phenotypes and variable distribution of effector and suppressor subsets in various stages of inflammation commend caution in interpretation of quantitative and qualitative aberrations as markers of disease severity in adoptive transfer experiments.
Keywords: adoptive transfer, diabetes, effector cells, immunophenotype, non-obese diabetic mice, regulatory cells
Introduction
The non-obese diabetic (NOD) mouse is widely used as a model to simulate the autoimmune disorder in human type 1 diabetes.1–3 The mechanisms underlying this inflammatory disease are not fully understood, yet it has been recognized that multiple cellular and molecular mechanisms participate in islet destruction.4–6 Autoreactive T cells in NOD mice develop at a very early age,7 their cytotoxic activity is elaborated in the pancreatic lymph nodes,8–10 and protracted inflammation results in destruction of the β-cells and insufficiency of insulin output. The aberrations in mechanisms of immune regulation in NOD mice have been extensively studied and discussed.1–3 One of the proposed mechanisms is escape of autoreactive T cells from negative selection and dominant activity of pathogenic cells without true deficit in regulatory mechanisms.11–13 Persistence of autoreactive cells might be caused by reduced sensitivity to negative regulation and apoptosis,14,15 which declines progressively with age.11,16,17 Alternatively, uncontrolled inflammatory insulitis might be caused by defective suppressor activity at disease onset,7 which persists and intensifies with age in NOD mice and in people with diabetes.16,18–20 Although some studies found that deficient suppression results from quantitative decline in numbers of regulatory T (Treg) cells with age,21,22 other studies could not confirm this mechanism.11,23–25
In our previous studies we found relatively stable levels of effector and suppressor cells in NOD mice at various ages, with a surge in Treg cells before the onset of overt hyperglycaemia.26 Because late stages of inflammatory insulitis are characterized by exacerbation of cytotoxic cell activity,27 we were interested to determine some of the characteristics of T cells in NOD mice. Diabetogenic and regulatory cells in NOD mice are submitted to negative regulation by Fas cross-linking in vitro28,29 and in vivo.30–32 In this study we assessed the patterns of expansion of diabetogenic effector and suppressor cells in vivo using a model of adoptive transfer into immunocompromised NOD.SCID (severe combined immunodeficiency) mice. Simultaneous reconstitution through spontaneous and homeostatic expansion under conditions of lymphopenia is expected to amplify possible differences in the behaviour of T cells.33–35 Furthermore, inherent and induced lymphopenia are conditions associated with predisposition to evolution of effector mechanisms that increase the susceptibility to anti-self reactivity and diabetic autoimmunity.36
The phase of accelerated destructive insulitis27 in the presence of high levels of Treg cells26 questioned whether the pathogenic activity of diabetogenic cells increases in the final stages of inflammatory insulitis. Immunophenotyping of adoptively transferred NOD.SCID mice revealed that each one of the T-cell subsets reconstitutes all effector and suppressor lineages, without significant differences between pre-diabetic and new-onset diabetic NOD female mice. We then questioned whether the incidence of Treg cell phenotypes correlates with severity of destructive insulitis. The similarities in immune profiles of the reconstituted mice suggest that phenotyping of regulatory subsets is unreliable in assessment of the severity of adoptive disease transfer.
Materials and methods
Mice and diabetes monitoring
Mice used in this study were NOD and NOD.SCID mice purchased from Jackson Laboratories (Bar Harbor, ME). The inbred colonies were housed in a barrier facility. The Institutional Animal Care Committee approved all procedures. Blood glucose was monitored between 9:00 and 11:00 a.m. in tail blood samples at weekly intervals using a glucometer (Roche Diagnostics, Florence, SC). Diabetes was defined as two consecutive blood glucose measurements above 200 mg/dl.13,31
Cell isolation, characterization and staining
Spleen, mesenteric/pancreatic lymph nodes, thymus and pancreas were gently minced on a 40-μm nylon mesh in Hanks' balanced salt solution to prepare single-cell suspensions.31 The pancreas was dissected into small pieces and incubated with 20 μg/ml Collagenase P (Roche Diagnostics) for 30 min at 37°. Lymphocytes were isolated by centrifugation over Lympholyte-M (Cedarlane, Burlington, NC) and washed twice with 1% BSA. The CD4+ and CD4+ CD25− subsets were isolated using the CD4+ CD25+ Treg cell isolation kit, according to manufacturer's instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). Purities of the isolated subsets were > 97% for CD4+ CD25− and > 87% for CD4+ CD25+ T cells (FoxP3 expression in ∼ 85% of the isolated cells) (Fig. 1). Cells were labelled with 10 μm 5-(and 6-)-carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Carlsbad, CA).28
Figure 1.
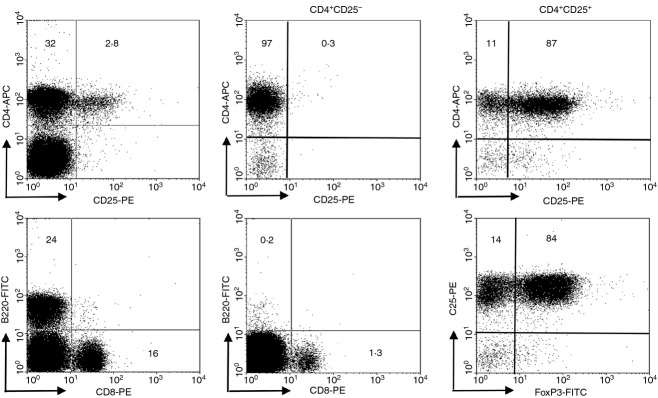
Phenotypic characterization of isolated T cells. Plots display the fractions of CD4+ T cells in reference to CD25 expression, CD8+ T cells and B lymphocytes before isolation (left panels). Isolation CD4+ CD25− T cells yields low contamination with CD4+ CD25+ T cells and CD8+ T cells (middle panels). The CD4+ CD25+ subset contains ∼ 10% CD4+ CD25− T cells and ∼ 85% express FoxP3 (right panels).
Adoptive transfer
NOD.SCID mice aged 5–6 weeks were injected with 2 × 107 splenocytes, 2·5 × 107 CD4+ CD25− T cells and in conjunction with 2·5 × 106 CD4+ CD25+ Treg cells (effector : suppressor ratio of 10 : 1).28,29 Blood glucose levels were monitored twice a week and confirmed upon appearance of hyperglycaemia exceeding 200 mg/dl. Mice were immunophenotyped within 3 days from onset of hyperglycaemia and euglycaemic mice were immunophenotyped at the experimental end-point of 25 weeks following adoptive transfer.
Flow cytometry
The yield of isolation was evaluated using fluorochrome-labelled primary antibodies: CD4 (clone RM 4-5), CD8 (clone 53-6.7), CD25 (clone PC61.5).31 FoxP3 was determined following permeabilization and intracellular staining with a phycoerythrin-labelled antibody (Foxp3 staining buffer set NRRF-30; eBioscience, San Diego, CA). Measurements were performed with a Vantage SE flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Positive staining was determined on a log scale, normalized with control cells stained with isotype control antibodies. Proliferation was determined from quantified CFSE dilution using ModFit software (Verity Software House, Topsham, ME).13
Statistical analysis
Data are presented as means ± standard deviations for each experimental protocol. Results in each experimental group were evaluated for reproducibility by linear regression of duplicate measurements. Differences between the experimental protocols were estimated with a post hoc Scheffe t-test and significance was considered at P < 0·05.
Results
Diabetogenic potential of T cells in pre-diabetic NOD mice
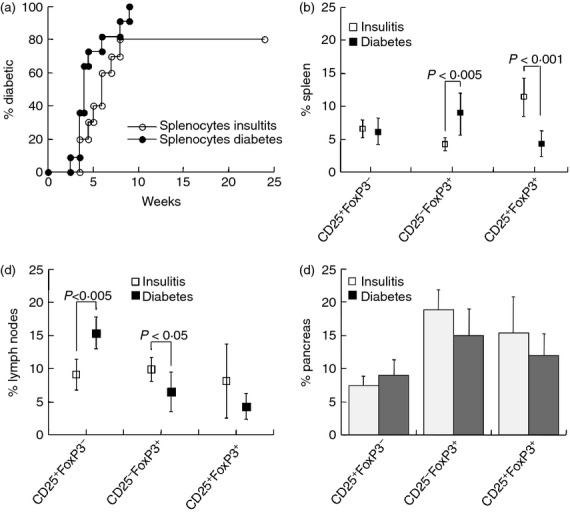
In our female colony overt hyperglycaemia appeared at 14 weeks and > 80% of the females were diabetic at 30 weeks of age.13,31 Initial experiments were designed to determine the relative diabetogenic potential of immune cells from pre-diabetic NOD females (age 11 weeks). Adoptive transfer of 2·5 × 107 CD4+ CD25− T cells induced diabetes in 80% of the NOD.SCID recipients, and co-adoptive transfer of CD25− : CD25+ T cells (at a 10 : 1 ratio) resulted in disease evolution in all recipients (Fig. 2a). In contrast, adoptive transfer of CD4+ CD25+ Treg cells failed to induce diabetes and the pancreata of recipients displayed very low levels of inflammation.
Figure 2.

Adoptive transfer of immune cells into NOD.SCID mice. (a) Incidence of hyperglycaemia in NOD.SCID mice following adoptive transfer of 2·5 × 107 CD4+ CD25− T cells (n = 26) and in conjunction with 2·5 × 106 CD4+ CD25+ Treg cells (n = 10) harvested from pre-diabetic NOD females aged 11 weeks. Immune profiles of diabetic and euglycaemic NOD.SCID mice following adoptive transfer of CD4+ CD25− (n = 5) and CD4+ CD25+ Treg cells (n = 8) were determined in the thymus, spleen, mesenteric lymph nodes and pancreatic infiltrates: (b) CD4+ CD25+ FoxP3−, (c) CD4+ CD25− FoxP3+ and (d) CD4+ CD25+ FoxP3+ T cells.
To determine the relative activities of these cell subsets, NOD.SCID mice were immunophenotyped at onset of hyperglycemia and at 25 weeks of age in euglycemic mice. The first general impression was the outstanding capacity of either one of these subsets to reconstitute all CD4+ T-cell lineages, with similar levels of expression of CD25 (Fig. 2b,d) and FoxP3 (Fig. 2c,d). These Treg cell markers were variably expressed under conditions of simultaneous reconstitution of the lymphopenic organs of NOD.SCID mice. For example, adoptive transfer of CD4+ CD25+ Treg cells (> 85% expressing FoxP3) resulted in dominant fractions of CD25+ FoxP3− T cells in the thymus, exceeding the levels induced by adoptive transfer of CD4+ CD25− T cells (P < 0·005, Fig. 2b).
The immune profiles of NOD.SCID mice grafted with various cell types emphasized several similarities and differences. Both fractions of CD25− FoxP3+ (Fig. 2c) and CD25+ FoxP3+ naturally occurring Treg cell phenotype (nTreg, Fig. 2d) were similarly reconstituted in the spleens and mesenteric lymph nodes by adoptive transfer of CD4+ CD25− T cells with and without Treg cells, and irrespective of glycaemic control. Significant differences were primarily related to adoptive transfer of CD4+ CD25+ nTreg cells compared with CD4+ CD25− effector cells : high CD25+ FoxP3− T cells in the thymus (P < 0·005, Fig. 2b) and high CD25− FoxP3+ in spleens and lymph nodes (P < 0·05, Fig. 2c). In addition, pancreata of euglycaemic mice following adoptive transfer of CD4+ CD25− effector cells displayed low levels of CD25− FoxP3+ Treg cells (P < 0·001 versus diabetic, Fig. 2c) and CD25+ FoxP3+ nTreg cells (P < 0·005 versus diabetic, Fig. 2d). This was a paradoxical result if low Treg cell fractions were expected to correlate with disease eruption.
Relative diabetogenic activity of T cells in pre-diabetic and diabetic NOD females
Appearance of peripheral blood hyperglycaemia is preceded by a phase of aggressive destructive inflammation27 despite a surge in Treg cells,26 questioning whether it is caused by increased cytotoxic activity of pathogenic cells. We therefore assessed differences in reconstituting activity of T cells from pre-diabetic NOD females aged 11 weeks (insulitis) and NOD.SCID mice with new-onset diabetes. Attempting to integrate mean disease onset and incidence, the diabetogenic potential was determined as 1 / (mean onset time) (MOT−1), where failure to develop the disease by 25 weeks of follow up was considered as MOT−1 = 0 (Table 1). This parameter revealed a higher diabetogenic potential of splenocytes from diabetic NOD mice compared with those from pre-diabetic NOD mice (P < 0·05). Whereas CD4+ CD25+ did not induce inflammatory insulitis (n = 12), adoptive transfer of CD25− T cells and mixtures of CD25− : CD25+ T cells (10 : 1 ratio) showed no significant differences between pre-diabetic and diabetic NOD females (Table 1). Evidently, the diabetogenic activity is largely confined within the CD4+ CD25− T-cell subset. Higher diabetogenic potential was achieved by adoptive transfer of 2 × 107 splenocytes than 2·5 × 107 CD4+ CD25− T cells, as expected from the additional contribution of CD8+ T cells and antigen-presenting cells to elaboration of the immune reaction.
Table 1.
NOD.SCID mice were adoptively transferred with 2 × 107 splenocytes, 2·5 × 107 CD4+ CD25− T cells and 2·5 × 106 CD4+ CD25+ regulatory T cells from pre-diabetic (insulitis) and new-onset diabetic NOD female mice
| Insulitis | Diabetes | ||||||
|---|---|---|---|---|---|---|---|
| Incidence (%) | Mean onset time (weeks) | Diabetogenic potential | Incidence (%) | Mean onset time (weeks) | Diabetogenic potential | P-value | |
| Splenocytes | 80 | 6·0 ± 2·3 | 0·16 ± 0·1 | 100 | 4·6 ± 2·1 | 0·26 ± 0·09 | 0·05 |
| CD25+ | 0 | 0 | |||||
| CD25− | 81 | 9·1 ± 3·6 | 0·1 ± 0·08 | 70 | 7·3 ± 1·2 | 0·09 ± 0·07 | |
| CD25− CD25+ | 100 | 13·1 ± 2·5 | 0·08 ± 0·02 | 100 | 13·2 ± 2·5 | 0·09 ± 0·02 | |
Immune profiles of the reconstituted mice are dissociated from diabetes induction
A standing question is whether destructive insulitis is the result of unleashed activity of diabetogenic cells, functional insufficiency of regulatory subsets or gradual progression of inflammation. The most significant difference was the higher efficacy of disease transfer (Table 1) by splenocytes from new-onset diabetic NOD females compared with pre-diabetic mice (insulitis at 11 weeks, Fig. 3a). Comparative immunophenotyping showed significant dissociation in relative reconstitution of the spleen and mesenteric lymph nodes. Adoptive transfer of cells from diabetic donors was associated with increased fractions of CD25− FoxP3+ and decreased fractions of CD25+ FoxP3+ nTreg cells in the spleen (Fig. 3b). In contrast, CD25+ FoxP3− effector subsets were increased and FoxP3+ Treg cell subsets were decreased in the lymph nodes of mice reconstituted with splenocytes from new-onset diabetic mice (Fig. 3c). Although the variations were similar in the lymph nodes and pancreatic infiltrates, the small variations were statistically insignificant in the latter (Fig. 3d). These data depict marked variations in composition of the spleens, lymph nodes and pancreata of the reconstituted NOD.SCID mice, without clear evidence that differences in immune profiles affect disease induction by splenocytes from pre-diabetic and diabetic NOD females.
Figure 3.
Differential reconstitution with splenocytes. NOD.SCID mice were infused with 2 × 107 splenocytes from pre-diabetic (insulitis, n = 10) and new-onset diabetic NOD female (n = 11) mice. (a) Onset of hyperglycaemia. Fractional expression of CD25 and FoxP3 is shown in the spleen (b), mesenteric lymph nodes (c) and pancreatic infiltrates (d).
Considering that the net suppressor activity of Treg cells is preserved with age,11,12 we assessed the differences in lymphoid reconstitution by CD4+ CD25− T cells in reference to the glycaemic state of the NOD.SCID recipients. The basis for comparative analysis of cells from pre-diabetic and new-onset diabetic mice was the similar efficiency of adoptive disease transfer by CD4+ CD25− T cells (Table 1). Recipients of effector T cells from diabetic NOD donors presented reduced fractions of CD25+ FoxP3− T cells in the mesenteric lymph nodes (Fig. 4a) and pancreatic infiltrates (Fig. 4d) compared with recipients of cells from pre-diabetic donors (P < 0·01). At the same time, mice reconstituted with effector T cells from diabetic NOD mice displayed elevated CD25− FoxP3+ fractions in the mesenteric lymph nodes (Fig. 4b), which were not consistent with the profiles of the pancreatic infiltrates (Fig. 4e). Likewise, CD25+ FoxP3+ nTreg cell phenotypes were largely dissociated in the mesenteric lymph nodes (Fig. 4c) and pancreatic infiltrates (Fig. 4f). Altogether these data emphasize dissociation between immune phenotypes and glycaemic control in the reconstituted lymphopenic recipients, indicating an overall poor predictive value of immune profiling on the severity of adoptively transferred destructive insulitis.
Figure 4.
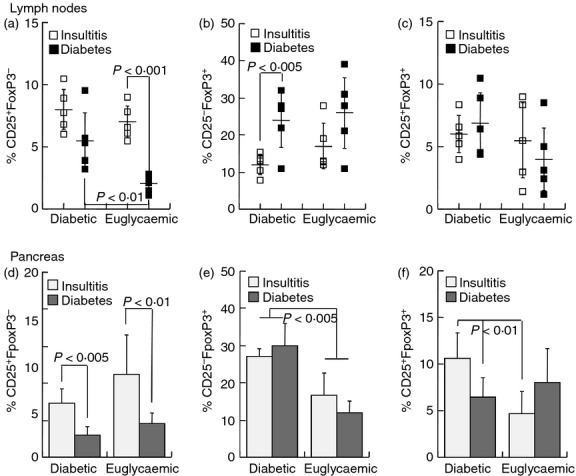
Immunophenotyping of diabetic and euglycaemic NOD.SCID recipients of T cells from pre-diabetic and diabetic NOD females. NOD.SCID mice were infused with 2·5 × 107 CD4+ CD25− T cells from pre-diabetic (age 11 weeks, n = 26) and new-onset diabetic NOD female (n = 17) mice. Data represent fractional CD25 and FoxP3 expression in the lymph nodes (a–c) and pancreatic infiltrates (d–f) in new-onset diabetic NOD.SCID mice and in euglycaemic mice at the experimental end-point of 25 weeks: (a,d) CD25+ FoxP3−, (b,e) CD25− FoxP3+, (c,f) CD25+ FoxP3+.
T-cell homing in lymphopenic recipients
To determine the distribution of adoptively transferred T cells in the lymphopenic recipients, cells labelled with an intracellular dye (CFSE) were harvested from the lymphoid organs. At 24 hr post-infusion, approximately 7%, 15% and 3% of the infused cells were detected in the spleen, mesenteric lymph nodes and pancreata of the NOD.SCID recipients, respectively (Fig. 5a). Soon after homing, these cells engaged in fast cycling, particularly in the mesenteric lymph node (P < 0·01 versus spleen, Fig. 5b). An impressive result was the acute up-regulation of CD25 in pancreas-homed cells during the second post-infusion day (Fig. 5c), an activation marker that reflects the expansion of pre-activated T cells in response to repeated antigen encounter.
Figure 5.
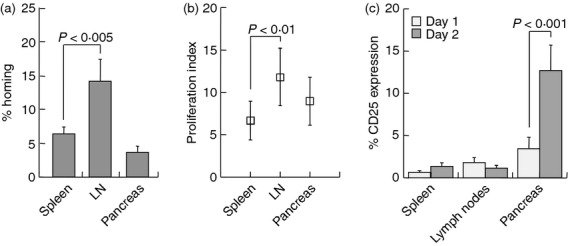
Migration and activity of grafted T cells. NOD.SCID mice were injected with CFSE-labelled 2·5 × 107 CD4+ CD25− T cells and were killed after 1 and 2 days for analysis of the spleen, mesenteric lymph nodes (LN) and pancreas. (a) Homing of donor T cells to these organs within 1 day, expressed as per cent of the donor inoculum. (b) Proliferation of donor T cells after 1 day, as determined from CFSE dilution and quantified with the ModFit software. (c) Expression of CD25 expressed as per cent of donor T cells homed to the various organs after 1 and 2 days.
Discussion
Adoptive transfer of effector cells into NOD.SCID mice is a prevalent experimental model for the evaluation of the pathogenic potential of various cell subsets. We used conditions of lymphopenic expansion to maximize possible differences in the behaviour of T cells from NOD females. Comparative analysis of various cell subsets assigns diabetogenic potential to the CD4+ CD25− subset, with minor variations in diabetogenic activity before and after onset of overt hyperglycaemia. We found that profiling of effector regulatory subsets does not reflect the severity of inflammation.
The infused cells migrate to all lymphoid organs including spleen, mesenteric lymph nodes and pancreas, accounting for 20–25% of the inoculum within the first 24 hr. T cells are non-selectively trapped in the liver and lungs, and home with high efficiency to the bone marrow,37 in addition to migration to the acellular lymphoid organs of NOD.SCID mice. Effector T cells of NOD females displayed remarkable responses of homeostatic and spontaneous expansion in the lymphoid organs and pancreas (range of proliferation index 7–12), concomitant with acute up-regulation of CD25 as an activation marker in the target tissue. Such early in situ stimulation indicates that pathogenic cells in NOD mice do not necessitate priming or extended and repeated exposure to antigens for reactivation, and consequently destructive insulitis to the extent of insulin deficiency is accomplished within 4–9 weeks in NOD.SCID mice. Although diabetogenic cells represent a minute fraction of T cells in NOD mice, it has been long recognized that small numbers of cells are sufficient to cause disease relapse and induce destructive insulitis.38 This is probably mediated by dominant activity of NOD T cells in response to islet antigens within the competitive clonal expansion under conditions of lymphopenia-induced proliferation.39,40 Unopposed expansion41,42 is per se a condition prone to sensitization of effector T cells and their conversion into cytotoxic cells,43,44 explaining the detrimental consequences of radiation-induced lymphopenia on therapeutic approaches to type 1 diabetes.36,45,46
Within the multiple and redundant mechanisms of immune cells with capacity to exert destructive insulitis,5,6 CD4+ T cells alone displayed sufficient activity to attack and destroy the pancreatic islets.4 Although the emphasis was on isolation of CD4+ T-cell subsets, impurities of 3% possibly representing CD8+ T cells may have contributed to adoptive disease transfer, evident from the superior diabetogenic activity of unfractionated splenocytes.4,47 Ideal assessment of CD4+ T cells would require absolute purification, though consistent contamination is unlikely to affect the comparative analysis presented here. The most impressive feature observed in these studies was the outstanding capacity of either one of the CD4+ T-cell subsets to reconstitute, with relatively minor variations, all components of this lymphoid compartment. CD4+ CD25− cells (purity exceeding 99%) converted to adopt regulatory phenotypes, corroborating the effective peripheral conversion of naive T cells to suppressor function.48–50 Reciprocally, adoptively transferred CD4+ CD25+ nTreg cells (> 85% Foxp3+) down-regulated both markers. The impurity of this subset was at maximum in the order of several per cent, but even if effector contaminants expanded vigorously under lymphopenic conditions,24 the effector : suppressor ratio was not low enough to prevent inflammation of the islets. Although Treg cells expand under lymphopenic conditions also,51 their slower proliferative and activation responses usually lag behind those of effector cells and postpone the onset of inflammation but fail to control its evolution.52 In fact, lymphopenia-induced expansion of cytotoxic cells overrides and abolishes the protective activities of Treg cells.46,53
Regulatory cells also migrate efficiently to the mesenteric lymph nodes and pancreata of NOD and NOD.SCID mice and proliferate in situ20,31,32 in response to antigenic stimulation,54 as emphasized by the delay of disease onset following co-administration of effector and regulatory cells at physiological ratios (P < 0·05 versus effector cells alone). Ineffective prevention of adoptive disease transfer by infusion of Treg cells at a physiological ratio corroborates previous reports,16 and differs from other studies using smaller numbers of effector T cells.20,22 These differences suggest that the potential efficacy of Treg cells depends on the pathogenic burden at the site of inflammation.
The most prominent and somewhat consistent difference under conditions of homeostatic expansion in NOD.SCID mice was the lower level of CD25 expression in CD4+ T cells from diabetic mice, which was not apparent in the baseline phenotypic characterization of our colony.13,26,31,32,53 Low fractional CD25 expression was detected in CD4+ T cells in the mesenteric lymph nodes and islet infiltrates irrespective of blood glucose levels. Consistently, dominant CD25− FoxP3+ fractions suggest relative deficiency in expression of the α-chain of the interleukin-2 receptor. Notably, CD4+ CD25− FoxP3+ cells represent a subset of peripheral progenitors that can convert into either effector or suppressor cells.55,56 This subset was found at increased frequencies following immunomodulation in NOD mice13,31,32 and displayed suppressive activity in vitro.57 It has been shown that nTreg cells down-regulate expression and shed the α-chain of the interleukin-2 receptor under particular conditions without impairing their suppressor activity.58–60 A decline in CD25 expression has been attributed to reduced interleukin-2 output from inflammatory cells in the late stages of destructive insulitis,61–63 and may represent a physiological feature of termination of the inflammatory reaction due to extinction of β-cell mass.64 Reversal of the predisposition of T cells from mice with new-onset diabetes to display lower levels of CD25 by co-adoptive transfer with other cell types (splenocytes and CD4+ CD25+ Treg cells) confirm that CD25 is submitted to environmental influences and is not an intrinsic characteristic of diabetogenic cells.
Altogether the data presented in this study indicate that immune phenotyping is of poor predictive value in evaluation of the severity of inflammatory insulitis and glycaemic control in adoptive transfer experiments. Apparently, increased diabetogenic activity of splenocytes from diabetic female NOD mice was associated with preferential reconstitution of CD4+ CD25−T cells and reduced FoxP3+ Treg cells. However, detailed analysis of the pancreatic infiltrates of mice reconstituted with splenocytes and CD4+ CD25− effectors showed significant dissociation and even inverse frequencies of FoxP3+ T cells in the mesenteric lymph nodes and pancreas. Considering that inflammatory insulitis was present in NOD.SCID mice that sustained euglycaemia after adoptive transfer of CD4+ CD25− T cells, it is tempting to speculate that the elevated Treg cell fractions reflect an effort of the lymph nodes to counteract islet inflammation. A similar surge in FoxP3+ Treg was observed in pancreata and regional lymph nodes of aged euglycaemic NOD females in advanced stages of destructive insulitis before the onset of hyperglycemia.26 In fact, the dissociation between immunophenotypes and glycaemic control in adoptively transferred NOD.SCID mice emphasizes the poor predictive value of Treg cell measurements in the assessment of disease severity, and warrant careful interpretation.
Numerous immune aberrations have been suggested to underlie the evolution of spontaneous autoimmune insulitis in NOD mice, including deficient suppression and hyperactivity of effector cells. Each one of these subsets has the plastic capacity to reconstitute all components of the T-cell compartment in lymphoid organs of immunocompromised mice, but only CD4+ CD25− T cells adoptively transfer inflammatory insulitis. We found no evidence of differences in expansion, reconstitution and diabetogenic activity of T cells from new-onset diabetic mice compared with pre-diabetic female NOD mice. Effector T cells in NOD mice display neither aberrant sensitivity to negative regulation by Fas-mediated apoptosis,28 nor different susceptibility from nTreg cells.29 Considering stable fractions of naive and cytotoxic T cells in NOD mice as a function of age24,26 and effective suppression by regulatory subsets,11,12 their particular characteristics will be revealed only by the development of techniques to identify islet-specific T cells.
Acknowledgments
This work was supported by grants from the Frankel Trust for Experimental Bone Marrow Transplantation. The excellent technical assistance of Mrs Ela Zuzovsky and Mrs Ana Zemlianski is gratefully acknowledged.
Disclosures
The authors have no conflict of interest to disclose.
References
- 1.Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 1997;7:727–38. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- 2.Adorini L, Gregori S, Harrison LC. Understanding autoimmune diabetes: insights from mouse models. Trends Mol Med. 2002;8:31–8. doi: 10.1016/s1471-4914(01)02193-1. [DOI] [PubMed] [Google Scholar]
- 3.Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol. 2011;33:67–87. doi: 10.1007/s00281-010-0204-1. [DOI] [PubMed] [Google Scholar]
- 4.Pearl-Yafe M, Kaminitz A, Yolcu ES, Yaniv I, Stein J, Askenasy N. Pancreatic islets under attack: cellular and molecular effectors. Curr Pharm Des. 2007;13:749–60. doi: 10.2174/138161207780249155. [DOI] [PubMed] [Google Scholar]
- 5.Thomas HE, McKenzie MD, Angstetra E, Campbell PD, Kay TW. Beta cell apoptosis in diabetes. Apoptosis. 2009;14:1389–404. doi: 10.1007/s10495-009-0339-5. [DOI] [PubMed] [Google Scholar]
- 6.Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 2010;10:501–13. doi: 10.1038/nri2787. [DOI] [PubMed] [Google Scholar]
- 7.Askenasy EM, Askenasy N. Is autoimmune diabetes caused by aberrant immune activity or defective suppression of physiological self-reactivity? Autoimmun Rev. 2013;12:633–7. doi: 10.1016/j.autrev.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Hoglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet ell antigens in the pancreatic lymph nodes. J Exp Med. 1999;189:331–9. doi: 10.1084/jem.189.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gagnerault MC, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for priming of cell reactive. T cells in NOD mice. J Exp Med. 2002;196:369–77. doi: 10.1084/jem.20011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pearl-Yafe M, Iskovich S, Kaminitz A, Stein J, Yaniv I, Askenasy N. Does physiological β cell turnover initiate autoimmune diabetes in the regional lymph nodes? Autoimmun Rev. 2006;5:338–43. doi: 10.1016/j.autrev.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 11.You S, Belghith M, Cobbold S, et al. Autoimmune diabetes onset results from qualitative rather than quantitative age-dependent changes in pathogenic T-cells. Diabetes. 2005;54:1415–22. doi: 10.2337/diabetes.54.5.1415. [DOI] [PubMed] [Google Scholar]
- 12.D'Alise AM, Auyeung V, Feuerer M, et al. The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proc Natl Acad Sci USA. 2008;105:19857–62. doi: 10.1073/pnas.0810713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yarkoni S, Kaminitz A, Sagiv Y, Askenasy N. Targeting of IL-2 receptor with a caspase fusion protein disrupts autoimmunity in prediabetic and diabetic NOD mice. Diabetologia. 2010;53:356–68. doi: 10.1007/s00125-009-1604-4. [DOI] [PubMed] [Google Scholar]
- 14.Arreaza G, Salojin K, Yang W, et al. Deficient activation and resistance to activation-induced apoptosis of CD8+ T cells is associated with defective peripheral tolerance in nonobese diabetic mice. Clin Immunol. 2003;107:103–15. doi: 10.1016/s1521-6616(03)00049-4. [DOI] [PubMed] [Google Scholar]
- 15.Decallonne B, van Etten E, Giulietti A, et al. Defect in activation induced cell death in non-obese diabetic (NOD) T lymphocytes. J Autoimmun. 2003;20:219–26. doi: 10.1016/s0896-8411(03)00025-8. [DOI] [PubMed] [Google Scholar]
- 16.Gregori S, Giarratana N, Smiroldo S, Adorini L. Dynamics of pathogenic and suppressor T cells in autoimmune diabetes development. J Immunol. 2003;171:4040–7. doi: 10.4049/jimmunol.171.8.4040. [DOI] [PubMed] [Google Scholar]
- 17.Yang W, Hussain S, Mi QS, Santamaria P, Delovitch TL. Perturbed homeostasis of peripheral T cells elicits decreased susceptibility to anti-CD3-induced apoptosis in prediabetic nonobese diabetic mice. J Immunol. 2004;173:4407–16. doi: 10.4049/jimmunol.173.7.4407. [DOI] [PubMed] [Google Scholar]
- 18.Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-β-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–8. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- 19.Gregg RK, Jain R, Schoenleber SJ, et al. A sudden decline in active membrane-bound TGF-β impairs both T regulatory cell function and protection against autoimmune diabetes. J Immunol. 2004;173:7308–16. doi: 10.4049/jimmunol.173.12.7308. [DOI] [PubMed] [Google Scholar]
- 20.Tritt M, Sgouroudis E, d'Hennezel E, Albanese A, Piccirillo CA. Functional waning of naturally occurring CD4+ regulatory T-cells contributes to the onset of autoimmune diabetes. Diabetes. 2008;57:113–23. doi: 10.2337/db06-1700. [DOI] [PubMed] [Google Scholar]
- 21.Alard P, Manirarora JN, Parnell SA, Hudkins JL, Clark SL, Kosiewicz MM. Deficiency in NOD antigen-presenting cell function may be responsible for suboptimal CD4+ CD25+ T-cell-mediated regulation and type 1 diabetes development in NOD mice. Diabetes. 2006;55:2098–105. doi: 10.2337/db05-0810. [DOI] [PubMed] [Google Scholar]
- 22.Pop SM, Wong CP, Culton DA, Clarke SH, Tisch R. Single cell analysis shows decreasing FoxP3 and TGFβ1 coexpressing CD4+ CD25+ regulatory T cells during autoimmune diabetes. J Exp Med. 2005;201:1333–46. doi: 10.1084/jem.20042398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berzins SP, Venanzi ES, Benoist C, Mathis D. T-cell compartments of prediabetic NOD mice. Diabetes. 2003;52:327–34. doi: 10.2337/diabetes.52.2.327. [DOI] [PubMed] [Google Scholar]
- 24.Mellanby RJ, Thomas D, Phillips JM, Cooke A. Diabetes in non-obese diabetic mice is not associated with quantitative changes in CD4+ CD25+ Foxp3+ regulatory T cells. Immunology. 2007;121:15–28. doi: 10.1111/j.1365-2567.2007.02546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brusko T, Wasserfall C, McGrail K, et al. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes. 2007;56:604–12. doi: 10.2337/db06-1248. [DOI] [PubMed] [Google Scholar]
- 26.Kaminitz A, Mizrahi K, Askenasy N. Surge in regulatory cells does not prevent onset of hyperglycemia in NOD mice. Autoimmunity. 2014;47:105–12. doi: 10.3109/08916934.2013.866103. [DOI] [PubMed] [Google Scholar]
- 27.Kaminitz A, Stein J, Yaniv I, Askenasy N. The vicious cycle of apoptotic β-cell death in type 1 diabetes. Immunol Cell Biol. 2007;85:582–9. doi: 10.1038/sj.icb.7100093. [DOI] [PubMed] [Google Scholar]
- 28.Kaminitz A, Askenasy EM, Yaniv I, Stein J, Askenasy N. Apoptosis of purified CD4+ T cell subsets is dominated by cytokine deprivation and absence of other cells in new onset diabetic NOD mice. PLoS ONE. 2010;5:e15684. doi: 10.1371/journal.pone.0015684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaminitz A, Yolcu ES, Askenasy EM, Stein J, Yaniv I, Shirwan H, Askenasy N. Effector and naturally occurring regulatory T cells display no abnormalities in activation induced cell death in NOD mice. PLoS ONE. 2011;6:e21630. doi: 10.1371/journal.pone.0021630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franke DD, Yolcu ES, Alard P, Kosiewicz MM, Shirwan H. A novel multimeric form of FasL modulates the ability of diabetogenic T cells to mediate type 1 diabetes in an adoptive transfer model. Mol Immunol. 2007;44:2884–92. doi: 10.1016/j.molimm.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaminitz A, Yolcu ES, Stein J, Yaniv I, Shirwan H, Askenasy N. Killer Treg restore immune homeostasis and suppress autoimmune diabetes in prediabetic NOD mice. J Autoimmun. 2011;37:39–47. doi: 10.1016/j.jaut.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Kaminitz A, Yolcu ES, Mizrahi K, Shirwan H, Askenasy N. Killer Treg cells ameliorate inflammatory insulitis in non-obese diabetic mice through local and systemic immunomodulation. Int Immunol. 2013;25:485–94. doi: 10.1093/intimm/dxt016. [DOI] [PubMed] [Google Scholar]
- 33.Wicker LS, Miller BJ, Mullen Y. Transfer of autoimmune diabetes mellitus with splenocytes from nonobese diabetic (NOD) mice. Diabetes. 1986;35:855–60. doi: 10.2337/diab.35.8.855. [DOI] [PubMed] [Google Scholar]
- 34.Boitard C, Yasunami R, Dardenne M, Bach JF. T cell-mediated inhibition of the transfer of autoimmune diabetes in NOD mice. J Exp Med. 1989;169:1669–80. doi: 10.1084/jem.169.5.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yagi H, Matsumoto M, Kunimoto K, Kawaguchi J, Makino S, Harada M. Analysis of the roles of CD4+ and CD8+ T cells in autoimmune diabetes of NOD mice using transfer to NOD athymic nude mice. Eur J Immunol. 1992;22:2387–93. doi: 10.1002/eji.1830220931. [DOI] [PubMed] [Google Scholar]
- 36.Askenasy EM, Askenasy N, Askenasy JJ. Does lymphopenia preclude restoration of immune homeostasis? The particular case of type 1 diabetes. Autoimmun Rev. 2010;9:687–90. doi: 10.1016/j.autrev.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 37.Askenasy N, Farkas DL. In vivo imaging studies of the effect of recipient conditioning, donor cell phenotype and antigen disparity on homing of haematopoietic cells to the bone marrow. Br J Haematol. 2003;120:505–15. doi: 10.1046/j.1365-2141.2003.04114.x. [DOI] [PubMed] [Google Scholar]
- 38.Serreze DV, Johnson EA, Chapman HD, et al. Autoreactive diabetogenic T-cells in NOD mice can efficiently expand from a greatly reduced precursor pool. Diabetes. 2001;50:1992–2000. doi: 10.2337/diabetes.50.9.1992. [DOI] [PubMed] [Google Scholar]
- 39.La Gruta NL, Driel IR, Gleeson PA. Peripheral T cell expansion in lymphopenic mice results in a restricted T cell repertoire. Eur J Immunol. 2000;30:3380–6. doi: 10.1002/1521-4141(2000012)30:12<3380::AID-IMMU3380>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 40.Troy AE, Shen H. Cutting edge: homeostatic proliferation of peripheral T lymphocytes is regulated by clonal competition. J Immunol. 2003;170:672–6. doi: 10.4049/jimmunol.170.2.672. [DOI] [PubMed] [Google Scholar]
- 41.Le Campion A, Gagnerault MC, Auffray C, et al. Lymphopenia-induced spontaneous T-cell proliferation as a cofactor for autoimmune disease development. Blood. 2009;114:1784–93. doi: 10.1182/blood-2008-12-192120. [DOI] [PubMed] [Google Scholar]
- 42.Shvets A, Chakrabarti R, Gonzalez-Quintial R, Baccala R, Theofilopoulos AN, Prud'homme GJ. Impaired negative regulation of homeostatically proliferating T cells. Blood. 2009;113:622–5. doi: 10.1182/blood-2008-03-139964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Surh CD, Sprent J. Homeostatic T cell proliferation: how far can T cells be activated to self-ligands? J Exp Med. 2000;21:F9–14. doi: 10.1084/jem.192.4.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shklovskaya E, Fazekas de St. Groth B. Severely impaired clonal deletion of CD4+ T cells in low-dose irradiated mice: role of T cell antigen receptor and IL-7 receptor signals. J Immunol. 2006;177:8320–30. doi: 10.4049/jimmunol.177.12.8320. [DOI] [PubMed] [Google Scholar]
- 45.Yaniv I, Ash S, Farkas DL, Askenasy N, Stein J. Consideration of strategies for hematopoietic cell transplantation. J Autoimmun. 2009;33:255–9. doi: 10.1016/j.jaut.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 46.Ash S, Yarkoni S, Askenasy N. Lymphopenia is detrimental to therapeutic approaches to type 1 diabetes using regulatory T cells. Immunol Res. 2014;58:101–5. doi: 10.1007/s12026-013-8476-x. [DOI] [PubMed] [Google Scholar]
- 47.Mandrup-Poulsen T. Beta cell death and protection. Ann NY Acad Sci. 2003;1005:32–42. doi: 10.1196/annals.1288.005. [DOI] [PubMed] [Google Scholar]
- 48.Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25– naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. CD25– T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol. 2004;173:7259–68. doi: 10.4049/jimmunol.173.12.7259. [DOI] [PubMed] [Google Scholar]
- 50.Liang S, Alard P, Zhao Y, Parnell S, Clark SL, Kosiewicz MM. Conversion of CD4+ CD25– cells into CD4+ CD25+ regulatory T cells in vivo requires B7 costimulation, but not the thymus. J Exp Med. 2005;201:127–37. doi: 10.1084/jem.20041201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bourgeois C, Stockinger B. CD25+CD4+ regulatory T cells and memory T cells prevent lymphopenia-induced proliferation of naive T cells in transient states of lymphopenia. J Immunol. 2006;177:4558–66. doi: 10.4049/jimmunol.177.7.4558. [DOI] [PubMed] [Google Scholar]
- 52.Askenasy N. Hematopoietic transplants for disease suppression and cure in type 1 diabetes. Curr Stem Cell Res Ther. 2013;8:333–9. doi: 10.2174/1574888x113089990001. [DOI] [PubMed] [Google Scholar]
- 53.Kaminitz A, Mizrahi K, Yaniv I, Stein J, Askenasy N. Immunosuppressive therapy exacerbates autoimmunity in NOD mice and diminishes the protective activity of regulatory T cells. J Autoimmun. 2010;35:145–52. doi: 10.1016/j.jaut.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 54.Tonkin DR, He J, Barbour G, Haskins K. Regulatory T cells prevent transfer of type 1 diabetes in NOD mice only when their antigen is present in vivo. J Immunol. 2008;181:4516–22. doi: 10.4049/jimmunol.181.7.4516. [DOI] [PubMed] [Google Scholar]
- 55.Zelenay S, Lopes-Carvalho T, Caramalho I, Moraes-Fontes MF, Rebelo M, Demengeot J. Foxp3+ CD25– CD4 T cells constitute a reservoir of committed regulatory cells that regain CD25 expression upon homeostatic expansion. Proc Natl Acad Sci USA. 2005;102:4091–6. doi: 10.1073/pnas.0408679102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci USA. 2009;106:1903–8. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yarkoni S, Prigozhina TB, Slavin S, Askenasy N. IL-2-targeted therapy ameliorates the severity of graft-versus-host disease: ex vivo selective depletion of host-reactive T cells and in vivo therapy. Biol Blood Marrow Transplant. 2012;18:523–35. doi: 10.1016/j.bbmt.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 58.Stephens LA, Mason D. CD25 is a marker for CD4+ thymocytes that prevent autoimmune diabetes in rats, but peripheral T cells with this function are found in both CD25+ and CD25– subpopulations. J Immunol. 2000;165:3105–10. doi: 10.4049/jimmunol.165.6.3105. [DOI] [PubMed] [Google Scholar]
- 59.Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4+CD25+ suppressor T cells in vivo. Nat Immunol. 2002;3:33–41. doi: 10.1038/ni743. [DOI] [PubMed] [Google Scholar]
- 60.Pedersen AE, Lauritsen JP. CD25 shedding by human natural occurring CD4+CD25+ regulatory T cells does not inhibit the action of IL-2. Scand J Immunol. 2009;70:40–3. doi: 10.1111/j.1365-3083.2009.02268.x. [DOI] [PubMed] [Google Scholar]
- 61.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–51. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 62.Yarkoni S, Kaminitz A, Sagiv Y, Yaniv I, Askenasy N. Involvement of IL-2 in homeostasis of regulatory T cells: the IL-2 cycle. BioEssays. 2008;30:875–88. doi: 10.1002/bies.20812. [DOI] [PubMed] [Google Scholar]
- 63.Tang Q, Adams JY, Penaranda C, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–97. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yolcu ES, Ash S, Kaminitz A, Sagiv Y, Askenasy N, Yarkoni S. Apoptosis as a mechanism of T-regulatory cell homeostasis and suppression. Immunol Cell Biol. 2008;86:650–8. doi: 10.1038/icb.2008.62. [DOI] [PubMed] [Google Scholar]