

Abstract
The therapeutic index and cellular mechanism of action of [Pt(BDIQQ)]Cl, a monocationic, square-planar platinum(II) complex, are reported. [Pt(BDIQQ)]Cl was used to treat several cell lines, including wild type and cisplatin-resistant ovarian carcinoma cells (A2780 and A2780CP70) and non-proliferating lung carcinoma cells (A549). [Pt(BDIQQ)]Cl selectively kills cancer over healthy cells and exhibits no cross-resistance with cisplatin. The mechanism of cell killing was established through detailed cell-based assays. [Pt(BDIQQ)]Cl exhibits dual-threat capabilities, targeting nuclear DNA and mitochondria simultaneously. [Pt(BDIQQ)]Cl induces DNA damage, leading to p53 enrichment, mitochondrial membrane potential depolarisation, and caspase-mediated apoptosis. [Pt(BDIQQ)]Cl also accumulates in the mitochondria, resulting in direct mitochondrial damage. Flow cytometric studies demonstrated that [Pt(BDIQQ)]Cl has no significant effect on cell cycle progression. Remarkably, p53-status is a not a determinant of [Pt(BDIQQ)]Cl activity. In p53-null cells, [Pt(BDIQQ)]Cl induces cell death through mitochondrial dysfunction. Cancers with p53-null status could therefore be targeted using [Pt(BDIQQ)]Cl.
Introduction
Platinum-based drugs, namely cisplatin, carboplatin, and oxaliplatin are regularly used to treat solid tumours.1,2 Cisplatin is highly effective against testicular and ovarian cancers while carboplatin and oxaliplatin are routinely used to treat lung, ovarian, and colorectal tumours.3-7 Although many details of the molecular mechanism of cytotoxicity of platinum drugs have been elucidated, nuances continue to be discovered. The current consensus is that cisplatin and its derivatives bind genomic DNA, forming DNA cross-links that block DNA synthesis/transcription and cell cycle progression that ultimately induce tumour cell death.8-12 The shortcomings associated with platinum therapy,13-15 such as inherent, acquired, or cross-resistance, toxic side effects, and high incidence of relapse, highlight the need for new anticancer drugs with novel mechanisms of action that can target platinum-resistant tumours, reduce general toxicity, and broaden the spectrum of activity.
Platinum(II) compounds that bind to DNA non-covalently show promising antineoplastic properties and are now recognised as viable alternatives to conventional platinum drugs, which form irreversible covalent DNA adducts.16-21 In particular, complexes with π-conjugated ligands such as bipyridine, terpyridine, and phenanthroline can intercalate between adjacent base pairs in double stranded DNA.22-24 These metallointercalators display antiproliferative properties, presumably arising from their ability to unwind or bend DNA. Although these compounds unequivocally interact with DNA in vitro, the significance of DNA as an important cellular target has been questioned.25,26 Indeed, the true mechanism of cytotoxic action of some compounds capable of acting as metallointercalators involves cytoskeletal, mitochondrial, and cytoplasmic proteins rather than DNA. Here, we have investigated the cytotoxic potential of a platinum(II) complex bearing the tetradentate ligand, BDIQQH. We recently reported the synthesis and photophysical properties of [Pt(BDIQQ)]Cl.27 In aqueous buffer, [Pt(BDIQQ)]Cl forms non-emissive aggregates, but in the presence of DNA, the aggregates disperse yielding single molecules capable of intercalating between base pairs. Upon intercalation, a 150-fold turn-on in emission intensity occurs. Encouragingly, [Pt(BDIQQ)]Cl displays potency against human lung carcinoma (A549) and cervical adenocarcinoma (HeLa) cells in the micromolar range, similar to or better than that of cisplatin. In the present studied we have delineated the mechanism of cytotoxic activity of [Pt(BDIQQ)]Cl as well as its therapeutic potential.
Results and discussion
The cytotoxicity of [Pt(BDIQQ)]Cl against HeLa and A549 cells was previously reported.27 To evaluate the potential of [Pt(BDIQQ)]Cl as a chemotherapeutic agent, an expanded panel of cell lines was tested. The cytotoxicity of [Pt(BDIQQ)]Cl against five cancerous human cell lines (U2OS, MCF-7, HT-29, A2780, and A2780CP70) and one normal fibroblast cell line (MRC-5) was assessed using the colourimetric MTT assay. The IC50 values (concentration required to induce 50% inhibition) are summarised in Table 1. [Pt(BDIQQ)]Cl displayed micromolar potency against all cancerous cell lines tested, similar to or better than that of cisplatin, and reduced toxicity against healthy cells. Notably, [Pt(BDIQQ)]Cl exhibited higher potency by an order of magnitude towards the lung carcinoma cell line A549 than for a normal lung fibroblast cell line, MRC-5. [Pt(BDIQQ)]Cl kills cisplatin-sensitive and -resistant ovarian carcinoma cells, A2780 and A2780CP70, equally well and thus exhibits no cross-resistance with cisplatin. To gauge therapeutic potential, cell viability studies were conducted with quiescent A549 cells. The IC50 value of [Pt(BDIQQ)]Cl in non-proliferating A549 cells was significantly higher (p > 0.005, t test) than in proliferating A549 cells, indicative of selective potency toward fast growing cancer cells. This feature is a highly attractive characteristic that augurs well for in vivo and clinical applications.
Table 1.
IC50values (μM) of [Pt(BDIQQ)]Cl and cisplatin against various cancerous and healthy cell lines. The errors represent standard deviations.
| Cell line | Cancer type | [Pt(BDIQQ)]Cl | cisplatin |
|---|---|---|---|
| A549 | Lung carcinoma | 1.2 ± 0.2a | 5.2 ± 1.0a |
| HeLa | Cervical adenocarcinoma | 2.8 ± 1.7a | 2.3 ± 1.4a |
| MCF-7 | Breast adenocarcinoma | 6.4 ± 1.1 | 7.4 ± 2.1 |
| HT-29 | Colorectal adenocarcinoma | 13.6 ± 3.3 | 15.03 ± 2.1 |
| U2OS | Bone osteosarcoma | 3.6 ± 0.1 | 4.6 ± 0.6 |
| A2780 | Ovarian carcinoma | 3.2 ± 0.5 | 0.7 ± 0.2 |
| A2780CP70 | Ovarian carcinoma | 3.7 ± 0.1 | 9.6 ±1.6 |
| MRC-5 | Lung fibroblast | 12.2 ± 2.1 | 5.3 ± 0.6 |
| A549 (quiescent) | Lung carcinoma | 9.5 ± 1.3 | 14.2 ± 0.2 |
IC50 values taken from Ref. 27.
The anti-proliferative properties of the free ligand, BDIQQH, against a panel of cell lines were determined (Table S1). BDIQQH displayed poor toxicity against all cell lines (IC50> 40 μM), one order of magnitude lower than that of [Pt(BDIQQ)]Cl. This result suggests that the platinum(II) centre plays an important role in the potency exhibited by [Pt(BDIQQ)]Cl.
Cellular uptake studies were performed to investigate the cell permeability and localisation of [Pt(BDIQQ)]Cl. A549 and A2780 cells were incubated with [Pt(BDIQQ)]Cl (10 μM) for 12 h and the platinum content was determined in the cytoplasmic, nuclear, membrane, and mitochondrial fractions using graphite furnace atomic absorption spectrometry (GF-AAS). The data, reported as ng of platinum per million cells, are shown in Figure 1. [Pt(BDIQQ)]Cl is readily absorbed by cells (ca. 100 ng/ million cells) and distributed evenly between the nucleus and cytoplasm. An appreciable amount of internalised [Pt(BDIQQ)]Cl is also detected in the mitochondria. Therefore cellular toxicity could result from mechanisms associated not only with nuclear DNA, but also cytoplasmic or mitochondrial dysfunction. A large portion of the administered dose was observed in the cell membrane, which we ascribe to the lipophillicity of [Pt(BDIQQ)]Cl.
Figure 1.
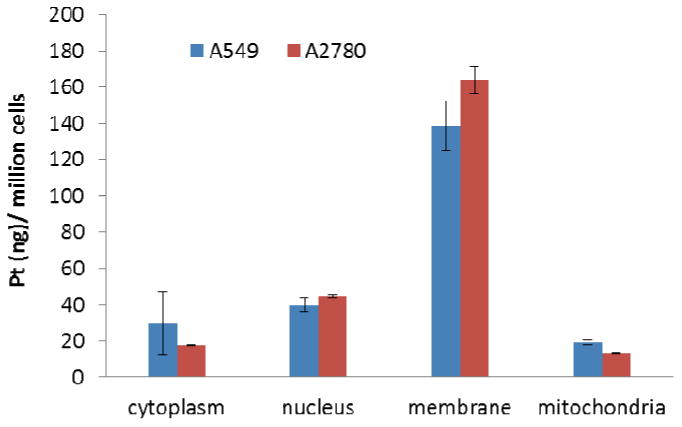
Cellular uptake following [Pt(BDIQQ)]Cl treatment (10 μM for 12 h). The amount of platinum (ng) per million A549 lung carcinoma and A2780 ovarian carcinoma cells is shown for the cytoplasmic, nuclear, membrane, and mitochondrial fractions. The errors represent standard deviations.
To gain insight into the mechanism of action of [Pt(BDIQQ)]Cl, detailed cell-based studies were carried out. Because [Pt(BDIQQ)]Cl interacts strongly with DNA27 and enters the nucleus, the expression of biomarkers related to the DNA damage pathway were investigated. DNA damage leads to phosphorylation of H2AX, a histone variant, at the Ser139 position by PI3K kinases.28 A549 cells treated with [Pt(BDIQQ)]Cl (1-5 μM for 72 h) displayed a marked increase in expression of the phosphorylated form of H2AX (γH2AX), indicative of DNA damage (Figure 2a). To further validate DNA damage, nuclear DNA from [Pt(BDIQQ)]Cl- and cisplatin-treated A549 and A2780 cells was extracted and analysed by GF-AAS. The data are shown in Figure 2b and expressed in terms of pmol of platinum per μg of DNA. Genomic DNA from cells treated with [Pt(BDIQQ)]Cl contained 10-fold higher levels of platinum than those treated with cisplatin. Collectively the data prove that [Pt(BDIQQ)]Cl is able to target and damage nuclear DNA. To shed light on how DNA damage occurs, gel electrophoresis studies were conducted with pUC18 plasmid DNA. At low [Pt(BDIQQ)]Cl concentrations (0.1-5 μM) a clear increase in the amount of open circular DNA was observed and at higher concentrations (7.5-20 μM) an increase in the amount of supercoiled plasmid DNA was detected (Figure S1). This classical gel shift pattern is indicative of DNA unwinding, induced most likely by intercalation of [Pt(BDIQQ)]Cl into the DNA duplex.
Figure 2.
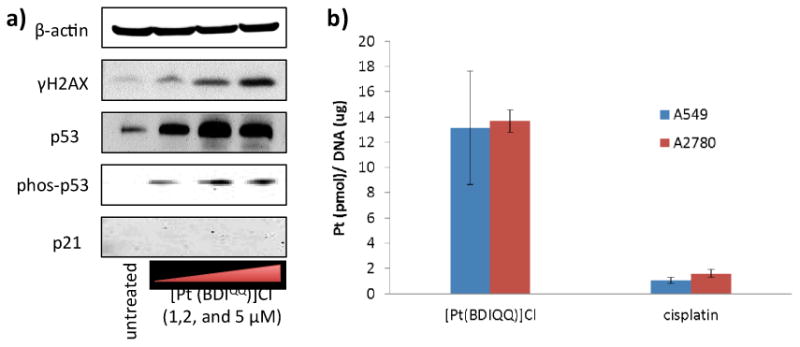
(a) Immunoblotting analysis of proteins related to the DNA damage response and p53 induction pathways. Protein expression in A549 cells was observed following treatment with [Pt(BDIQQ)]Cl (1–5 μM for 72 h). Whole cell lysates were resolved by SDS-PAGE and analysed by immunoblotting against γH2AX, p53, phosp53, p21, and β-actin (loading control). (b) Platinum content in genomic DNA extracted from A549 and A2780 cells dosed with [Pt(BDIQQ)]Cl and cisplatin (10 μM for 12 h).
DNA damage can lead to a multitude of chemical signals including p53 upregulation. In response to DNA damage, apical kinases ATM and ATR phosphorylate p53 at the Ser15 position.29 This alteration inhibits the interaction between p53 and its negative regulator, MDM2, and thus promotes p53 accumulation.30 Incubation of A549 cells with [Pt(BDIQQ)]Cl led to upregulation of p53 and its phosphorylated form, phospho-p53 (Ser15), suggesting that p53 plays a part in the cellular response evoked by [Pt(BDIQQ)]Cl (Figure 2a). Upon activation, p53 can bind DNA and mediate transcription of genes, the products of which control cell cycle arrest, DNA repair, and apoptosis.31-33 Therefore, the expression of downstream effectors of p53 such as p21 (cyclin-dependent kinase inhibitory protein) and BAX (pro-apoptotic protein)34 were investigated. The incremental addition of [Pt(BDIQQ)]Cl did not activate p21 expression, suggesting that the expression of cell cycle controllers like p21 is not a major determinant in the cellular response induced (Figure 2a). To confirm this result, DNA flow cytometric studies were carried out. A549 cells were dosed with [Pt(BDIQQ)]Cl (2 μM) and the cell cycle profile was determined by PI staining at 24 h, 48 h, and 72 h. By comparison to untreated control, [Pt(BDIQQ)]Cl treatment did not trigger cell cycle arrest, even after 72 h incubation (Figure S2). This observation confirmed that cell cycle controllers are not involved in the cellular response. In contrast, cisplatin treatment (5 μM) induced S- and G2/M-phase cell cycle arrest (Figure S2).
[Pt(BDIQQ)]Cl treatment increased BAX levels (Figure S3). When overexpressed, BAX inserts into the outer mitochondrial membrane and oligomerises to form pores that induce mitochondrial outer membrane permeabilisation (MOMP).35 Cytochrome c then enters the cytoplasm (from the mitochondria) and initiates apoptosis.36 The integrity of the mitochondrial membrane upon [Pt(BDIQQ)]Cl treatment was probed by flow cytometry, using the JC-1 assay (5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethyl benzimidazolyl carbocyanine iodide). JC-1 is a cationic lipophilic dye that accumulates in the mitochondria of viable cells as red-emitting aggregates.37,38 If the mitochondrial membrane potential is depleted, the JC-1 aggregates disintegrate, forming monomers with weak red emission. A549 and A2780 cells dosed with a well-known mitochondrial membrane depolariser, carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (1 μM for 48 h), exhibited reduced red fluorescence compared to untreated cells (Figure 2a). [Pt(BDIQQ)]Cl treated (5 μM for 48 h) cells also exhibited reduced red fluorescence, indicative of mitochondrial membrane damage (Figure 2a & b). Mitochondrial membrane disruption can result from p53 and/or BAX activity,39-41 both of which are upregulated upon [Pt(BDIQQ)]Cl treatment. Alternatively, mitochondrial dysfunction can be triggered directly by the action of [Pt(BDIQQ)]Cl on this organelle. [Pt(BDIQQ)]Cl is lipophilic and cationic, and it therefore can readily accumulate in the negatively charged mitochondrial matrix and induce MOMP. Indeed, cellular uptake studies (presented above) indicate that [Pt(BDIQQ)]Cl is able to localise in the mitochondria. To confirm the intracellular targets of [Pt(BDIQQ)]Cl, A549 cells were incubated with a non-lethal dose of [Pt(BDIQQ)]Cl (0.5 μM for 24 h) and then treated with nuclei and mitochondria dyes, Hoechst 33258 (7.5 μM for 30 min) and MitoTracker Green (250 nM for 30 min) respectively. Cisplatin dosed cells (2 μM for 12 h) were also studied as a control. Untreated cells displayed the expected staining pattern but cells incubated with [Pt(BDIQQ)]Cl displayed non-specific nuclei and mitochondria staining, indicative of disruption of typical nuclear DNA and mitochondria structure (Figure 2c). Therefore [Pt(BDIQQ)]Cl targets both nuclear DNA and mitochondria. Cells treated with cisplatin exhibited poor nuclear DNA staining, but mitochondrial staining remained largely unaltered (Figure S4). This result is expected because cisplatin selectively targets nuclear DNA over the mitochondria.
Having established the intracellular targets of [Pt(BDIQQ)]Cl and identified the initial cellular response, we sought to understand how mitochondrial membrane depolarisation is induced. To determine whether or not [Pt(BDIQQ)]Cl-mediated mitochondrial membrane depolarisation is p53-dependent, the JC-1 assay was conducted with HCT116 p53+/+ (p53 wild type) and HCT116 p53-/- (p53 null) cells. [Pt(BDIQQ)]Cl treated (5 μM for 48 h) HCT116 p53+/+ and HCT116 p53-/- cells exhibited similar levels of mitochondrial membrane depolarisation (Figure S5), suggesting that [Pt(BDIQQ)]Cl-induced mitochondrial dysfunction was p53-independent. Mitochondria extracted from HCT116 p53+/+ and HCT116 p53-/- cells dosed with [Pt(BDIQQ)]Cl (10 μM for 12 h) contained substantial amounts of platinum (ca. 5-20 ng of platinum per million cells, Figure S6), confirming that [Pt(BDIQQ)]Cl can mediate direct mitochondrial damage irrespective of p53-status. To establish the role of p53 in toxicity, cytotoxicity studies were conducted with HCT116 p53+/+ and HCT116 p53-/- cells. The IC50 value of [Pt(BDIQQ)]Cl was statistically similar for HCT116 p53+/+ and HCT116 p53-/- cells, indicating that toxicity is p53-independent (Figure S7). The activity of [Pt(BDIQQ)]Cl in HCT116 p53-/- cells is likely to originate from direct mitochondrial damage, a p53-independent process. Notably, the potency of cisplatin is only marginally dependent on p53-status (Figure S7). The tumour suppressor protein, p53, is mutated, defective, or inactivated in several cancers,42 therefore compounds like [Pt(BDIQQ)]Cl, which act independently of p53, hold significant therapeutic potential.
To identify the mode of cell death prompted by [Pt(BDIQQ)]Cl treatment, the dual staining Annexin V/PI flow cytometry assay was conducted. Most clinically employed drugs induce apoptosis.43 During apoptosis, the cell membrane architecture undergoes significant changes, for instance, phosphatidylserine residues that usually reside on the cytosolic side of the cell membrane become exposed on the cell exterior. Phosphatidylserine residues can be detected by Annexin V.44 A549 cells incubated with [Pt(BDIQQ)]Cl (5 μM) for 72 h displayed populations undergoing early- and late-stage apoptosis (17 and 5% respectively) (Figure S8 and Table S2). In support of these observations, immunoblotting studies revealed that [Pt(BDIQQ)]Cl treated A549 cells expressed several proteins characteristic of apoptosis, namely, cleaved caspase 3, 7, and 9, as well as poly ADP ribose polymerase (PARP) (Figure S3).
To confirm apoptosis as the mode of cell death, the morphology and nuclear staining pattern of A549 cells in the presence of toxic amounts of [Pt(BDIQQ)]Cl (20 μM for 12 h) were probed using microscopy. Upon [Pt(BDIQQ)]Cl treatment, some cells displayed membrane blebbing (Figure S9), indicative of apoptosis. Furthermore, some nuclear stained [Pt(BDIQQ)]Cl-treated cells exhibited intense regions within the nucleus, characteristic of chromatin condensation. These features are typical of apoptosis. Taken together, the microscopy data confirm that [Pt(BDIQQ)]Cl is able to induce apoptosis in A549 cells.
Conclusion
In summary, we present the cytotoxic mechanism of action of [Pt(BDIQQ)]Cl. Encouragingly, [Pt(BDIQQ)]Cl exhibits selective toxicity for cancer cells over normal fibroblast cells, and no cross-resistance with cisplatin. Additionally, [Pt(BDIQQ)]Cl selectivity kills proliferating A549 cells over non-proliferating A549 cells. Taken together, the cytotoxicity data are very appealing in terms of anticancer therapy. [Pt(BDIQQ)]Cl acts in a dual-threat manner, concurrently targeting genomic DNA and the mitochondria. [Pt(BDIQQ)]Cl damages genomic DNA, enriches p53 and BAX levels, and subsequently induces mitochondrial-mediated apoptosis. Owing to its lipophilic and cationic properties, [Pt(BDIQQ)]Cl also accumulates in mitochondria and causes direct mitochondrial damage. The latter mechanism proceeds independently of p53 and therefore allows [Pt(BDIQQ)]Cl to exhibit equal toxicity in p53-null and p53-postive cells. Because p53 activity is abrogated in many cancers, the p53-independence of [Pt(BDIQQ)]Cl is highly desirable. Overall, our findings clearly indicate [Pt(BDIQQ)]Cl to be a promising candidate for additional pre-clinical studies.
Experimental Details
Materials
[Pt(BDIQQ)]Cl was prepared as previously reported.27
Cell Lines and Cell Culture Conditions
A549 lung adenocarcinoma, A2870 ovarian carcinoma, A2870/CP70 cisplatin-resistant ovarian carcinoma, MCF-7 breast adenocarcinoma, HT-29 colorectal adenocarcinoma, U2OS bone osteosarcoma, and MRC-5 normal human fetal lung fibroblast cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). HCT116 p53+/+ and HCT116 p53-/- colorectal carcinoma cells were kindly donated by Laura Trudel (MIT). A549, U2OS, MCF-7, HT-29, and MRC-5 cell lines were maintained in Dulbecco's Modified Eagle's Medium (DMEM-low glucose) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. The A2780 and A2780/CP70 cell lines were cultured in RPMI (glutamine-free) media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. The HCT116 p53+/+ and HCT116 p53-/- cell lines were grown in McCoy's 5A modified media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. All cells were grown at 310 K in a humidified atmosphere containing 5% CO2.
Cytotoxicity MTT assay
The colourimetric MTT assay was used to determine the toxicity of [Pt(BDIQQ)]Cl, BDIQQH, and cisplatin. Cells (2 × 103) were seeded in each well of a 96-well plate. After incubating the cells overnight, various concentrations of test compound (0.3-100 μM) were added and incubated for 72 h (total volume 200 μL). [Pt(BDIQQ)]Cl and BDIQQH were prepared as 2-5 mM solutions in DMSO and diluted using media. The final concentration of DMSO in each well was 0.5% and this amount was present in the untreated control as well. Cisplatin was prepared as a 5 mM solution in PBS and diluted further using media. After 72 h, the medium was removed, 200 μL of a 0.4 mg/mL solution of MTT in DMEM, RPMI or, McCoy's 5A was added, and the plate was incubated for an additional 1-2 h. The DMEM/MTT, RPMI/MTT, or McCoy's 5A/MTT mixture was aspirated and 200 μL of DMSO was added to dissolve the resulting purple formazan crystals. The absorbance of the solution wells was read at 550 nm. Absorbance values were normalized to DMSO-containing control wells and plotted as concentration of test compound versus % cell viability. IC50 values were interpolated from the resulting dose dependent curves. The reported IC50 values are the average from at three independent experiments, each of which consisted of six replicates per concentration level.
Cellular Uptake
To measure the cellular uptake of [Pt(BDIQQ)]Cl, ca. 20 million A549 and A2780 cells were treated with 10 μM of [Pt(BDIQQ)]Cl at 37 °C for 12 h. Then the media was removed, the cells were washed with PBS solution (1 mL × 3), harvested, and centrifuged. The cellular pellet was suspended in an appropriate volume of PBS to obtain a homogeneous cell suspension (eg. 100 μL). The Thermo Scientific NE-PER Nuclear and Cytoplasmic Extraction Kit and Mitochondria Isolation Kit were used to extract the separate cytoplasmic, nuclear, membrane, and mitochondrial fractions. The fractions were mineralized with 65% HNO3 and then completely dried at 120 °C. The solid extracts were re-dissolved in 4% HNO3 and analysed using graphite furnace atomic absorption spectroscopy (Perkin-Elmer AAnalyst600 GF-AAS). Cellular platinum levels were expressed as ng of Pt per million cells. Results are presented as the mean of three determinations for each data point.
Genomic DNA Platinum Content
To measure the amount of platinum on genomic DNA, ca. 1 million A549 and A2780 cells were treated with 10 μM of [Pt(BDIQQ)]Cl or cisplatin at 37 °C for 12 h. Then the media were removed, the cells were washed with PBS solution (1 mL × 3), harvested, and centrifuged. The nuclei were extracted using the Thermo Scientific NE-PER Nuclear and Cytoplasmic Extraction Kit. The nuclear pellets were suspended in DNAzol (1 mL, genomic DNA isolation reagent, MRC). The genomic DNA was precipitated with ethanol (0.5 mL), washed with 75% ethanol (0.75 mL × 3), and re-dissolved in 200 μL of 8 mM NaOH. The DNA concentrations were determined by UV–visible spectroscopy, and platinum, was quantified by GFAAS. The reported values are the average of at least three independent experiments with the error reported as the standard deviation.
Immunoblotting Analysis
A549 cells (5 × 105 cells) were incubated with [Pt(BDIQQ)]Cl (concentrations, μM) for 72 h at 37 °C. Cells were washed with PBS, scraped into SDS-PAGE loading buffer (64 mM Tris-HCl (pH 6.8)/ 9.6% glycerol/ 2% SDS/ 5% β-mercaptoethanol/ 0.01% Bromophenol Blue), and incubated at 95 °C for 10 min. Whole cell lysates were resolved by 4-20 % sodium dodecylsulphate polyacylamide gel electrophoresis (SDS-PAGE; 200 V for 25 min) followed by electro transfer to polyvinylidene difluoride membrane, PVDF (350 mA for 1 h). Membranes were blocked in 5% (w/v) non-fat milk in PBST (PBS/0.1% Tween 20) and incubated with the appropriate primary antibodies (Cell Signalling Technology and Santa Cruz). After incubation with horseradish peroxidase-conjugated secondary antibodies (Cell Signalling Technology), immuno complexes were detected with the ECL detection reagent (BioRad) and analyzed using an Alpha Innotech ChemiImager™ 5500 fitted with a chemiluminescence filter.
Flow Cytometry
In order to monitor the cell cycle, flow cytometry studies were carried out. A549 cells (5 × 105 cells) were incubated with and without the test compounds for 24, 48, and 72 h at 37 °C. Cells were harvested from adherent cultures by trypsinization and combined with all detached cells from the incubation medium to assess total cell viability. Following centrifugation at 1000 rpm for 5 min, cells were washed with PBS and then fixed with 70% ethanol in PBS. Fixed cells were collected by centrifugation at 2500 rpm for 3 min, washed with PBS, and centrifuged as before. Cellular pellets were re-suspended in 50 μg/mL propidium iodide (Sigma) in PBS for nucleic acids staining and treated with 100 μg/mL RNaseA (Sigma). DNA content was measured on a FACSCalibur-HTS flow cytometer (BD Biosciences) using laser excitation at 488 nm and 20,000 events per sample were acquired. Cell cycle profiles were analysed using the ModFit software.
Apoptosis Studies
For this work the Annexin V-FITC Early Apoptosis Detection Kit (Cell Signaling Technology) was used. The manufacture's protocol was followed to carry out the experiments. Briefly, untreated and treated A549 cells (1 × 105) were suspended in 1× annexin binding buffer (96 μL) (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4), then 1 μL FITC annexin V and 12.5 μL propidium iodine (10 μg/ mL) were added to each sample and incubated on ice for 15 min. Subsequently, more binding buffer (150 μL) was added while gently mixing. The samples were kept on ice prior to being read on the FACSCalibur-HTS flow cytometer (BD Biosciences) (20,000 events per sample were acquired). Cell populations were analysed using the FlowJo software (Tree Star).
JC-1 Assay
The JC-1 Mitochondrial Membrane Potential Assay Kit (Cayman) was used. The manufacture's protocol was followed to carry out this experiment. Briefly, to untreated and treated A549 cells grown in 6-well plates (at a density of 5 × 105 cells/ mL) was added the JC-1 staining solution (100 μL/ mL of cell media). The cells were incubated for 30 min, harvested, and analysed by using the FACSCalibur-HTS flow cytometer (BD Biosciences) (20,000 events per sample were acquired). The FL2 channel was used to assess mitochondrial depolarisation. Cell populations were analysed using the FlowJo software (Tree Star).
Fluorescence Microscopy
Non-lethal conditions: A549 cells (1×104) were incubated with and without [Pt(BDIQQ)]Cl (0.5 μM for 24 h) and cisplatin (2 μM for 12 h). The media were then removed and the cells were washed with additional media (2 mL × 2). After incubation of the cells with more media containing Hoechst 33258 (7.5 μM) and MitoTracker Green (250 nM), the nuclear and mitochondrial regions were imaged using a fluorescent microscope. Apoptotic conditions: A549 cells (1×104) were incubated with and without [Pt(BDIQQ)]Cl (20 μM for 12 h). The media were then removed and the cells were washed with additional media (2 mL × 2). After incubation of the cells with more media containing Hoechst 33258 (7.5 μM), the nuclear region was imaged using a fluorescent microscope. Fluorescence imaging experiments were performed using a Zeiss Axiovert 200M inverted epifluorscence microscope with a Hamamatsu EM-CCD digital camera C9100 and a MS200 XY Piezo Z stage (Applied Scientific Instruments, Inc.). An X-Cite 120 metal halide lamp (EXFO) was used as the light source. Zeiss standard filter sets 49 and 38 HE were employed for imaging the nuclear and mitochondrial sensors. The microscope was operated with Volocity software (version 6.01, Improvision). The exposure time for acquisition of fluorescence images was kept constant for each series of images at each channel. Images corresponding to all localization studies were deconvoluted using Volocity restoration algorithms.
Gel Electrophoresis
The DNA-binding ability of [Pt(BDIQQ)]Cl was determined by monitoring the conversion of supercoiled plasmid DNA (form I) to open circular DNA (form I0) using agarose gel electrophoresis. To probe the effect of [Pt(BDIQQ)]Cl, solutions containing pUC18 DNA (nucleotide: 60 μM) and 0.1, 0.25, 0.5, 1, 2, 5, 7.5, 10, 15, and 20 μM of [Pt(BDIQQ)]Cl with a total reaction volume of 15 μL were incubated at 37 °C for 6 h. After this time period, loading buffer (5 μL, containing 0.25% bromophenol blue, 0.25% xylene cyanol and 60% glycerol) was added and the reaction mixtures were immediately loaded onto 1% agarose gels. The DNA fragments were separated by applying 20 V for 16 h. Tris-acetate EDTA (TAE) was used as the running buffer. The gels were stained in TAE solution containing ethidium bromide (0.3 μg/mL) for 3 h and de-stained with ddH2O for 3 h. The DNA bands were analysed under UV light using a Flor-S reader (BioRad).
Supplementary Material
Figure 3.
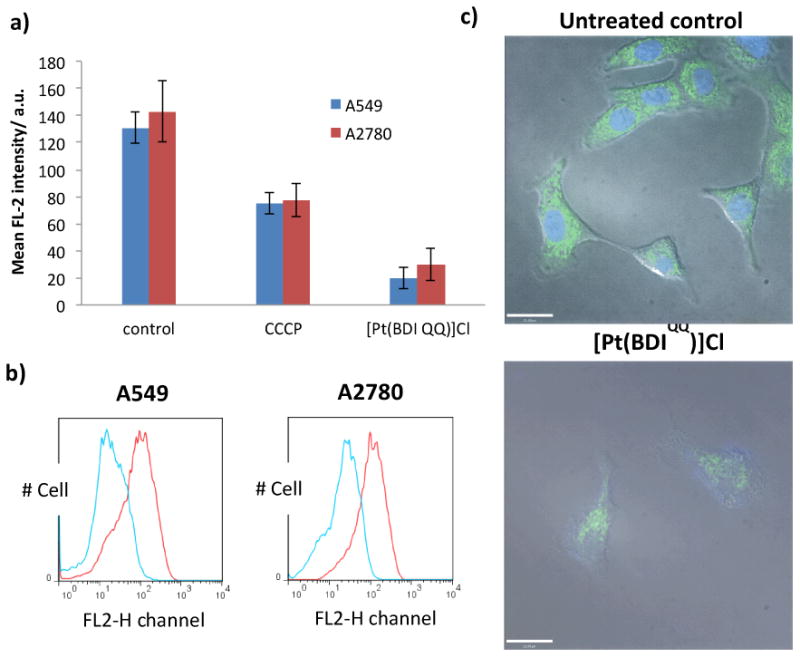
(a) The mean intensity of red fluorescence emitted by JC-1 stained A549 and A2780 cells in the absence and presence of [Pt(BDIQQ)]Cl (5 μM for 48 h) or carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (1 μM for 48 h). The errors represent standard deviations. (b) Histogram displaying red fluorescence emitted by JC-1 stained A549 and A2780 cells prior to (red line) and upon treatment with [Pt(BDIQQ)]Cl (5 μM for 48 h) (blue line). (c) Fluorescence microscopy images of A549 cells untreated and treated with [Pt(BDIQQ)]Cl (0.5 μM for 24 h), and then stained with Hoechst 33258 and MitoTracker Green. Scale bar = 21 μm.
Acknowledgments
This work was funded by the National Cancer Institute under grant R01-CA034992. K.S. received support from a Misrock Fellowship.
References
- 1.Fricker SP. Dalton Trans. 2007:4903–4917. doi: 10.1039/b705551j. [DOI] [PubMed] [Google Scholar]
- 2.Kelland L. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 3.Canetta R, Rozencweig M, Carter SK. Cancer Treat Rev. 1985;12(Suppl A):125–136. doi: 10.1016/0305-7372(85)90027-1. [DOI] [PubMed] [Google Scholar]
- 4.Giacchetti S, Perpoint B, Zidani R, Le Bail N, Faggiuolo R, Focan C, Chollet P, Llory JF, Letourneau Y, Coudert B, Bertheaut-Cvitkovic F, Larregain-Fournier D, Le Rol A, Walter S, Adam R, Misset JL, Levi F. J Clin Oncol. 2000;18:136–147. doi: 10.1200/JCO.2000.18.1.136. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg RM, Sargent DJ, Morton RF, Fuchs CS, Ramanathan RK, Williamson SK, Findlay BP, Pitot HC, Alberts SR. J Clin Oncol. 2004;22:23–30. doi: 10.1200/JCO.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 6.Rothenberg ML, Oza AM, Bigelow RH, Berlin JD, Marshall JL, Ramanathan RK, Hart LL, Gupta S, Garay CA, Burger BG, Le Bail N, Haller DG. J Clin Oncol. 2003;21:2059–2069. doi: 10.1200/JCO.2003.11.126. [DOI] [PubMed] [Google Scholar]
- 7.Wong E, Giandomenico CM. Chem Rev. 1999;99:2451–2466. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- 8.Jung Y, Lippard SJ. Chem Rev. 2007;107:1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 9.Lippard SJ. Science. 1982;218:1075–1082. doi: 10.1126/science.6890712. [DOI] [PubMed] [Google Scholar]
- 10.Lippert B. Verlag Helvetica Chimica Acta. Wiley-VCH, Zürich Weinheim; New York: 1999. Cisplatin : chemistry and biochemistry of a leading anticancer drug. [Google Scholar]
- 11.Todd RC, Lippard SJ. Metallomics : integrated biometal science. 2009;1:280–291. doi: 10.1039/b907567d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang D, Lippard SJ. Nat Rev Drug Discovery. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 13.Brabec V, Kasparkova J. Drug Resist Updates. 2005;8:131–146. doi: 10.1016/j.drup.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 14.McWhinney SR, Goldberg RM, McLeod HL. Mol Cancer Ther. 2009;8:10–16. doi: 10.1158/1535-7163.MCT-08-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siddik ZH. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 16.Brodie CR, Collins JG, Aldrich-Wright JR. Dalton Trans. 2004:1145–1152. doi: 10.1039/b316511f. [DOI] [PubMed] [Google Scholar]
- 17.Fisher DM, Bednarski PJ, Grunert R, Turner P, Fenton RR, Aldrich-Wright JR. ChemMedChem. 2007;2:488–495. doi: 10.1002/cmdc.200600211. [DOI] [PubMed] [Google Scholar]
- 18.Fisher DM, Fenton RR, Aldrich-Wright JR. Chem Commun. 2008:5613–5615. doi: 10.1039/b811723c. [DOI] [PubMed] [Google Scholar]
- 19.Garbutcheon-Singh KB, Leverett P, Myers S, Aldrich-Wright JR. Dalton Trans. 2013;42:918–926. doi: 10.1039/c2dt31323e. [DOI] [PubMed] [Google Scholar]
- 20.Kemp S, Wheate NJ, Buck DP, Nikac M, Collins JG, Aldrich-Wright JR. J Inorg Biochem. 2007;101:1049–1058. doi: 10.1016/j.jinorgbio.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 21.Krause-Heuer AM, Grunert R, Kuhne S, Buczkowska M, Wheate NJ, Le Pevelen DD, Boag LR, Fisher DM, Kasparkova J, Malina J, Bednarski PJ, Brabec V, Aldrich-Wright JR. J Med Chem. 2009;52:5474–5484. doi: 10.1021/jm9007104. [DOI] [PubMed] [Google Scholar]
- 22.Jennette KW, Lippard SJ, Vassiliades GA, Bauer WR. Proc Natl Acad Sci. 1974;71:3839–3843. doi: 10.1073/pnas.71.10.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long EC, Barton JK. Acc Chem Res. 1990;23:271–273. [Google Scholar]
- 24.Wu YS, Koch KR, Abratt VR, Klump HH. Arch Biochem Biophys. 2005;440:28–37. doi: 10.1016/j.abb.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 25.Davis KJ, Carrall JA, Lai B, Aldrich-Wright JR, Ralph SF, Dillon CT. Dalton Trans. 2012;41:9417–9426. doi: 10.1039/c2dt30217a. [DOI] [PubMed] [Google Scholar]
- 26.Garbutcheon-Singh KB, Myers S, Harper BW, Ng NS, Dong Q, Xie C, Aldrich-Wright JR. Metallomics : integrated biometal science. 2013;5:1061–1067. doi: 10.1039/c3mt00023k. [DOI] [PubMed] [Google Scholar]
- 27.Hope JM, Wilson JJ, Lippard SJ. Dalton Trans. 2013;42:3176–3180. doi: 10.1039/c2dt32462h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 29.Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shieh SY, Ikeda M, Taya Y, Prives C. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 31.Enoch T, Norbury C. Trends Biochem Sci. 1995;20:426–430. doi: 10.1016/s0968-0004(00)89093-3. [DOI] [PubMed] [Google Scholar]
- 32.Gottlieb TM, Oren M. Semin Cancer Biol. 1998;8:359–368. doi: 10.1006/scbi.1998.0098. [DOI] [PubMed] [Google Scholar]
- 33.Tokino T, Nakamura Y. Crit Rev Oncol Hematol. 2000;33:1–6. doi: 10.1016/s1040-8428(99)00051-7. [DOI] [PubMed] [Google Scholar]
- 34.Murphy M, Mabruk MJ, Lenane P, Liew A, McCann P, Buckley A, Billet P, Leader M, Kay E, Murphy GM. Br J Dermatol. 2002;147:110–117. doi: 10.1046/j.1365-2133.2002.04749.x. [DOI] [PubMed] [Google Scholar]
- 35.Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B, Andrews DW. EMBO J. 2005;24:2096–2103. doi: 10.1038/sj.emboj.7600675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang X, Wang X. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 37.Reers M, Smith TW, Chen LB. Biochemistry. 1991;30:4480–4486. doi: 10.1021/bi00232a015. [DOI] [PubMed] [Google Scholar]
- 38.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD, Chen LB. Proc Natl Acad Sci. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. Mol Cell Biol. 2004;24:6728–6741. doi: 10.1128/MCB.24.15.6728-6741.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marchenko ND, Zaika A, Moll UM. J Biol Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 41.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 42.Vogelstein B, Lane D, Levine AJ. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 43.Fischer U, Schulze-Osthoff K. Cell Death Differ. 2005;12(Suppl 1):942–961. doi: 10.1038/sj.cdd.4401556. [DOI] [PubMed] [Google Scholar]
- 44.Quinn PJ. Subcell Biochem. 2002;36:39–60. doi: 10.1007/0-306-47931-1_3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.