

Abstract
A tripartite association of Rab11a with both Rab11-FIP2 and MYO5B regulates recycling endosome trafficking. We sought to define the intermolecular interactions required between Rab11-FIP2 with MYO5B. Using a random mutagenesis strategy, we identified point mutations at S229P or G233E in Rab11-FIP2 that caused loss of interaction with MYO5B in yeast 2-hybrid assays as well as loss of interaction of Rab11-FIP2(129-356) with MYO5B tail when expressed in HeLa cells. Single mutations or the double S229P/G233E mutation failed to alter the association of full-length Rab11-FIP2 with MYO5B tail in HeLa cells. While EGFP-Rab11-FIP2 wild type co-localized with endogenous MYO5B staining in MDCK cells, EGFP-Rab11-FIP2(S229P/G233E) showed a significant decrease in localization with endogenous MYO5B. Analysis of Rab11a-containing vesicle movement in live HeLa cells demonstrated that when the MYO5B/Rab11-FIP2 association is perturbed by mutation or by Rab11-FIP2 knockdown, vesicle movement is increased in both speed and track length, consistent with an impairment of MYO5B tethering at the cytoskeleton. These results support a critical role for the interaction of MYO5B with Rab11-FIP2 in stabilizing the functional complex with Rab11a, which regulates dynamic movements of membrane recycling vesicles.
Keywords: Rab11, MYO5B, Rab11-FIP2, membrane recycling
Introduction
Rab proteins act as cell compartment-specific trafficking regulators by specifying the intracellular sites for assembly of multiprotein complexes that regulate the discrete functions of particular trafficking pathways (1). These multiprotein assemblies are involved in vesicle budding, vesicle apposition and fusion as well as vesicle movement along cytoskeletal elements. The interaction of Rab proteins with molecular motors is of particular interest, because it accounts for a critical mechanism for the regulation of the movement of vesicles within cells along organelle-specific paths. The three members of Class V myosins, myosin VA, VB and VC, (MYO5A, MYO5B AND MYO5C) all are localized to membrane subsets based on interactions with Rab small GTPases. Thus, Rab27a interacts with MYO5A and this interaction is required for normal movement of melanosomes and for function of secretory lysosomes in general (2-5). Similarly, the Rab11 family of small GTPases (Rab11a, Rab11b and Rab25) can all interact directly with MYO5A and MYO5B (6, 7). All three class V myosins can interact with Rab8a and Rab10, although the interaction of Rab10 with MYO5A and MYO5B is dependent on the presence of an alternatively-spliced 30 amino acid exon (Exon D) (8).
While Rab proteins may designate the site for assembly of multiprotein complexes, other protein interactions likely define functional properties. Thus, in addition to the direct interactions of Rab proteins with class V myosins, in the case of MYO5A and MYO5B, adapter molecules exist for further coordination. Melanophilin associates with both Rab27a and MYO5A and the interaction requires the presence of the MYO5A Exon F (9-12). MyRIP can interact with MYO5A and Rab27a, but the interaction is not dependent on the presence of Exon F in MYO5A (12-14). Similarly, Rab11-FIP2 can interact with both Rab11a and MYO5B (15). Rab11-FIP2 is a member of a family of Rab11a binding proteins that all contain a conserved carboxyl-terminal Rab11-binding domain (16, 17). Among the Rab11-FIP proteins, only Rab11-FIP2 can interact with MYO5B. We have previously demonstrated that the interaction of MYO5B with Rab11-FIP2 occurs through a region of Rab11-FIP2 that does not include either the amino terminal C2-domain or the carboxyl terminal Rab11-binding domain (15). We have now sought to define discrete residues within Rab11-FIP2 that are responsible for interaction with MYO5B. These studies have identified two residues within Rab11-FIP2 that regulate interaction with MYO5B. The overall results indicate that either loss of Rab11-FIP2 or destabilization of the interaction between Rab11-FIP2 and MYO5B leads to an increase in the movement of Rab11a-containing vesicles by releasing vesicles from a non-motile tethered pool. These results demonstrate a function for the Rab11-FIP2/MYO5B complex in resisting the transit of Rab11a-containing vesicle along microtubules and support the role of MYO5B as a dynamic tether.
Results
Characterization of MYO5B binding residues in Rab11-FIP2
Over the past decade, a number of studies have suggested that the assembly of both Rab11-FIP2 and MYO5B with Rab11a creates a critical regulatory complex for recycling of a number of diverse cargoes (15, 18-20). Our previous studies demonstrated that the central region of Rab11-FIP2, Rab11-FIP2(129-356), which lacked both the amino terminal C2 domain and the carboxyl terminal Rab11 binding domain, colocalized with MYO5B tail when both were co-expressed in HeLa cells (15). Nevertheless, the identity of critical residues for MYO5B association with Rab11-FIP2 had not been determined. We therefore sought to examine which amino acids in Rab11-FIP2 could account for the interaction of the key regulators. We performed a random mutagenesis of Rab11-FIP2(129-356) in pBD-GAL-Cam by transforming the plasmid into the mutator strain XL-1 Red and growing the bacteria for 73 hours to produce a predicted one mutation per 1000 nucleotides. The plasmids isolated from this culture served as a library of random mutants of Rab11-FIP2(129-356) and were cotransformed into yeast with pAD-GAL-MYO5B tail. Colonies with loss of X-gal reactivity were isolated and the plasmids were rescued and sequenced. As expected, the majority of revertent clones were due to non-sense truncation mutants. However, we did isolate two point mutants at S229P and G233E, which caused a loss of yeast 2-hybrid interaction. When we introduced the S229P mutation into full-length Rab11-FIP2, the protein also lost interaction with MYO5B tail (Figure 1), while the G233E mutation failed to disrupt the interaction with full-length MYO5B. A dual mutation with S229P/G233E in full length Rab11-FIP2 also lost interaction with MYO5B in yeast 2-hybrid assays.
Figure 1. Yeast 2-hybrid assay of interactions of Rab11-FIP2 mutants with MYO5B tail.

A) The pBD bait vectors were co-expressed in yeast with pAD-GAL MYO5B tail vectors and cells were plated on dual drop out plates. The colony lifts were cracked in liquid nitrogen and then incubated in X-gal to determine the presence of 2-hybrid interactions. The results were considered positive if blue color developed within three hours. The results were consistent over three separate assays. B) Quantitative β-gal liquid assays were performed on Rab11-FIP2-BD wild type and mutant constructs against MYO5B-AD and Abs420 was measured. Triplicate samples were done each time and three separate experiments were performed. MYO5B-AD alone was used for background subtraction. All Abs420 were averaged and plotted and ANOVA with Bonferroni were performed. FIP2(S229P) and FIP2(SPGE) were significantly decreased from FIP2 and FIP2(129-356) (S229P) and FIP2(129-356) (G233E) were significantly decreased from FIP2(129-356) (*p<0.001).
Rab11-FIP2 mutations alter its interaction with MYO5B
To evaluate the efficacy of Rab11-FIP2 mutations, we studied the ability of MYO5B tail to sequester expressed Rab11-FIP2 in HeLa cells. We have previously demonstrated that MYO5B tail can sequester both wild type Rab11-FIP2 and Rab11-FIP2(129-356) (15). Both Rab11-FIP2(129-356) (S229P) and Rab11-FIP2(129-356) (G233E) failed to localize with mCherry-MYO5B tail and were distributed through the cytosol (Figure 2, Supplemental Figure 1). For each GFP-Rab11-FIP2 construct, we imaged 10 cells and scored them blindly for colocalization with mCherry-MYO5B tail. GFP-Rab11-FIP2 was classified as either colocalized, partial, or separate from mCherry-MYO5B tail. These classifications were graphed and the distributions were analyzed by Chi Square analysis. For both GFP-Rab11-FIP2(S229P) and GFP-Rab11-FIP2(G233E), the distributions were significantly different than wild type (p<0.001). These results confirm that these two amino acids are critical for MYO5B binding to Rab11-FIP2.
Figure 2. Mutations in Rab11-FIP2(129-356) block interaction with MYO5B tail in vivo.
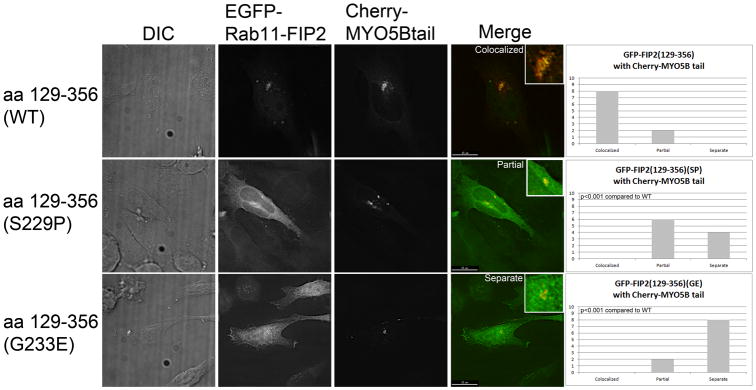
HeLa cells were co-transfected with an mCherry chimera of MYO5B tail in combination with EGFP-chimeras of Rab11-FIP2(129-356), Rab11-FIP2(129-356) (S229P) or Rab11-FIP2(129-356) (G233P). Dual color overlays are shown at the right with EGFP in green and mCherry in red.
We next evaluated the effects of putative MYO5B binding region Rab11-FIP2 mutations on the behavior of full length Rab11-FIP2 co-expressed with MYO5B tail. As previously reported, full-length wild type Rab11-FIP2 was sequestered efficiently with MYO5B tail in HeLa cells (Figure 3). Furthermore, the S229P and G233E mutants of wild type Rab11-FIP2, as well as the double S229P/G233E mutant, all still sequestered with mCherry-MYO5B tail when co-expressed in HeLa cells. For the 10 cells imaged and scored for GFP-Rab11-FIP2, GFP-Rab11-FIP2(S229P), GFP-Rab11-FIP2(G233E), and GFP-Rab11-FIP2(S229P/G233E), we found no statistical difference in the distributions with mCherry-MYO5B tail.
Figure 3. Pull in of full length Rab11-FIP2 by MYO5B tail is independent of mutations.

HeLa cells were co-transfected with an mCherry chimera of MYO5B tail in combination with Venus-chimeras of wild type (WT) Rab11-FIP2, Rab11-FIP2(S229P), Rab11-FIP2(G233E) or Rab11-FIP2(S229P/G233E). Dual color overlays are shown at the right with Venusin green and mCherry in red.
Rab11-FIP2(S229P/G233E) shows decreased localization with endogenous MYO5B
While our studies demonstrated that full-length Rab11-FIP2(S229P/G233E) could localize with MYO5B tail, the results of these dual over-expression studies can be influenced by altered avidity due to the over-expression of both proteins. Therefore, to evaluate the in situ association of the Rab11-FIP2(S229P/G233E) with MYO5B, we utilized a newly-developed chicken anti-MYO5B antibody that recognizes endogenous MYO5B in MDCK cells by immunofluorescence (21). MDCK cells were transfected with either full length Venus-Rab11-FIP2 wild type or Venus-Rab11-FIP2(S229P/G233E) with simultaneous staining for endogenous MYO5B and Rab11a (Figure 4, Supplemental Figure 2). Quantitative analysis of the staining demonstrated that there was a significant decrease in colocalization of MYO5B with Rab11-FIP2(S229P/G233E) compared to wild type Rab11-FIP2(Figure 4A,B). Similar deficits were seen for localization of MYO5B with Rab11a in cells expressing Rab11-FIP2(S229P/G233E). In contrast no significant difference was observed for co-localization of the two Rab11-FIP2 proteins and Rab11a. We have previously noted that expression of Rab11-FIP2(129-512), which lacks the amino terminal C2-domain, causes a prominent inhibition of Rab11a-dependent recycling in MDCK cells (22, 23). We therefore also evaluated the effects of the S229P/G233E mutations in the context of the Rab11-FIP2(129-512) truncation (Figure 4C,D). As with the full length construct, the S229P/G233E mutation elicited a decrease in the accumulation of MYO5B with Rab11-FIP2(129-512) and also decreased colocalization of MYO5B with Rab11a. Also similar to the full length constructs, the mutations had no effect on the association of Rab11a with the truncated Rab11-FIP2(129-512). These studies confirm that the S229P/G233E mutations lead to a decrease in the effective association of endogenous MYO5B with Rab11-FIP2.
Figure 4. Effects of Rab11-FIP2 expression on the distribution of endogenous MYO5B.
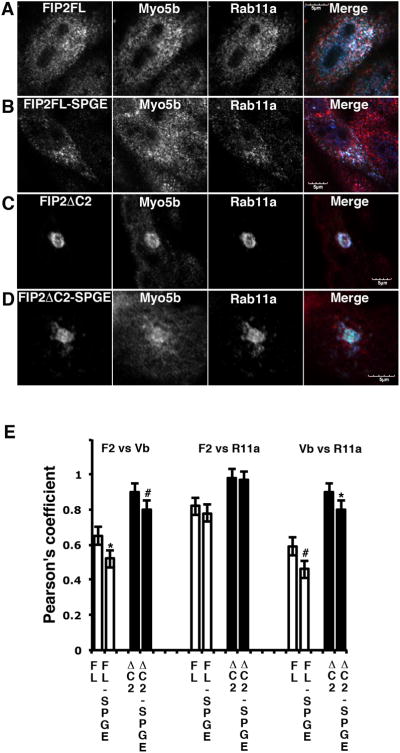
MDCK cells were transfected with an Venus chimeras of A. wild type (WT) Rab11-FIP2, B. Rab11-FIP2(S229P/G233E), C. Rab11-FIP2(129-512) or D. Rab11-FIP2(129-512) (S229P/G233E) and then immunostained for endogenous MYO5B and endogenous Rab11a. Triple color overlays are shown at the right with Venus in green and MYO5B immunostaining in red with Rab11a immunostaining in blue. E. Quantitation of colocalization (Pearson's coefficients) between expressed Venus-FIP2 constructs and endogenous MYO5B or Rab11a is shown in E. *p<0.05; #p<0.01. Results are from at least 6 cells in 3 separate experiments.
Additionally, to confirm that the S229P/G233E point mutants in Rab11-FIP2 do not interfere with Rab11-FIP2 binding to Rab11a, we performed an in vitro binding assay. Using bacterially expressed Rab11-FIP2 or Rab11-FIP2(S229P/G233E), we found that recombinant Rab11a was able to bind similarly to both forms of Rab11-FIP2(Supplemental Figure 3).
Rab11-FIP2 mutations and Rab11-FIP2 knockdown alter the movements of Rab11a-containing vesicles
While our data indicated that a Rab11a/Rab11-FIP2/MYO5B complex might be assembled even in the face of altered interactions between Rab11-FIP2 and MYO5B, we hypothesized that the functional integrity of that complex might be compromised. Thus, we sought to investigate whether mutations in Rab11-FIP2 would alter the behavior of Rab11a-containing vesicles. To determine whether the Rab11-FIP2(S229P/G233E) mutant influences Rab11a vesicle movement, we conducted live cell imaging of HeLa cells expressing mCherry-Rab11a in combination with either Venus-Rab11-FIP2 wild type or the Venus-Rab11-FIP2(S229P/G233E) double mutant. We tracked a minimum of 20,000 individual Rab11a-containing vesicles moving over time in at least 11 cells for each condition and focused on two specific parameters of vesicle movement (Figure 5A). First, we evaluated track displacement length as the distance between the first and last points on the track. Second, we examined track speed mean as the average speed of the vesicle over the entire track. We observed a significant increase in Rab11a vesicle track speed from 0.27μm/s in cells expressing wild type Rab11-FIP2 to 0.34μm/sin the presence of the Rab11-FIP2(S229P/G233E) (p<0.0001;Figure 5A, Supplemental Figure 5 and Supplemental Videos 1,2). This increase in observed speed in the presence of Rab11-FIP2(S229P/G233E)was also associated with a significant increase in track displacement for Rab11a vesicles (5.50μm vs.6.57μm in wild type-expressing cells, p<0.0001). These combined results suggest that the Rab11a vesicles, in the presence of the Rab11-FIP2(S229P/G233E) double mutant, move both more rapidly and farther compared with Rab11a vesicles in the presence of wild type Rab11-FIP2.
Figure 5. Particle tracking data of Rab11a labeled particles in live HeLa cells.

A) Time lapse, widefield movies of mCherry-Rab11a particles were collected using a Deltavision deconvolution microscope. Images were taken every 2 seconds for 2 minutes total. Track speed mean and track displacement length were calculated in at least 11 cells for each condition. A) mCherry-Rab11a vesicle speed increased significantly from 0.27 ± 0.001 μm/s in cells expressing wild type Rab11-FIP2 to 0.34 ± 0.001 μm/sin the presence of the Rab11-FIP2(S229P/G233E). Rab11a track displacement length increased significantly from 5.50 ± 0.04 μm to 6.57 ± 0.03 μm in wild type-expressing cells vs. Rab11-FIP2 mutant cells. B) mCherry-Rab11a vesicle speed increased significantly from 0.28 ± 0.001_μm/s in scrambled control to 0.33 ± 0.001 μm/s Rab11-FIP2 knockdown cells. For rescue, mCherry-Rab11a vesicle speed decreased from 0.33 ± 0.001 μm/s in Rab11-FIP2 knockdown cells to 0.27 ± 0.001_μm/s in Rab11-FIP2 knockdown cells plus Venus-Rab11-FIP2 wild type. Vesicle speed increased from 0.27 ± 0.001 μm/s in Rab11-FIP2 knockdown plus wild type to 0.31 ± 0.001 μm/s in knockdown plus Rab11-FIP2(S229P/G233E) mutant. mCherry-Rab11a track displacement length increased significantly from 4.63 ± 0.036 μm in scrambled control to 6.42 ± 0.039 μm in Rab11-FIP2 knockdown cells. mCherry-Rab11a displacement significantly decreased from 6.42 ± 0.039 μm in Rab11-FIP2 knockdown plus Venus-Rab11-FIP2 wild type to 4.56 ± 0.029 mm in Rab11-FIP2 knockdown alone. Track displacement length in Rab11-FIP2 knockdown plus Venus-Rab11-FIP2(S229P/G233E) was significantly increased from 4.56 ± 0.029 mm to 5.58 ± 0.026 μm in Rab11-FIP2 knockdown plus Venus-Rab11-FIP2. Results are presented as the mean ± SEM. * p<0.001
Since we found a significant increase in Rab11a vesicle speed and displacement in the presence of Rab11-FIP2(S229P/G233E) compared to wild type, we examined Rab11a vesicle speed and displacement in the absence of Rab11-FIP2. We created stable HeLa cell lines expressing either a scrambled shRNA control or shRNA against the Rab11-FIP23′-untranslated region (UTR) which elicited a prominent reduction in Rab11-FIP2 protein expression (Supplemental Figure 4). Using the same parameters as in the overexpression experiments, we found that Rab11-FIP2 knockdown cells displayed a significant increase in mCherry-Rab11a vesicle speed over scrambled shRNA control (0.33 μm/s vs. 0.28 μm/s in control, p<0.001) (Figure 5B, Supplemental Figure 5, and Supplemental Videos 3,4). Similarly, track displacement length was increased significantly in Rab11-FIP2 knockdown cells compared to control cells expressing scrambled shRNA (6.42 μm vs. 4.63 μm in control, p<0.001).
Next, we examined whether re-expression of either Venus-Rab11-FIP2 or Venus-Rab11-FIP2(S229P/G233E) in the knockdown cells could normalize the increased Rab11a vesicle movement phenotype. Rab11-FIP2 knockdown cells expressing Venus-Rab11-FIP2 wild type showed a significant decrease in vesicle speed compared to Rab11-FIP2 knockdown cells (0.27 μm/s vs. 0.33 μm/s, p<0.001) (Figure 5B, Supplemental Figure 5 and Supplemental Videos 4,5). Also, track displacement length was significantly decreased in Rab11-FIP2 knockdown expressing Venus-Rab11-FIP2 wild type compared to Rab11-FIP2 knockdown alone (4.56 μm vs. 6.42 μm, p<0.001). These results demonstrate that re-expression of Rab11-FIP2 can reverse the increasedRab11a vesicle movement phenotype elicited with Rab11-FIP2 knockdown.
In contrast, cells expressing Venus-Rab11-FIP2(S229P/G233E) in the context of endogenous Rab11-FIP2 knockdown still displayed a significant increase in vesicle speed over re-expression of Venus-Rab11-FIP2 wild type in knockdown cells (0.31 μm/s vs. 0.27 μm/s in wild type, p<0.001) (Figure 5B, Supplemental Figure 5 and Supplemental Videos 4,6). Additionally, mCherry-Rab11a track displacement length in Rab11-FIP2 knockdown cells expressing Venus-Rab11-FIP2(S229P/G233E) showed a significant increase over Rab11-FIP2 knockdown cells expressing Venus-Rab11-FIP2 wild type in (5.58 μm vs. 4.56 μm in wild type, p<0.001). These results demonstrate that Venus-Rab11-FIP2(S229P/G233E) could not rescue the aberrant phenotype of Rab11-FIP2 knockdown cells to the extent observed with Venus-Rab11-FIP2 wild type. Therefore, knockdown of Rab11-FIP2 alters Rab11a-containing vesicle movement in a similar manner to that observed with overexpression of Venus-Rab11-FIP2(S229P/G233E). These findings show that the mutations in the MYO5B binding domain of Rab11-FIP2 result in more rapid vesicle movement along microtubules.
To examine the underlying cause for changes in Rab11a vesicle movement, we sought to evaluate in greater depth the parameters of vesicle displacement. We determined that the distribution of vesicle movement parameters, including track speed mean, track displacement length, and instantaneous vesicle speed, were changed in Rab11-FIP2 knockdown compared to control. The overall distributions were significantly different among all four conditions (Chi square, p<0.0001 for track displacement length, p<0.0001 for track speed mean, and p<0.0001 for instantaneous vesicle speed) (Figure 6A,C,E). The distribution in all three parameters was shifted such that the population of less motile vesicles was decreased in Rab11-FIP2 knockdown compared to control cells. For track displacement length, 67.9% of mCherry-Rab11a vesicles traveled less than 5μm in control cells, while this population was decreased to 56.3% in Rab11-FIP2 knockdown cells (Figure 6B). Furthermore, rescue with Venus-Rab11-FIP2 brought this slow-moving vesicle population back to 69.4%, while rescue with Venus-Rab11-FIP2(S229P/G233E) remained low at 61.8%. Similarly, the percentage of vesicles moving at mean speeds of less than 0.25μm/sec was decreased in Rab11-FIP2 knockdown (40.6%) compared to control (53.5%) (Figure 6D). Also, this phenotype was rescued with Venus-Rab11-FIP2(57.3%) but not with Venus-Rab11-FIP2(S229P/G233E) (45.9%). All of these parameters suggested that the increase in vesicle speed observed with loss of Rab11-FIP2 expression or loss of Rab11-FIP2 interaction with MYO5B resulted from a decrease in non-motile vesicles.
Figure 6. Particle tracking distribution of Rab11a labeled vesicle speed and distance in live HeLa cells.

mCherry-Rab11a vesicles were tracked by Imaris software in Figure 6 and data was graphed as distributions. X axis labels in B,D, and F denote Bin Centers which are the midpoints of each category. A) Track displacement length was distributed over 0-55 μm and grouped into 5 μm bins. Chi square analysis was done over the four groups (Scrambled control, Rab11-FIP2 knockdown, Rab11-FIP2 knockdown + Venus-Rab11-FIP2, Rab11-FIP2 knockdown + Venus-Rab11-FIP2(S229P/G233E)) with p<0.0001 across all groups. B) Track displacement length bar graph of mCherry-Rab11a vesicles moving 5 μm distance or less in each of the 4 conditions. Z-test for statistical significance between populations was calculated and * = p<0.001. C) Track speed mean was distributed over 0-1.3 μm/sec and grouped into 0.05 μm/sec bins. Chi square analysis was done over the four groups (Scrambled control, Rab11-FIP2 knockdown, Rab11-FIP2 knockdown + Venus-Rab11-FIP2, Rab11-FIP2 knockdown + Venus-Rab11-FIP2(S229P/G233E)) with p<0.0001 across all groups. D) Track speed mean bar graph of mCherry-Rab11a vesicles moving 0.25 μm/sec speed or less in each of the 4 conditions. Z-test for statistical significance between populations was calculated and * = p<0.001. E) Vesicle speed (instantaneous) was distributed over 0-2.78 μm/sec and grouped into 0.05 μm/sec bins. Chi square analysis was done over the four groups (Scrambled control, Rab11-FIP2 knockdown, Rab11-FIP2 knockdown + Venus-Rab11-FIP2, Rab11-FIP2 knockdown + Venus-Rab11-FIP2(S229P/G233E)) with p<0.0001 across all groups. F) Vesicle speed (instantaneous) bar graph of mCherry-Rab11a vesicles moving 0.05μm/sec speed or less in each of the 4 conditions. Z-test for statistical significance between populations was calculated and * = p<0.001.
Next, we compared the instantaneous speed of individual vesicles at each point along the tracks (Figure 6F). This measurement indicates the vesicle speed at every point along the track. We found that the population of mCherry-Rab11a vesicles moving less than 0.05 μm/sec was decreased in Rab11-FIP2 knockdown cells compared to scrambled control cells (24.9% in KD, 32.4% in control). Rescue of this phenotype with Venus-Rab11-FIP2 increased this population back to 37.4%, while the population in cells rescued with Venus-Rab11-FIP2(S229P/G233E) was 27.5%. Because our measurements were performed on vesicles of approximately 0.4 μm in diameter, this population of vesicles moving less than 0.05 μm/sec represents a population of non-motile vesicles or points at which vesicles are stalled. Therefore, this population of non-motile or stalled vesicles revealed by perturbing the MYO5B/Rab11-FIP2 interaction suggests that MYO5B has reduced its ability to tether these Rab11a-containing vesicles.
Finally, we analyzed this population of non-motile vesicles to determine if they were concentrated in specific regions of the cell or if they were randomly distributed. By color coding the vesicles by speed we found that different vesicle speeds were randomly distributed across the cell for all test conditions (Figure 7). Thus, in scrambled control, FIP2 knockdown, FIP2 knockdown + FIP2(WT) and FIP2 knockdown + FIP2(SPGE) we observed slower moving vesicles coded in blue across the entire cell and faster moving vesicles coded in green, yellow, and red also distributed across the entire cell. We saw no concentration of slow or fast moving vesicle populations an any discrete cell regions.
Figure 7. Non-motile vesicle populations are distributed randomly throughout the cell.

mCherry-Rab11a vesicles were tracked by Imaris software for vesicle speed and displayed across the cell. The vesicles were color-coded for speed with blue being the slowest and red being the fastest. Each image is representative of at least 3 cells.
Discussion
The assembly of multiprotein complexes is critical to the function of Rab small GTPases in the regulation of specific membrane trafficking pathways. In the case of Rab11a, where multiple Rab11-binding proteins are known and are present in the same cell, many protein assemblies are possible (22). While relatively little is known about how formation of specific complexes regulates particular aspects of trafficking, the tripartite complex of Rab11a with both MYO5B and Rab11-FIP2 is one of the best-characterized interacting protein complexes. Rab11a binds to the carboxyl terminal region of Rab11-FIP2 through an amphipathic alpha-helical domain that is conserved in Rab11-FIP proteins (16, 17). Limited structural studies of the carboxyl terminus of Rab11-FIP2 have shown that the terminal 100 amino acids of the protein form a dimer that interacts with two molecules of Rab11a (24-26). The binding site for Rab11a with MYO5B is more complex and direct interaction was established in yeast 2-hybrid screening studies as well as FRET studies in vivo (7, 27). More recently we have been able to obtain complexes of Rab11a with the globular tail of MYO5B through co-expression of the proteins in bacteria (28). In those studies, mutation of two key amino acids in MYO5B abolishes interaction of Rab11a with MYO5B (28).
We have now determined key residues within Rab11-FIP2 that are critical for interactions between Rab11-FIP2 and MYO5B. It is clear that MYO5B tail can interact directly with Rab11-FIP2 in both yeast 2-hybrid assays and in vivo interaction assays (29). We have utilized the interaction in yeast 2-hybrid assays to identify two residues that influence the interaction between the two proteins. The two residues, S229 and G233, reside in a flexible region distal to the amino-terminal C2 domain. Mutation of these amino acids in the minimal Rab11-FIP2 MYO5B binding region (amino acids 129-356) caused a loss in association with MYO5B. Interestingly, when the mutations were introduced into full length Rab11-FIP2, we did not observe a loss of interaction with MYO5B. However, we did observe a loss of colocalization between Rab11-FIP2 and endogenous MYO5B in MCDK cells, indicating that these residues are important for maintaining the tripartite complex.
Thus, by implication, the interaction of MYO5B and Rab11-FIP2 is likely involved in stabilizing the complex or in functional aspects of the Rab11a/Rab11-FIP2/MYO5B complex. Our data suggest that MYO5B acts as a dynamic tether and that disruption of the MYO5B/Rab11-FIP2 association leads to increased Rab11a-containing vesicle motility. By analyzing the association of endogenous MYO5B with the dual mutant form of Rab11-FIP2, we observed a decrease in MYO5B association with Rab11-FIP2(S229P/G233E) with no change in association of Rab11-FIP2(S229/G233E) with Rab11a. This observation is consistent with the concept that inhibition of the interaction of MYO5B with Rab11-FIP2 would destabilize the trafficking complex. It remains unclear where MYO5B acts as a true processive motor for vesicle movement along actin filaments versus a dynamic tether resisting movement or altering vesicle directionality along microtubules. Recent studies have suggested that the MYO5B motor acts to resist the movement of recycling system vesicles along microtubules (30). Consistent with this concept, our studies showed that either mutation or knockdown of Rab11-FIP2 caused Rab11a-containing vesicles to move more quickly over greater distances, pointing to a role of MYO5B as a dynamic tether.
Previous work however, implicated MYO5B as a tether specifically in peripheral endocytic compartments, whereas our work analyzes the role of MYO5B/Rab11-FIP2 in the entire population of Rab11a-containing vesicles. Furthermore, our analyses identify a pool of non-motile or stalled vesicles. We have shown that this non-motile pool is smaller when the MYO5B/Rab11-FIP2 interaction is perturbed, pointing to the critical role of the MYO5B/Rab11-FIP2/Rab11a tripartite complex in the overall motility of Rab11a containing vesicles. Furthermore, our distribution analysis reveals a stopping and starting behavior of these vesicles, which suggests that the MYO5B tethering is dynamic. Similarly, several studies have recently suggested a model where myosin V motors can participate in a “tug-of-war” with either kinesin or dynein and can resist movement of organelles along microtubules by processive anchoring to F-actin filaments (31, 32). Our data support this hypothesis as well, since release of the MYO5B/Rab11-FIP2 resistance leads to increased Rab11a-containing vesicle movement as measured by an increase in vesicle speed and distance traveled. Our analyses show that this increased vesicle movement is due to a smaller population of non-motile vesicles in conditions where the MYO5B/Rab11-FIP2 interaction is disrupted and thus supports a role for MYO5B as a tether. Furthermore, this tethering function as seen in populations of non-motile vesicles was randomly distributed across the entire cell. It should also be noted that MYO5B may also serve as a true processive motor in certain cell regions. Thus, MYO5B and Rab11-FIP2 are thought to be critical for trafficking of AMPA receptors into dendritic spines (33). The difference in function could reflect a role for MYO5B as a resistor of long-distance vesicle transport along microtubules, compared to directed transport across shorter distances along F-actin filaments. In any case, the present results support a critical role for the binding of Rab11-FIP2 with MYO5B within a functional complex with Rab11a to regulate dynamic movements of membrane recycling vesicles.
In conclusion, our data define two important residues (S229 and G233) involved in the interaction of Rab11-FIP2 with MYO5B and show that this interaction is important for the stability and tethering function of the Rab11a/Rab11-FIP2/MYO5B tripartite complex. Our findings strengthen the hypothesis that MYO5B is a dynamic tether and indicate a role for this tether in resisting the movement of Rab11a-containing vesicles, distinct from any capacity as a processive motor.
Materials and Methods
Constructs
EGFP-Rab11-FIP2 constructs were described previously and Rab11-FIP2 sequences were subcloned into Venus-C2 plasmids (16). MYO5B and MYO5B tail constructs were described previously (7, 27). The Venus- versions of Rab11-FIP2 and Rab11-FIP2(129-356) with both single and double mutants were made by standard Quikchange methods (Stratagene).
Antibodies
The Rabbit anti-Rab11a and the chicken anti-MYO5B were as previously described (21, 34). We further validated the anti-MYO5B antibody by probing lysates of control and MYO5B knockdown in MDCK cells (Supplemental Figure 6) (21). Secondary antibodies were from Jackson ImmunoResearch. Three different Rab11-FIP2 antibodies were used in combination for western blotting: a polyclonal rabbit antibody from Sigma-Aldrich (HPA037726), a polyclonal rabbit antibody from Proteintech Group (18136-1-AP), and a polyclonal goat antibody. The polyclonal goat anti-Rab11-FIP2 was produced against a KLH-conjugated peptide (NRQDYFDYESTN) corresponding to amino acids 395-406 in the human Rab11-FIP2 sequence (Strategic Biosolutions, Ramona, CA). The antiserum was affinity purified utilizing the non-conjugated peptide. Rabbit anti-VDAC was from Abcam (ab15895).
XL-1 Red mutagenesis
To create random mutations in the Rab11-FIP2 nucleotide sequence, we used XL-1 Red competent cells from Stratagene and followed the Stratagene instructions to create a “mini-library” of Rab11-FIP2 mutants to be used in a yeast 2-hybrid screen. We followed the standard competent cell transformation for XL-1 Red until the plating step. At this step, we took the 100 μl of XL-1 Red cells transformed with Rab11-FIP2(129-356) in pBD-GAL-Cam (Stratagene, referred to as BD) and added 100 ml LB with antibiotic. We grew this 100 ml at 37°C for 73 hours. 73 hours was chosen based on calculations of the length of the Rab11-FIP2(129-356)-BD plasmid (approximately 7200 nt) and the goal of one mutation per 1000 nucleotides. Fresh antibiotic was added every 24 hours. After 73 hours, the 100 ml culture was used for a maxiprep (Qiagen). We utilized this plasmid stock as a library of random mutations in Rab11-FIP2(129-356)-BD.
Yeast 2-hybrid screen
To find mutants of Rab11-FIP2(129-356)-BD that could not interact with MYO5B tail in pAD-GAL (Stratagene, referred to as BD or AD), we performed a modified yeast 2-hybrid screen. A target line of MYO5B tail-AD was created. Using this yeast line, we transformed 2.5 μg of our Rab11-FIP2(129-356)-BD mutant library. Transformed yeast were plated onto 10 plates under -Trp-Leu-Ade-His selection and incubated at 30°C for 4 days. On day 4, blue-white screening was performed on plates to look for loss of Rab11-FIP2(129-356)-BD and MYO5B tail-AD interaction. Colonies were transferred to filter paper, frozen in liquid nitrogen twice, and then incubated with X-gal (20 mg/ml). Because the interaction between Rab11-FIP2(129-356)-BD wild type and MYO5B tail-AD is a robust blue signal within 2-3 hours, we began looking for white colonies after three hours. Colonies that did not turn blue after three hours were propagated, yeast minipreps were performed to isolate the Rab11-FIP2(129-356)-BD DNA, and plasmids were sequenced to identify mutations in Rab11-FIP2(129-356)-BD. Of the 48 white colonies isolated, 29 were wild type in Rab11-FIP2(129-356), 14 were frameshift mutations resulting in a truncated protein, 3 were nonsense mutations, and 2 were missense mutations.
Binary yeast 2-hybrid assays
After obtaining the two missense mutations, we created these single point mutants (S229P and G233E) as well as the double point mutant (S229P/G233E) in full-length Rab11-FIP2-BD as well as Rab11-FIP2(129-356)-BD for binary yeast 2-hybrid assays against MYO5B tail-AD. These constructs were made by standard Quikchange methods (Stratagene). Binary yeast 2-hybrid assays were performed as previously described with the same Rab11-FIP2-BD constructs that exhibited no auto-activation (15).
Semi-quantitative Yeast 2-hybrid β-galactosidase assays
Individual colonies of dually expressing Y190 yeast were grown overnight at 30°C in YPDA and used to inoculate fresh cultures the next morning. These cultures were grown for 6 hours and OD600 was determined. All samples were adjusted to the same OD600 and then resuspended in a buffer of Z buffer/YPER/ONPG and incubated at 37°C for 16-24 hours. Reactions were stopped with 1 M sodium acetate and absorbance at 420 nm was read on a spectrophotometer. All samples were assayed in triplicate and each experiment was conducted three times. Arbitrary absorbance units were calculated by subtracting MYO5B tail-AD interaction with empty pBD vector as background and the values were averaged. An ANOVA test was performed across groups with post hoc comparison of significant means with Bonferroni's multiple comparison test for pairwise comparisons using PRISM software.
In vitro Binding Assay
Rab11-FIP2 (WT or S229P/G233E) in pET30a was expressed in bacteria, induced for 2 hours with IPTG, and lysed in SDS sample buffer (16). Equal amounts of uninduced and induced bacterial lysate were resolved by SDS-PAGE and stained with Coomassie blue. A second gel with equal amounts was used for Rab11a overlay. After transfer to nitrocellulose, 4 ug of recombinant Rab11a (preloaded with GTP) was incubated with the blot and washed with TBS + 0.05% Tween. The blot was incubated with rabbit anti-Rab11a antibody, washed, incubated with secondary antibody (Li-Cor 926-32213 donkey anti-rabbit IR800CW), washed, and signal was detected on a Li-Cor Odyssey imager.
HeLa and MDCK Cell Transfections and Staining
HeLa and MDCK cells were maintained and propagated by standard methods. Transient transfections were performed for approximately 18-24 hours with 200 ng total DNA using either Effectene transfection reagent (Qiagen) or PolyJet (SignaGen Laboratories). Cells were fixed for 20 min. in either 4% paraformaldehyde or, for endogenous MYO5B staining, with 200 mM Pipes, 2 mM EGTA, 2 mM MgSO4, 0.2% TritonX-100, 60% glycerol pH 7.0, washed 3 times in PBS, and then blocked/extracted for 30 minutes at RT in 10% normal donkey serum (Jackson Immuoresearch), 0.3% Triton X-100 in PBS. Coverslips were incubated for 2 hours at RT with primary antibodies diluted in 1% normal donkey serum, 0.05% Tween-20 in PBS. Coverslips were washed 3 times for 15 minutes at RT with 0.05% Tween-20 in PBS (PBS-T), then incubated for 1 hour at RT with secondary antibodies diluted as above, washed 3 times in PBS-T, once in PBS, then rinsed in water and mounted with ProLong Gold plus DAPI (Invitrogen).
Deconvolution Imaging and Quantitation
HeLa cells were fixed and imaged with a 100× oil immersion objective (1.4 numerical aperture) on a DeltaVision deconvolution microscope (Applied Precision, Issaquah, WA) and a CoolSNAP HQ2 camera (Photometrics, Tucson, AZ). Single plane, widefield images were collected for DIC, FITC (GFP/Venus), and TRITC (mCherry). Images were collected at 512 × 512 pixels. Exposures were typically 0.2 to 0.5 seconds and were sufficient to achieve a minimum 5:1 signal-to-noise ratio. Individual raw files were deconvolved using Applied Precision Softworx deconvolution package. For each GFP-Rab11-FIP2 construct, 10 cells were imaged. These cells were chosen based on a low expression level of GFP-Rab11-FIP2. Images were then deidentified and scored blindly. Images were scored as to the colocalization of GFP-Rab11-FIP2 with Cherry-MYO5B tail as Colocalized, Partial, or Separate. These distributions were plotted as a bar graph. Distributions and Chi Square analyses were performed with PRISM software for each pair.
Confocal Imaging and Quantitation
Confocal images were taken either on a Zeiss LSM 510 inverted confocal microscope using a 40×/1.30 Plan-Neofluar oil objective or on an Olympus FV-1000 inverted confocal microscope using a 60×/1.42 Plan APO oil objective. Both microscopes are maintained in the Vanderbilt Cell Imaging Shared Resource. Images were converted into tifs and processed for figures in Adobe Photoshop. Quantitation of co-localization within the perinuclear region of the cells was performed using the JACoP plug-in of ImageJ (35).
shRNA
Stable knockdown lines were created with lentiviral shRNA constructs from Sigma (Rab11-FIP2, # TRCN0000144035). Briefly, the shRNA vector of interest along with two packaging vectors (pMDG and pAX2, gift from P. Spearman, Emory University) were transfected into HEK cells with Effectene (Qiagen) to produce virus (36). The next day the cells were washed and refed. Virus was harvested after 48 hours, filtered, and immediately added to HeLa cells along with fresh media and polybrene (5 μg/ml) for infection. After 24 hours, cells were rinsed and re-fed with fresh media and puromycin at 1 μg/ml for selection. Knockdown was confirmed by western blot with antibodies to FIP2.
Live cell imaging and particle tracking
HeLa cells were maintained in RPMI media with 10% fetal bovine serum. For microscopy, cells were grown in 35-mm glass bottom Matek dishes for at least one day. Cells were then transfected using Effectene (Qiagen) and 200ng of DNA for each construct used. HeLa cells were transfected with mCherry-Rab11a in combination with either Venus-Rab11-FIP2 wild type or Venus-Rab11-FIP2-SPGE for at least 16 hours. Cells were then imaged in a 37°C, 5% CO2 chamber using a Deltavision deconvolution microscope (Applied Precision). Single plane, widefield images were collected at 100 × in the mCherry-Rab11a (TRITC) channel every 2 seconds for 2 minutes at exposures of 0.3 to 0.5 seconds, which were sufficient to achieve a 5 to 1 signal to noise ratio. Individual raw files were deconvolved using Applied Precision Softworx deconvolution package. Imaris software particle tracking from Bitplane was used to analyze track displacement length, track speed mean, and instantaneous track speed. The Imaris program defines these parameters as follows: Track displacement length is the distance between first and last position of each vesicle. Track speed mean is the average of track speed. If the track has no merges or splits, then the average speed is given by the track length divided by the time between first and last object in the track. Speed is the instantaneous speed of the object. Particles were chosen with an average diameter of 0.4 μm and a maximum distance between frames of 2 μm. A gap size of 2 was incorporated so that a particle could leave the field for two frames, be detected by the software, and still be incorporated into the track. The data were compiled manually using PRISM software to calculate average values for each parameter. Data were analyzed using an ANOVA with post hoc analysis with a Bonferroni correlation for pairwise comparisons. Data are reported as the mean ± SEM. Distributions and Chi Square analyses were performed with PRISM software. Chi Square analyses were performed across all four groups to calculate significance. For bar graphs in Figure 7, bin centers along the X-axis denote the midpoints of each grouping and Z-tests were calculated to show statistical significance across populations. Studies were repeated at least three times with the following numbers of cells and vesicles for track displacement length and track speed mean: Venus-Rab11-FIP2 overexpression, 11 cells, 34,355 vesicles; Venus-Rab11-FIP2(S229P/G233E) overexpression, 19 cells, 74,155 vesicles; Scrambled, 11 cells, 23,332 vesicles; Rab11-FIP2 knockdown, 16 cells, 32,082 vesicles; Rab11-FIP2 knockdown + Venus-Rab11-FIP2, 19 cells, 36,411 vesicles; Rab11-FIP2 knockdown + Rab11-FIP2(S229P/G233E), 30 cells, 61,562 vesicles. For instantaneous track speed, the number of events measured was 355,255 for Scrambled; 581,293 for Rab11-FIP2 knockdown; 617,791 for Rab11-FIP2 knockdown + Venus-Rab11-FIP2; and 1,036,532 for Rab11-FIP2 knockdown + Venus-Rab11-FIP2(S229P/G233E).
Supplementary Material
GFP-Rab11-FIP2 expression levels in HeLa cells. A GFP 20× field of each GFP-Rab11-FIP2 construct expressed in HeLa cells along with mCherry-MYO5B tail was taken to demonstrate the variety of expression levels. Each GFP-Rab11-FIP2(129-356) was captured with the same exposure time, and each GFP-Rab11-FIP2 full-length was captured with the same exposure time. Arrows indicate representative cells chosen for images in this study based on low expression levels.
Colocalization of GFP-Rab11-FIP2 with endogenous MYO5B and endogenous Rab11a. Double merge images of panels from Figure 5 are shown. MDCK cells were transfected with Venus chimeras of A. wild type (WT) Rab11-FIP2, B. Rab11-FIP2(S229P/G233E), C. Rab11-FIP2(129-512) or D. Rab11-FIP2(129-512) (S229P/G233E) and then immunostained for endogenous MYO5B and endogenous Rab11a. Double merges for all pairs are shown.
In vitro binding assay of Rab11-FIP2 and Rab11-FIP2(S229P/G233E) to Rab11a. Rab11-FIP2(WT or S229P/G233E) was expressed in bacteria, induced for 2 hours with IPTG, and lysed in SDS sample buffer. Equal amounts of uninduced (U) and induced (I) bacterial lysates were resolved by SDS-PAGE and stained with Coomassie blue (left). A second gel with equal amounts was used for Rab11a overlay (right). After transfer to nitrocellulose, 4 ug of recombinant Rab11a (preloaded with GTP) was incubated with the blot and bindings was detected with anti-Rab11a antibodies. Quantitation of Rab11-FIP2 bands (WT or S229P/G233E) in Coomassie and Western Blot using Image Studio software by Li-Cor showed equal binding of Rab11a to WT and S229P/G233E when adjusted for loading.
Rab11-FIP2 knockdown in HeLa cells. HeLa cell lysates expressing either scrambled shRNA control or Rab11-FIP2-UTR shRNA were analyzed by western blot with antibodies to Rab11-FIP2 to evaluate knockdown of protein. Anti-VDAC served as a loading control. White arrowheads point to Rab11-FIP2 bands.
mCherry-Rab11a tracks in pseudocolor by track length. mCherry-Rab11a movement was tracked in HeLa cell lines using Imaris software. Example cells display all tracks detected and then pseudocolored based on track displacement length. Examples are representative and are set to the same scale.
Validation of MYO5B antibody. MDCK cell lysates were prepared from control and MYO5B knockdown cell lines using CHAPS detergent. Equal amounts of protein were loaded and resolved by SDS-PAGE. After transfer to nitrocellulose, blots were probed with chicken anti-MYO5B antibody (left) and anti-alpha-tubulin (right) as a loading control. Secondary antibodies were chicken HRP and rabbit HRP, respectively. Compared to loading control, MYO5B protein was greatly reduced in MYO5B knockdown cells compared to control.
mCherry-Rab11a movement. mCherry-Rab11a vesicles in HeLa cells were imaged over time on a Deltavision deconvolution microscope. Images were acquired every 2 seconds for a total of 2 minutes. Files were converted to Quicktime and are set at 2 frames per second for viewing. Movies are representative of at least 11 cells per condition. Supplemental Movie 1 shows mCherry-Rab11a in the presence of Venus-Rab11a-FIP2. Supplemental Movie 2 shows mCherry-Rab11a in the presence of Venus-Rab11a-FIP2(S229P/G233E). Supplemental Movie 3 shows mCherry-Rab11a in scrambled shRNA control cells. Supplemental Movie 4 shows mCherry-Rab11a in Rab11-FIP2-3′UTR knockdown cells. Supplemental Movie 5 shows mCherry-Rab11a in Rab11-FIP2-3′UTR knockdown cells with re-expressed Venus-Rab11-FIP2 wild type. Supplemental Movie 6 shows mCherry-Rab11a in Rab11-FIP2-3′UTR knockdown cells with re-expressed Venus-Rab11-FIP2(S229P/G233E).
*.
This work was supported by NIH National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants DK48370, DK070856 and DK070856-S1 (to J.R.G.) and F32 GM082146 to J.C.S. Confocal fluorescence imaging was performed through the use of the VUMC Cell Imaging Shared Resource, supported by National Institute of Health (NIH) Grants CA-68485, DK-20593, DK-58404 and HD15052. Live cell and deconvolution imaging studies were performed in the Vanderbilt Digital Histology Shared Resource.
References
- 1.Schwartz SL, Cao C, Pylypenko O, Rak A, Wandinger-Ness A. Rab GTPases at a glance. J Cell Sci. 2007;120:3905–3910. doi: 10.1242/jcs.015909. [DOI] [PubMed] [Google Scholar]
- 2.Wu X, Rao K, Bowers MB, Copeland NG, Jenkins NA, Hammer JA. Rab27a enables myosin Va-dependent melanosome capture by recruiting the myosin to the organelle. JCell Sci. 2001;114:1091–1100. doi: 10.1242/jcs.114.6.1091. [DOI] [PubMed] [Google Scholar]
- 3.Hume AN, Collinson LM, Rapak A, Gomes AQ, Hopkins CR, Seabra MC. Rab27a regulates the peripheral distribution of melanosomes in melanocytes. JCell Biol. 2001;152:795–808. doi: 10.1083/jcb.152.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahadoran P, Aberdam E, Mantoux F, Busca R, Bille K, Yalman N, de Sain-Basile G, Cararoli-Marano R, Ortonne JP, Ballotti R. Rab27a: A key to melanosome transport in human melanocytes. JCell Biol. 2001;152:843–850. doi: 10.1083/jcb.152.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strom M, Hume AN, Tarafder AK, Barkagianni E, Seabra MC. A family of Rab27a-binding proteins: Melanophilin links Rab27a and myosin Va functions in melanosome transport. J Biol Chem. 2002;277:25423–25430. doi: 10.1074/jbc.M202574200. [DOI] [PubMed] [Google Scholar]
- 6.Lapierre LA, Kumar R, Hales CM, Navarre J, Bhartur SG, Burnette JO, Provance DW, Jr, Mercer JA, Bahler M, Goldenring JR. Myosin vb is associated with plasma membrane recycling systems. Mol Biol Cell. 2001;12:1843–1857. doi: 10.1091/mbc.12.6.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roland JT, Kenworthy AK, Peranen J, Caplan S, Goldenring JR. Myosin Vb Interacts with Rab8a on a Tubular Network Containing EHD1 and EHD3. Mol Biol Cell. 2007;18:2828–2837. doi: 10.1091/mbc.E07-02-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roland JT, Lapierre LA, Goldenring JR. Alternative splicing in class V myosins determines association with Rab10. J Biol Chem. 2009;284:1213–1223. doi: 10.1074/jbc.M805957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hume AN, Tarafder AK, Ramalho JS, Sviderskaya EV, Seabra MC. A coiled-coil domain of melanophilin is essential for Myosin Va recruitment and melanosome transport in melanocytes. Mol Biol Cell. 2006;17:4720–4735. doi: 10.1091/mbc.E06-05-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hume AN, Ushakov DS, Tarafder AK, Ferenczi MA, Seabra MC. Rab27a and MyoVa are the primary Mlph interactors regulating melanosome transport in melanocytes. J Cell Sci. 2007;120:3111–3122. doi: 10.1242/jcs.010207. [DOI] [PubMed] [Google Scholar]
- 11.Ramalho JS, Lopes VS, Tarafder AK, Seabra MC, Hume AN. Myrip uses distinct domains in the cellular activation of myosin VA and myosin VIIA in melanosome transport. Pigment Cell Melanoma Res. 2009;22:461–473. doi: 10.1111/j.1755-148X.2009.00567.x. [DOI] [PubMed] [Google Scholar]
- 12.Waselle L, Coppola T, Fukuda M, Iezzi M, El-Amraoui A, Petit C, Regazzi R. Involvement of the Rab27 binding protein Slac2c/MyRIP in insulin exocytosis. Mol Biol Cell. 2003;14:4103–4113. doi: 10.1091/mbc.E03-01-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuroda TS, Fukuda M. Identification and biochemical analysis of Slac2-c/MyRIP as a Rab27A-, myosin Va/VIIa-, and actin-binding protein. Methods Enzymol. 2005;403:431–444. doi: 10.1016/S0076-6879(05)03038-7. [DOI] [PubMed] [Google Scholar]
- 14.Kuroda TS, Itoh T, Fukuda M. Functional analysis of slac2-a/melanophilin as a linker protein between Rab27A and myosin Va in melanosome transport. Methods Enzymol. 2005;403:419–431. doi: 10.1016/S0076-6879(05)03037-5. [DOI] [PubMed] [Google Scholar]
- 15.Hales CM, Vaerman JP, Goldenring JR. Rab11 family interacting protein 2 associates with Myosin Vb and regulates plasma membrane recycling. J Biol Chem. 2002;277:50415–50421. doi: 10.1074/jbc.M209270200. [DOI] [PubMed] [Google Scholar]
- 16.Hales CM, Griner R, Hobdy-Henderson KC, Dorn MC, Hardy D, Kumar R, Navarre J, Chan EKL, Lapierre LA, Goldenring JR. Identification and characterization of a family of Rab11-interacting proteins. JBiolChem. 2001;276:39067–39075. doi: 10.1074/jbc.M104831200. [DOI] [PubMed] [Google Scholar]
- 17.Prekeris R, Davies JM, Scheller RH. Identificaition of a novel Rab11/25 binding domain in eferin and Rip proteins. JBiolChem. 2001;276:38966–38970. doi: 10.1074/jbc.M106133200. [DOI] [PubMed] [Google Scholar]
- 18.Fan GH, Lapierre LA, Goldenring JR, Sai J, Richmond A. Rab11-family interacting protein 2 and myosin Vb are required for CXCR2 recycling and receptor-mediated chemotaxis. Mol Biol Cell. 2004;15:2456–2469. doi: 10.1091/mbc.E03-09-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nedvetsky PI, Stefan E, Frische S, Santamaria K, Wiesner B, Valenti G, Hammer JA, 3rd, Nielsen S, Goldenring JR, Rosenthal W, Klussmann E. A Role of myosin Vb and Rab11-FIP2 in the aquaporin-2 shuttle. Traffic. 2007;8:110–123. doi: 10.1111/j.1600-0854.2006.00508.x. [DOI] [PubMed] [Google Scholar]
- 20.Gidon A, Bardin S, Cinquin B, Boulanger J, Waharte F, Heliot L, de la Salle H, Hanau D, Kervrann C, Goud B, Salamero J. A Rab11A/Myosin Vb/Rab11-FIP2 Complex Frames Two Late Recycling Steps of Langerin from the ERC to the Plasma Membrane. Traffic. 2012;13:815–833. doi: 10.1111/j.1600-0854.2012.01354.x. [DOI] [PubMed] [Google Scholar]
- 21.Lapierre LA, Avant KM, Caldwell CM, Oztan A, Apodaca G, Knowles BC, Roland JT, Ducharme NA, Goldenring JR. Phosphorylation of Rab11-FIP2 regulates polarity in MDCK cells. Mol Biol Cell. 2012;23:2302–2318. doi: 10.1091/mbc.E11-08-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hales CM, Griner R, Hobdy-Henderson KC, Dorn MC, Hardy D, Kumar R, Navarre J, Chan EK, Lapierre LA, Goldenring JR. Identification and characterization of a family of Rab11-interacting proteins. J Biol Chem. 2001;276:39067–39075. doi: 10.1074/jbc.M104831200. [DOI] [PubMed] [Google Scholar]
- 23.Ducharme NA, Williams JA, Oztan A, Apodaca G, Lapierre LA, Goldenring JR. Rab11-FIP2 regulates differentiable steps in transcytosis. Am J Physiol Cell Physiol. 2007;293:C1059–1072. doi: 10.1152/ajpcell.00078.2007. [DOI] [PubMed] [Google Scholar]
- 24.Jagoe WN, Jackson SR, Lindsay AJ, McCaffrey MW, Khan AR. Purification, crystallization and preliminary X-ray diffraction studies of Rab11 in complex with Rab11-FIP2. Acta crystallographica. 2006;62:692–694. doi: 10.1107/S1744309106023074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei J, Fain S, Harrison C, Feig LA, Baleja JD. Molecular dissection of Rab11 binding from coiled-coil formation in the Rab11-FIP2 C-terminal domain. Biochemistry. 2006;45:6826–6834. doi: 10.1021/bi052655o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jagoe WN, Lindsay AJ, Read RJ, McCoy AJ, McCaffrey MW, Khan AR. Crystal structure of rab11 in complex with rab11 family interacting protein 2. Structure. 2006;14:1273–1283. doi: 10.1016/j.str.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 27.Lapierre LA, Kumar R, Hales CM, Navarre J, Bhartur SG, Burnette JO, Mercer JA, Bahler M, Goldenring JR. Myosin Vb is associated with and regulates plasma membrane recycling systems. MolBiolCell. 2001;12:1843–1857. doi: 10.1091/mbc.12.6.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roland JTE, Bryant DM, Datta A, Itzen A, Mostov KE, Goldenring JR. Rab GTPase-Myo5B complexes control membrane recycling and epithelial polarization. Proc Natl Acad Sci U S A. 2011;108:2789–2794. doi: 10.1073/pnas.1010754108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hales CM, Vaerman JP, Goldenring JR. Rab11-Family interacting protein 2(Rab11-FIP2) assocaites with myosin Vb and regulates plasma membrane recycling. J Biol Chem. 2002;277:50415–50421. doi: 10.1074/jbc.M209270200. [DOI] [PubMed] [Google Scholar]
- 30.Provance DW, Jr, Addison EJ, Wood PR, Chen DZ, Silan CM, Mercer JA. Myosin-Vb functions as a dynamic tether for peripheral endocytic compartments during transferrin trafficking. BMC Cell Biol. 2008;9:44. doi: 10.1186/1471-2121-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schroeder HW, 3rd, Mitchell C, Shuman H, Holzbaur EL, Goldman YE. Motor number controls cargo switching at actin-microtubule intersections in vitro. Curr Biol. 2010;20:687–696. doi: 10.1016/j.cub.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schroeder HW, 3rd, Hendricks AG, Ikeda K, Shuman H, Rodionov V, Ikebe M, Goldman YE, Holzbaur EL. Force-dependent detachment of kinesin-2 biases track switching at cytoskeletal filament intersections. Biophysical journal. 2012;103:48–58. doi: 10.1016/j.bpj.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Edwards JG, Riley N, Provance DW, Jr, Karcher R, Li XD, Davison IG, Ikebe M, Mercer JA, Kauer JA, Ehlers MD. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ducharme NA, Hales CM, Lapierre LA, Ham AJ, Oztan A, Apodaca G, Goldenring JR. MARK2/EMK1/Par-1Balpha phosphorylation of Rab11-family interacting protein 2 is necessary for the timely establishment of polarity in Madin-Darby canine kidney cells. Mol Biol Cell. 2006;17:3625–3637. doi: 10.1091/mbc.E05-08-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. Journal of microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 36.Qi M, Williams JA, Chu H, Chen X, Wang JJ, Ding L, Akhirome E, Wen X, Lapierre LA, Goldenring JR, Spearman P. Rab11-FIP1C and Rab14 direct plasma membrane sorting and particle incorporation of the HIV-1 envelope glycoprotein complex. PLoS Pathog. 2013;9:e1003278. doi: 10.1371/journal.ppat.1003278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GFP-Rab11-FIP2 expression levels in HeLa cells. A GFP 20× field of each GFP-Rab11-FIP2 construct expressed in HeLa cells along with mCherry-MYO5B tail was taken to demonstrate the variety of expression levels. Each GFP-Rab11-FIP2(129-356) was captured with the same exposure time, and each GFP-Rab11-FIP2 full-length was captured with the same exposure time. Arrows indicate representative cells chosen for images in this study based on low expression levels.
Colocalization of GFP-Rab11-FIP2 with endogenous MYO5B and endogenous Rab11a. Double merge images of panels from Figure 5 are shown. MDCK cells were transfected with Venus chimeras of A. wild type (WT) Rab11-FIP2, B. Rab11-FIP2(S229P/G233E), C. Rab11-FIP2(129-512) or D. Rab11-FIP2(129-512) (S229P/G233E) and then immunostained for endogenous MYO5B and endogenous Rab11a. Double merges for all pairs are shown.
In vitro binding assay of Rab11-FIP2 and Rab11-FIP2(S229P/G233E) to Rab11a. Rab11-FIP2(WT or S229P/G233E) was expressed in bacteria, induced for 2 hours with IPTG, and lysed in SDS sample buffer. Equal amounts of uninduced (U) and induced (I) bacterial lysates were resolved by SDS-PAGE and stained with Coomassie blue (left). A second gel with equal amounts was used for Rab11a overlay (right). After transfer to nitrocellulose, 4 ug of recombinant Rab11a (preloaded with GTP) was incubated with the blot and bindings was detected with anti-Rab11a antibodies. Quantitation of Rab11-FIP2 bands (WT or S229P/G233E) in Coomassie and Western Blot using Image Studio software by Li-Cor showed equal binding of Rab11a to WT and S229P/G233E when adjusted for loading.
Rab11-FIP2 knockdown in HeLa cells. HeLa cell lysates expressing either scrambled shRNA control or Rab11-FIP2-UTR shRNA were analyzed by western blot with antibodies to Rab11-FIP2 to evaluate knockdown of protein. Anti-VDAC served as a loading control. White arrowheads point to Rab11-FIP2 bands.
mCherry-Rab11a tracks in pseudocolor by track length. mCherry-Rab11a movement was tracked in HeLa cell lines using Imaris software. Example cells display all tracks detected and then pseudocolored based on track displacement length. Examples are representative and are set to the same scale.
Validation of MYO5B antibody. MDCK cell lysates were prepared from control and MYO5B knockdown cell lines using CHAPS detergent. Equal amounts of protein were loaded and resolved by SDS-PAGE. After transfer to nitrocellulose, blots were probed with chicken anti-MYO5B antibody (left) and anti-alpha-tubulin (right) as a loading control. Secondary antibodies were chicken HRP and rabbit HRP, respectively. Compared to loading control, MYO5B protein was greatly reduced in MYO5B knockdown cells compared to control.
mCherry-Rab11a movement. mCherry-Rab11a vesicles in HeLa cells were imaged over time on a Deltavision deconvolution microscope. Images were acquired every 2 seconds for a total of 2 minutes. Files were converted to Quicktime and are set at 2 frames per second for viewing. Movies are representative of at least 11 cells per condition. Supplemental Movie 1 shows mCherry-Rab11a in the presence of Venus-Rab11a-FIP2. Supplemental Movie 2 shows mCherry-Rab11a in the presence of Venus-Rab11a-FIP2(S229P/G233E). Supplemental Movie 3 shows mCherry-Rab11a in scrambled shRNA control cells. Supplemental Movie 4 shows mCherry-Rab11a in Rab11-FIP2-3′UTR knockdown cells. Supplemental Movie 5 shows mCherry-Rab11a in Rab11-FIP2-3′UTR knockdown cells with re-expressed Venus-Rab11-FIP2 wild type. Supplemental Movie 6 shows mCherry-Rab11a in Rab11-FIP2-3′UTR knockdown cells with re-expressed Venus-Rab11-FIP2(S229P/G233E).