

Abstract
Accumulation of the microtubule-binding protein tau is a key event in several neurodegenerative disorders referred to as tauopathies, which include Alzheimer's disease, frontotemporal lobar degeneration, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Thus, understanding the molecular pathways leading to tau accumulation will have a major impact across multiple neurodegenerative disorders. To elucidate the pathways involved in tau pathology, we removed the gene encoding the beta-2 adrenergic receptors (β2ARs) from a mouse model overexpressing mutant human tau. Notably, the number of β2ARs is increased in brains of AD patients and epidemiological studies show that the use of beta-blockers decreases the incidence of AD. The mechanisms underlying these observations, however, are not clear. We show that the tau transgenic mice lacking the β2AR gene had a reduced mortality rate compared with the parental tau transgenic mice. Removing the gene encoding the β2ARs from the tau transgenic mice also significantly improved motor deficits. Neuropathologically, the improvement in lifespan and motor function was associated with a reduction in brain tau immunoreactivity and phosphorylation. Mechanistically, we provide compelling evidence that the β2AR-mediated changes in tau were linked to a reduction in the activity of GSK3β and CDK5, two of the major tau kinases. These studies provide a mechanistic link between β2ARs and tau and suggest the molecular basis linking the use of beta-blockers to a reduced incidence of AD. Furthermore, these data suggest that a detailed pharmacological modulation of β2ARs could be exploited to develop better therapeutic strategies for AD and other tauopathies.
INTRODUCTION
Tauopathies comprise a class of neurodegenerative disorders characterized histopathologically by the presence of filamentous inclusions composed mainly of hyperphosphorylated forms of tau, a microtubule binding protein (1). In addition to Alzheimer's disease (AD)—the most common neurodegenerative disorder, tauopathies, include conditions such as frontotemporal dementia, Pick's disease, progressive supranuclear palsy and corticobasal degeneration (1). Clinically, these disorders are characterized by profound behavioral, motor and cognitive alterations that eventually lead to patients being bedridden. While pathological accumulation of hyperphosphorylated tau and the consequent formation of neurofibrillary tangles (NFTs) is a critical event in all of these conditions, more needs to be done to elucidate the molecular mechanisms leading to tau hyperphosphorylation and aggregation and the concomitant behavioral deficits.
Aging is the single major risk factor for tauopathies, including AD and frontotemporal dementia. Thus, it is likely that molecular changes contributing to the aging process may facilitate tau accumulation and represent common mechanisms across different tauopathies. Evidence from epidemiological studies demonstrates that the use of non-selective β-adrenergic receptor antagonists correlates with a lower incidence of AD (2). Furthermore, genetic studies indicate that polymorphisms in the gene encoding the beta-2 adrenergic receptor (β2AR) are linked to a higher risk of late onset AD (2,3). Consistent with these data, chronic treatment with β2AR agonists increases amyloid-β (Aβ) load in transgenic mice (4), while the use of non-selective β-blockers conversely decreases acute stress-induced Aβ production (3). Despite this wealth of data from human and rodent studies, the link between β2ARs and tau remains evasive.
The β2ARs belong to the G-proteincoupled receptor superfamily and are comprised of seven transmembrane α-helices and are classically thought to signal via a Gs pathway (5). In response to its endogenous ligand norepinephrine, the receptor activation stimulates adenylyl cyclase to produce increased levels of cAMP further driving the activity of protein kinase A (PKA) and consequent phosphorylation of downstream intracellular targets (6). They play pleiotropic physiological roles and are expressed throughout the brain, abundantly in hippocampus and cortex (7)—the two brain regions responsible for higher cognitive functions. Indeed, the ability of the noradrenergic system to modulate cognition and behavioral function has long been recognized (8). Despite being involved in these fundamental processes, β2AR knockout mice are viable and fertile, suggesting that compensatory changes my overcome the lack of these receptors (9). Interestingly, in AD brains, β2AR concentrations are significantly reduced in the thalamus, nucleus basalis of Meynert and are conversely increased in the frontal cortex, hippocampus and putamen (10). Since hippocampus and cortex are two brain areas highly susceptible to the neurodegenerative insult in AD and FTLD, these observations led to β2AR becoming an important subject of investigation in the field of neurodegeneration (11–13).
Genetically modified mice are a great tool to study human disorders. Several animal models of tauopathies have been generated by overexpressing mutant human tau (14). In general, these mice show tau hyperphosphorylation and accumulation of NFTs; these neuropathological changes are often associated with severe motor impairments (14). These mice represent an important tool, not only to test preclinical therapies, but also to study in vivo mechanistic changes responsible for the underlying phenotype. Along these lines, here we sought to determine whether there is a direct link between β2ARs and tau pathology. Specifically, we used a genetic approach to remove the gene encoding the β2ARs from a transgenic mouse overexpressing mutant human tau. Understanding the role of β2ARs in the pathogenesis of tauopathies might lead to the identification of new molecular targets to therapeutically tackle these insidious neurodegenerative disorders.
RESULTS
To study the role of β2-adrenergic receptors (β2ARs) in tauopathies in vivo, we used a mouse model overexpressing human tau harboring the P301S mutation. These mice develop age-dependent accumulation of tau, which leads to profound motor deficits and reduced lifespan (15). By breeding tau P301S mice with β2AR knockout mice, which are viable, grossly normal and fertile (9), we were able to remove the gene encoding the β2ARs from the tau P301S mice. Specifically, we bred hemizygous P301S mice (P301S+/0) with homozygous β2ARs knockout mice (β2KO/KO). We then bred F1s littermates β2KO/wt; P301S+/0 to β2KO/wt; P301S0/0 mice. Fifty percent of the offspring were heterozygous for the β2ARs and thus were sacrificed immediately after genotyping (Fig. 1A). The remaining 50% of the mice had one of the following genotypes and were used for the experiments described in this study: (i) β2wt/wt; P301S+/0; (ii) β2KO/KO; P301S+/0; (iii) β2wt/wt; P301S0/0; (iv) β2KO/KO; P301S0/0. Herein, the β2wt/wt; P301S+/0 mice are referred to as P301S mice; the β2KO/KO; P301S+/0 as β2KO/P301S mice; the β2wt/wt; P301S0/0 as control (CTL) mice, and the β2KO/KO; P301S0/0 as β2KO mice (Fig. 1A).
Figure 1.
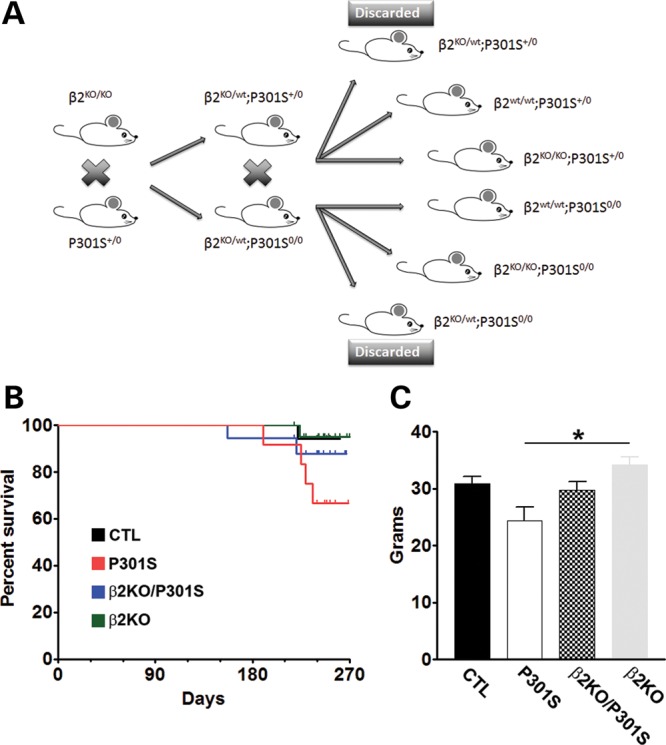
Removing the gene encoding the β2ARs increases the survival rate in P301S transgenic mice. (A) Breeding diagram illustrating the step-by-step generation of experimental mouse colony beginning with hemizygous P301S mice and homozygous β2KO mouse. (B) Percent survival of each group of experimental mice by age monthly. At the beginning of the experiment, we had n = 20/β2KO; n = 18/β2KO/P301S; n = 12/P301S; n = 17/CTL. While only 66.7% of the P301S mice were still alive at 8 months of age, 88.2% of the β2KO/P301S mice were alive at the same age. (C) Body weight of experimental mice at 8 months of age. Data are presented as means ± SEM and analyzed by ANOVA.
The P301S mice die prematurely due to weakness and paralysis of the rear limbs, and consequent inability to feed (15). Consistently, we found that at 8 months of age, only 66.7% of the P301S mice were still alive (Fig. 1B; Table 1). Notably, removing the β2ARs from the P301S mice improved the observed hazard rate by a ratio of 2.6 (P = 0.27, with the Cox PH model, adjusting for sex; Tables 2 and 3); indeed, 88.2% of the β2KO/P301S mice were still alive at 8 months of age (Fig. 1B; Table 1). When we examined the body weight of the mice that remained alive at 8 months of age, we found that there was a significant difference among the four groups of mice as indicated by one-way ANOVA (P = 0.001). To determine which group was responsible for the difference, we performed a Bonferroni's post hoc analysis and found that the body weight of the β2KO mice was significantly higher than that of P301S mice (P < 0.001; Fig. 1C). Taken together, these data demonstrate that removing the β2ARs from transgenic mice overexpressing human mutant tau has a positive effect on the general health and longevity of the tau mice, as shown by the reduction in mortality hazard rate.
Table 1.
Survival times of mice
| Genotype | Died | Sacd | Sum |
|---|---|---|---|
| CTL | 1 | 16 | 17 |
| β2KO | 1 | 20 | 21 |
| β2KO/P301S | 2 | 16 | 18 |
| P301S | 4 | 8 | 12 |
| Sum | 8 | 60 | 68 |
The survival times of mice from all genotypes were modeled using Cox proportional hazards regression with genotype as a factor and sex as an explanatory variable.
Table 2.
Cox proportional hazard regression (changes relative to CTL mice)
| Variable | Coef | SE_coef | Hazard_ratio | Lower_95 | Upper_95 | P-value |
|---|---|---|---|---|---|---|
| Genotype: β2KO | −1.9729 | 1.1183 | 0.139 | 0.0155 | 1.2446 | 0.0777 |
| Genotype: β2KO/P301S | −0.9599 | 0.878 | 0.3829 | 0.0685 | 2.1404 | 0.2743 |
| Genotype: CTL | −1.7609 | 1.1182 | 0.1719 | 0.0192 | 1.5385 | 0.1153 |
| Sex M | 0.2984 | 0.7182 | 1.3477 | 0.3298 | 5.5069 | 0.6777 |
Table 3.
Cox proportional hazard regression (changes relative to P301S mice)
| Variable | Coef | SE_coef | Hazard_ratio | Lower_95 | Upper_95 | P-value |
|---|---|---|---|---|---|---|
| Genotype: β2KO | −0.2121 | 1.4146 | 0.8089 | 0.0506 | 12.9428 | 0.8808 |
| Genotype: β2KO/P301S | 0.801 | 1.2347 | 2.2277 | 0.1981 | 25.0506 | 0.5165 |
| Genotype: P301S | 1.7609 | 1.1182 | 5.8175 | 0.65 | 52.067 | 0.1153 |
| Sex M | 0.2984 | 0.7182 | 1.3477 | 0.3298 | 5.5069 | 0.6777 |
To assess the effects of removing β2ARs on tau-induced motor deficits, we tested 8-month-old mice in a battery of behavioral tests focused on motor function. Mice were initially tested in the open field to assess general motor performance. We found that the spontaneous activity and gross motor function were similar among the four different groups, as assessed by the distance covered in the activity chamber and the average speed during the test (Fig. 2A and B, respectively). To further elucidate motor function, we tested the mice using the rotarod, which is widely employed for assessing motor coordination. Mice were given six trials per day for three consecutive days, during which the speed of the rotor was accelerated from 0 to 15 revolutions per minute (rpm) in 15 s and then maintained constant at 15 rpm for 75 additional seconds, for a total of 90 s per trial. Six, 90 s probe trials were conducted on day 4 on a constantly accelerating rod (1 rpm/s). Although statistically all four groups of mice performed similarly, we observed a strong trend showing diminished performance of P301S mice compared with the other three groups (Fig. 2C). Subsequently, we tested the mice in the hanging wire test, a test routinely used to measure motor strength in rodents (16). We found that the latency to fall for CTL mice was 43.61 ± 5.13 s; in contrast, the P301S mice were able to remain on the rod only 16.04 ± 7.76 s (Fig. 2D). Notably, removing the β2ARs from the P301S mice improved their performance in this task; indeed, the β2KO/P301S mice performed 2-fold better than the P301S mice and their average latency to fall was 32.38 ± 4.20 s. One-way ANOVA indicated that the difference among the groups was significant (P < 0.05) and a Bonferroni's post hoc analysis showed that the P301S mice performed significantly worse than the CTL mice, while the β2KO/P301S mice performed as well as the CTL mice (Fig. 2D). Given that the P301S were significantly impaired compared with CTL mice and that the β2KO/P301S mice performed as well as the CTL mice, we conclude that removing the β2ARs from tau transgenic mice significantly improved motor performance in the hanging wire test, indicative of motor strength specifically.
Figure 2.

Genetic suppression of β2AR rescues motor strength. Behavioral testing was performed on 8-month-old mice (n = 20/β2KO; n = 15/β2KO/P301S; n = 8/P301S; n = 16/CTL). (A and B) The open-field activity test was conducted to measure spontaneous activity and general motor function. No statistically significant differences were found among the four groups in the distance covered during the exploration time or the speed of movement. (C) To measure motor coordination, we used the accelerating rotarod and found no statistically significant changes among the four groups analyzed. (D) To measure motor strength, we used the hanging wire test and found that the P301S mice were significantly impaired compared with the CTL mice. In contrast, removing the β2ARs from these mice partially rescued this phenotype. Data are presented as means ± SEM and analyzed by ANOVA.
At the end of the behavioral tests, mice were 8-month-old; at this age, the P301S mice show robust tau accumulation and phosphorylation (15). To assess the effects of deleting β2ARs on brain tau pathology, sections from P301S and β2KO/P301S mice (n = 6/group) were immunostained with HT7, an anti-tau antibody that recognizes total tau, and AT8, an anti-tau antibody that recognizes tau phosphorylated at Ser202/Thr205. We focused our analysis on the basal ganglia, hippocampus and cortex, three brain regions highly susceptible to tau pathology in mouse and human brains (1). We found that HT7 (Fig. 3A, B, G) and AT8 (Fig. 3J, K, P) immunoreactivity was markedly reduced in the basal ganglia of β2KO/P301S mice compared with P301S mice. High magnification views of the basal ganglia highlight how in the P301S mice the vast majority of neurons show a robust HT7 and AT8 immunoreactivity. In contrast, in the β2KO/P301S mice very few neurons are positive for HT7 and AT8 (Fig. 3A, B, J, K). We observed a similar effect in the CA1 region of the hippocampus, where the HT7 (Fig. 3C, D, H) and AT8 (Fig. 3L, M, Q) immunoreactivity was greatly reduced in the β2KO/P301S mice compared with P301S mice. In contrast, we found no changes in HT7 (Fig. 3E-F, I) and AT8 (Fig. 3N-O, R) immunoreactivity in the neocortex. Although removing the β2ARs from the P301S mice leads to a robust reduction in tau immunoreactivity in the basal ganglia and hippocampus, further studies are needed to identify why changes in tau immunoreactivity appear to be region specific.
Figure 3.

Genetic suppression of β2ARs ameliorates neuropathology in hippocampus and basal ganglia of P301S mice. Photomicrographs of three commonly affected brain regions from P301S and β2KO/P301S mice. Sections were stained with the HT7 antibody, which recognizes total human tau, and AT8 antibody, which recognizes tau phosphorylated at Ser202/Thr205. (A and B) Representative basal ganglia sections demonstrate markedly reduced HT7 immunoreactivity in β2KO/P301S mice compared with P301S mice. (C and D) Representative sections of the hippocampal CA1 region. Effect similar to that in basal ganglia is observed with decreased HT7 immunoreactivity in β2KO/P301S mice. (E and F) Representative sections of neocortex demonstrates no difference in HT7 immunoreactivity between experimental and control mice. (G–I) The HT7 immunoreactivity was quantified using Image J (n = 6). The values in the y-axes are shown relative to P301S mice. (J and K) Representative basal ganglia sections demonstrate markedly reduced AT8 immunoreactivity in β2KO/P301S mice compared with P301S mice. (L and M) Representative sections of the hippocampal CA1 region. Effect similar to that in basal ganglia is observed with decreased AT8 immunoreactivity in β2KO/P301S mice. (N and O) Representative sections of neocortex demonstrate no difference in HT7 immunoreactivity between experimental and control mice. (P–R) The HT7 immunoreactivity was quantified using Image J (n = 6). The values in the y-axes are shown relative to P301S mice. Data are presented as means ± SEM and analyzed by Student's t-test.
To better quantify the changes in tau phosphorylation between P301S and β2KO/P301S mice, we measured tau steady-state levels in sarkosyl soluble and insoluble fractions by western blot. Notably, hyperphosphorylated tau is less soluble and more prone to aggregation; indeed, changes in tau phosphorylation and solubility have often been considered a marker of pathology severity and progression (17). In the soluble fraction, we found that endogenous tau levels were significantly reduced in the β2KO/P301S mice compared with the P301S mice, as detected by the Tau 5 antibody (P < 0.0001; Fig. 4A and B), which is highly selective for mouse tau over human tau (Supplementary Material, Fig. S1). Similarly, removing the gene encoding the β2ARs from the P301S mice significantly reduced the steady-state levels of human tau, as detected by the HT7 antibody (P = 0.04; Fig. 4A and C). In contrast, we found that the soluble steady-state levels of tau phosphorylated at Ser202/Thr205 (detected by the AT8 antibody) and at Thr181 (detected by the AT270 antibody) were similar between the two groups of mice (Fig. 4A D and E). In the sarkosyl insoluble fraction, we found a marked and significant decrease in the levels of total mouse and human tau in the β2KO/P301S mice compared with P301S mice (P < 0.01; Fig. 4F–H). In contrast to the soluble fraction, even the steady-state levels of insoluble phospho-tau were significantly reduced by removing the gene encoding the β2ARs from the P301S mice. Specifically, we found a dramatic reduction in the levels of tau phosphorylated at Ser202/Thr205 and Thr181 in the brains of the β2KO/P301S mice (P < 0.05; Fig. 4F, I and J). Taken together, these results clearly and unambiguously show that removing the gene encoding the β2ARs from transgenic mice overexpressing mutant human tau greatly reduces tau levels, phosphorylation and deposition.
Figure 4.

Genetic suppression of β2AR ameliorates tau levels, phosphorylation and aggregation. (A) Representative western blots of protein extracted from sarkosyl soluble fractions. (B–E) Quantitative analyses of the blots in panel A show that deletion of β2ARs decreases the steady-state levels of total endogenous (B) and human tau (C). However, levels of soluble tau phosphorylated at Ser202/Thr205 (D) and at Thr181 (E) are not affected by this genetic manipulation. (F) Representative western blots of protein extracted from sarkosyl insoluble fractions. (G–J) Quantitative analyses of the blots in (F) show that deletion of β2AR similarly decreases the steady-state levels of total endogenous (G) and human tau (H). Similarly, the AT8 (I) and AT270 (J) levels of tau were significantly reduced in β2KO/P301S mice compared with P301S mice. Quantifications of the western blots were performed by normalizing the protein of interest to β-actin, which was used as a loading control. n = 6/genotype. Data are presented as means ± SEM and analyzed by Student's t-test. *** indicates P < 0.001; ** indicates P < 0.01; * indicates P < 0.05.
To start understanding the mechanisms underlying the β2ARs-mediated reduction in tau phosphorylation, we focused on the two major tau kinases, CDK5 and GSK3β. Using western blot analysis, we found a small but significant reduction in the steady-state levels of total CDK5 in the β2KO/P301S mice compared with P301S mice (P = 0.01; Fig. 5A and B). CDK5 activity is facilitated by phosphorylation at tyrosine 15 (18). Consistent with a decrease in CDK5 activity in the β2KO/P301S compared with P301S mice, we found that the steady-state levels of CDK5 phosphorylated at tyrosine 15 were significantly reduced by removing the β2ARs from the P301S mice (P = 0.02; Fig. 5A and C). Unexpectedly, the levels of p35, an activator of CKD5, were not statistically different between the two groups (Fig. 5A and D). Along with CDK5, GSK3β is considered another major tau kinase. While changes in total levels of GSK3β do not correlate with the overall activity of the protein, the levels of GSK3β phosphorylated at serine 9 are inversely correlated with GS3Kβ activity (19). We therefore measured the steady-state levels of GSK3β phosphorylated at serine 9 and found that they were ∼50% higher in the brains of the β2KO/P301S mice than the P301S mice (P = 0.02; Fig. 5A and E). Collectively, these data show that the activities of the two major tau kinases were reduced by removing the β2ARs from the brains of the P301S mice, which is consistent with the previously described reduction in tau pathology (Figs. 3 and 4).
Figure 5.

CDK5 and GSK3β activities are reduced in β2KO/P301S mice. (A) Western blots of proteins extracted from the brains of P301S and β2KO/P301S mice. (B–E) Quantitative analyses of blots demonstrate that the levels of total CDK5 (B) and CDK5 phosphorylated at Tyr15 (C) are significantly higher in P301S mice compared with β2KO/P301S mice. However, the levels of p35 (D) remain unchanged. We also found that the levels of GSK3β phosphorylated at Ser9 (E) were significantly lower in the P301S mice compared with β2KO/P301S mice. Quantifications of the western blots were performed by normalizing the protein of interest to β-actin, which was used as a loading control. n = 6/genotype. Data are presented as means ± SEM and analyzed by Student's t-test. * indicates P < 0.05.
To further understand the mechanism linking the lack of β2ARs to the decrease in tau levels and phosphorylation, we used a candidate approach and assessed intracellular signaling pathways that have been linked to both β2ARs signaling and tau biology. We initially focused on AKT, as this kinase has been shown to be directly regulated by β2ARs signaling and can directly or indirectly (by activating other kinases) phosphorylate tau (20,21). We found that the steady-state levels of AKT were significantly different among the groups (P = 0.03 as calculated by one-way ANOVA; Fig. 6A and B). To find out which group was responsible for this difference, we performed a Tukey's multiple comparison test and found that the levels of AKT were significantly lower in the P301S mice than in WT mice (P < 0.05; Fig. 6A and B). However, AKT levels were not statistically different between β2KO/P301S mice and WT mice, suggesting that removing β2ARs from the P301S mice restored AKT levels to WT levels. AKT activity is regulated upon phosphorylation at several amino acids (22). Using a phospho-specific antibody, we found that the levels of AKT phosphorylated at Ser473 were similar among the three groups (P = 0.6; Fig. 6A and C). Other studies are needed to determine whether other epitopes in AKT are differentially phosphorylated among the three groups.
Figure 6.

Removing the β2ARs restores PKA levels in the P301S mice. (A) Representative western blots of proteins extracted from the brains of CTL, P301S and β2KO/P301S mice and probed with the indicated antibodies. (B and C) Quantitative analyses of the total and phospho-AKT blots show that total AKT levels were significantly reduced in the P301S mice compared with WT and β2KO/P301S mice. In contrast, the levels of AKT phosphorylated at Ser473 were similar among the three groups. (D and E) Quantitative analyses of the p54 and p46 JNK isoforms show that the levels of the p54 isoform were similar among the three groups, while p46 levels were significantly lower in CTL mice compared with the other two groups. (F and G) Quantitative analyses of the levels of the PKA regulatory subunit IIα indicate that the levels of the low molecular weight band of PKA were significantly different in the brains of P301S mice compared with the other two groups. Quantifications of the western blots were performed by normalizing the protein of interest to β-actin, which was used as a loading control. n = 6/genotype. Data are presented as means ± SEM and analyzed by one-way ANOVA. * indicates P < 0.05.
JNK and PKA are two other signaling kinases, whose activity can be regulated via β2ARs and have also been linked to tau metabolism (6,23–25). We found that the two major isoforms of JNK were differentially affected by the lack of β2ARs in the P301S mice. Specifically, using an antibody that recognizes JNK phosphorylated at Thr183 and Thr185, we found that the levels of phosphorylated p54 JNK isoform were similar among the three different groups (Fig. 6A and D). In contrast, we found that the levels of phosphorylated p46 JNK isoform were different among the three groups (P = 0.01; Fig. 6A and E). Post hoc analysis indicated that the WT mice had lower levels of phosphorylated p46 JNK than the P301S and β2KO/P301S mice (P < 0.05). However, the levels of phosphorylated p46 JNK were not statistically different between P301S and β2KO/P301S mice. These results suggest that changes in JNK phosphorylation are dependent on the presence of the tau transgene and most likely do not account for the decrease in tau pathology in the β2KO/P301S mice.
PKA is activated when cyclic AMP binds to its regulatory subunits, which then dissociate from the PKA catalytic subunit (26). To test for changes in PKA function, we measured the steady-state levels of PKA regulatory subunits, using an antibody specific against the PKA regulatory subunit IIα (PKA-rIIα). We found that the levels of a slow-running isoform of PKA-rIIα were similar among the three groups (P = 0.15; Fig. 6A and F). In contrast, we found that the levels of a smaller isoform of PKA-rIIα were significantly different among the three groups (P < 0.005; Fig. 6A, G). Tukey's multiple comparison test showed that the levels of PKA-rIIα were significantly higher in the P301S mice than the other two groups (P < 0.05). Notably, we found no statistically significant difference between WT and β2KO/P301S mice. These data clearly indicate that the elevated levels of PKA in the P301S mice were rescued by removing the gene encoding the β2ARs.
DISCUSSION
Tau is a soluble microtubule-binding protein, whose function is to promote microtubule assembly and stabilization (1). Pathological tau protein, by contrast, exhibits altered solubility properties, forms filamentous structures and is abnormally phosphorylated at selective residues (1). While more needs to be done to dissect the mechanisms linking tau accumulation to neurodegeneration, a primary role of tau in several neurodegenerative disorders, known as tauopathies, is unquestioned. AD, frontotemporal dementia with Parkinsonism linked to chromosome 17, Pick's disease, progressive supranuclear palsy and corticobasal degeneration are the major tauopathies (1).
A role for the β2ARs in neurodegeneration has been shown by several groups using complementary approaches. For example, early studies have reported that β2ARs are increased in several brain regions of AD patients, including prefrontal cortex and hippocampus (10,27). Consistently, epidemiological studies show that the use of beta-blockers decreases the incidence of developing AD (28). Moreover, polymorphisms in the human β2AR gene known to alter cellular trafficking and receptor desensitization (29,30) increase the risk of late onset AD (2,13,28,31). However, the relation between β2ARs and AD is not linear as others have reported decreased levels of β2ARs in various brain regions (32,33). The apparent discrepancy between these reports is unclear and further studies are needed to better integrate these observations.
Work in animal models has suggested possible mechanisms by which β2ARs can affect the risk of developing AD. For example, it has been shown that Aβ binds to β2ARs (34), which in turn increases γ-secretase activity and Aβ production (4), thereby creating a vicious cycle. We have previously shown that β2ARs mediate the Aβ-induced tau pathology. Specifically, we showed that deletion of the β2AR gene in an animal model of AD characterized by Aβ accumulation decreases tau phosphorylation (35). Remarkably, we and others have shown that in transgenic mice tau pathology is often highly dependent on Aβ levels (36–39). Thus, it remained to be established whether the β2ARs-mediated changes in tau were due to changes in Aβ metabolism or to a direct link between β2ARs and tau. Here, we report the first in vivo evidence of a direct link between β2ARs and tau. We show that removing the gene encoding the β2ARs from the brains of the P301S transgenic mice ameliorates motor deficits. Specifically, even though some of the P301S mice died prematurely, before 8 months of age, the results of the hanging wire test clearly showed a significant improvement in performance for the β2KO/P301S mice compared with the P301S mice. Notably, we also found a strong trend for motor improvement in the rotarod; however, the data did not reach statistical significance. Given that the P301S mice with the more robust motor deficits are the ones that died prematurely, it is tempting to speculate that if those mice had been included in the rotarod, the difference between the groups might have been larger.
The data presented here indicate that the most profound changes in tau immunoreactivity in the β2KO/P301S mice are in hippocampus and basal ganglia, brain regions that show robust tau pathology in the P301S mice (15); these changes also correlate with the distribution of β2ARs in the brain (7). However, the biochemical evidence showing the decrease in tau levels and phosphorylation was obtained from whole brain lysates. Thus it is tempting to speculate that the changes seen at a biochemical level could have been even more accentuated if one would use tissue from specific brain regions. Tau is a substrate for several kinases, including CDK5 and GSK3β, which are often considered the two major tau kinases (1). We presented biochemical evidence indicating that the β2AR-mediated changes in tau phosphorylation were linked to a decrease in the activity of CKD5 and GSK3β. Indeed, we showed that the levels of CDK5 phosphorylated at Tyr15, which correlate with its activity, were significantly decreased in the β2KO/P301S mice compared with P301S mice. Similarly, the levels of GSK3β phosphorylated at Ser9, which inversely correlate with its activity, were significantly increased after deleting the β2AR gene in the P301S mice. Additionally, our data suggest that the decrease in total tau in the β2KO/P301S mice compared with P301S mice may contribute to the overall decrease in tau phosphorylation. Further studies are needed to dissect the exact contribution of CDK5 and GSK3β over a general reduction in the total steady-state levels of tau.
β2ARs are activated by several environmental factors, including stress (40,41). This is highly germane to AD and other tauopathies as stress increases the risk of developing neurodegenerative disease in people and exacerbates neuropathology in animal models (42,43). Once activated, the β2ARs mainly signal via the cyclic AMP/PKA pathway (6). Activated PKA directly or indirectly modulates other intracellular pathways including MAP kinase signaling and AKT (6,21). Some of these pathways have been directly linked to tau phosphorylation. For example, PKA can directly phosphorylate tau (24,25). Indeed, several studies have shown that PKA-mediated phosphorylation of tau may be an early event leading to tau aggregation (44–48). Consistent with this observation, here we show that the levels of PKA strongly correlate with the β2ARs-mediated changes in tau phosphorylation in the brains of the P301S and β2KO/P301S mice. Thus, we suggest that removing the β2ARs from the brains of the P301S mice decreases PKA signaling, ultimately leading to reduced tau phosphorylation.
While β2ARs signaling is important for learning and memory (49–51), chronic activation is detrimental (52); more studies are needed to determine whether the temporal profile or the degree of β2ARs activation have differential effects on AD. Nevertheless, to our knowledge, this is the first investigation aimed at establishing whether removing the gene encoding the β2ARs has a direct effect on tau pathology in the absence of Aβ. Identifying whether there is a direct interaction between β2ARs and tau will not only lead to a better understanding of the role of β2ARs in AD, but it will also be crucial in determining the role of β2ARs in other tauopathies. In summary, our findings show that reducing β2ARs signaling greatly reduces tau pathology in a mouse model of tauopathies. These effects are most likely mediated by changes in key kinases known to phosphorylate tau. Our data suggest that a detailed pharmacological modulation of β2ARs could be exploited in order to develop better therapeutic strategies for AD and other tauopathies.
MATERIALS AND METHODS
Mice
The β2AR knockout and the P301S transgenic mice have been described previously (9,15). Mice were kept on 12 h light/dark cycle and body weight was determined at the end of the treatment. All animal procedures were approved by The Institutional Animal Care and Use Committee of The University of Texas Health Science Center at San Antonio.
Four limb hanging wire test
The hanging wire test was performed using a mesh feeder grid with its edges taped off to keep a mouse in a limited area and prevent it from climbing over. Each animal was placed on the wire mesh and lightly shaken three times to allow to grip onto the mesh. The grid was then flipped upside down, thereby inverting an animal over the open cage so that it had to hold on to the wire to avoid falling. Importantly, the distance between the hanging grid and the floor of the cage was maintained at ∼30 cm—short enough to prevent any animal injury during fall, and large enough to keep an animal from casually jumping off the grid without fearing to fall. Each mouse underwent three trials, 30 min apart. The first two trials served to familiarize the mouse with the testing conditions and train to grip. The subsequent probe trial was used to collect final latency to fall measurements. The latency time measurements began from the point when the grid was flipped over and ended with the animal falling into the cage under the grid. No attempt was made to force compliance during any of the various trials. In cases where an animal was unable to hang from the wire for at least 1 s, a fall time of zero was noted for that trial. The duration of training and probe trials was capped at 60 s, meaning no mouse was left to hang but was removed from the grid after this time passed.
Rotarod
This test was performed as previously described (53). Briefly, each mouse was trained for three consecutive days (six consecutive trials/day, at least 30 min apart) where the speed of the rotor was accelerated from 0 to 15 rpm in 15 s and then maintained constant at 15 rpm for 75 additional seconds, for a total of 90 s. Twenty-four hours after the last training session, the mice were tested in a probe trial consisting of six consecutive trials on a constantly accelerating rod (1 rpm/s). The latency to fall was then recorded, which is indicative of motor abilities and coordination.
Sarkosyl extraction and western blots
Mice were sacrificed by CO2 asphyxiation and their brains removed and sagittally bisected. Half of the brain was frozen in dry ice and used for biochemical evaluation, while the other half was dropped-fixed in 4% paraformaldehyde and used for histological and immunohistochemical evaluation. Frozen brains were homogenized in 1.4 ml of cold buffer H (10 mm Tris–HCl, 1 mm EGTA, 0.8 m NaCl, 10% sucrose, pH 7.4) using dounce homogenizer, after which they were centrifuged at 27 200 g for 20 min at 4°C. Supernatant was saved and pellet re-homogenized using 22 and 27 gauge needles in 1.4 ml cold buffer H and centrifuged at 150 000 g for 15 min at 4°C. Supernatant was saved and combined with the one from first spin. The total volume was adjusted to 1% (w/v) N-lauroylsarcosine and samples were incubated at 37°C with shaking for 90 min, after which they were centrifuged at 150 000 g for 35 min at 20°C. The resulting supernatant was collected and used for western blotting as sarkosyl soluble fraction. The pellet was re-suspended in 70 μl of 50 mm Tris–HCl, pH 7.4 and used as sarkosyl insoluble fraction. Proteins were resolved using precast SDS/PAGE gels as described previously (54). Blot quantification was conducted by the Image J software.
Immunohistochemistry
Fifty micrometer thick free-floating sections were obtained from fixed brains using a vibratome. Sections were stored in 0.02% sodium azide in PBS. For the experiment, sections were washed twice with TBS (100 mm Tris pH 7.5; 150 mm NaCl), 5 min each, followed by a 30 min incubation in 3% H2O2, to quench endogenous peroxidase activity. Next, sections were transferred into TBS-A (100 mm Tris pH 7.5; 150 mm NaCl; 0.1% Triton X-100) and TBS-B (100 mm Tris pH 7.5; 150 mm NaCl; 0.1% Triton X-100; 2% BSA) for 15 and 30 min, respectively, to block non-specific binding. Finally, the AT8 antibody (1 : 1000) was applied overnight at 4°C; antibody dilution was made in TBS-B. Sections were washed three times in TBS and incubated with a mouse secondary antibody for 1 h at room temperature and developed as described previously (55).
Statistical analyses
All data were analyzed using GraphPad Prism, GraphPad Software, San Diego, CA, USA, www.graphpad.com. Data were analyzed by one- or two-way ANOVA followed by Tukey or Bonferroni post hoc analysis. When applicable, data were analyzed by Student's t-test, as specified in the results section. The survival data were analyzed by using a Cox regression analysis.
FUNDING
This work was supported by grants to S.O. by the National Institutes of Health (R01 AG037637) and the American Federation for Aging Research. E.V.W. was supported by a training grant from the National Institutes of Health (F30 AG043248).
Supplementary Material
Acknowledgements
The authors thank Miss Lauren Hartman for outstanding technical support; the Nathan Shock Aging Center at San Antonio, TX and Dr Gelfond for analyzing the survival data.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Ballatore C., Lee V.M., Trojanowski J.Q. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg P.B., Mielke M.M., Tschanz J., Cook L., Corcoran C., Hayden K.M., Norton M., Rabins P.V., Green R.C., Welsh-Bohmer K.A., et al. Effects of cardiovascular medications on rate of functional decline in Alzheimer disease. Am. J. Geriatr. Psychiatry. 2008;16:883–892. doi: 10.1097/JGP.0b013e318181276a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu J.T., Tan L., Ou J.R., Zhu J.X., Liu K., Song J.H., Sun Y.P. Polymorphisms at the beta2-adrenergic receptor gene influence Alzheimer's disease susceptibility. Brain Res. 2008;1210:216–222. doi: 10.1016/j.brainres.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 4.Ni Y., Zhao X., Bao G., Zou L., Teng L., Wang Z., Song M., Xiong J., Bai Y., Pei G. Activation of beta2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat. Med. 2006;12:1390–1396. doi: 10.1038/nm1485. [DOI] [PubMed] [Google Scholar]
- 5.Kolinski M., Plazinska A., Jozwiak K. Recent progress in understanding of structure, ligand interactions and the mechanism of activation of the beta(2)-adrenergic receptor. Curr. Med. Chem. 2012;19:1155–1163. doi: 10.2174/092986712799320547. [DOI] [PubMed] [Google Scholar]
- 6.Insel P.A. beta(2)-Adrenergic receptor polymorphisms and signaling: do variants influence the ‘memory’ of receptor activation? Sci. Signal. 2011;4:pe37. doi: 10.1126/scisignal.2002352. [DOI] [PubMed] [Google Scholar]
- 7.Russo-Neustadt A., Cotman C.W. Adrenergic receptors in Alzheimer's disease brain: selective increases in the cerebella of aggressive patients. J. Neurosci. 1997;17:5573–5580. doi: 10.1523/JNEUROSCI.17-14-05573.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramos B.P., Arnsten A.F. Adrenergic pharmacology and cognition: focus on the prefrontal cortex. Pharmacol. Ther. 2007;113:523–536. doi: 10.1016/j.pharmthera.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chruscinski A.J., Rohrer D.K., Schauble E., Desai K.H., Bernstein D., Kobilka B.K. Targeted disruption of the beta2 adrenergic receptor gene. J. Biol. Chem. 1999;274:16694–16700. doi: 10.1074/jbc.274.24.16694. [DOI] [PubMed] [Google Scholar]
- 10.Kalaria R.N., Andorn A.C., Tabaton M., Whitehouse P.J., Harik S.I., Unnerstall J.R. Adrenergic receptors in aging and Alzheimer's disease: increased beta 2-receptors in prefrontal cortex and hippocampus. J. Neurochem. 1989;53:1772–1781. doi: 10.1111/j.1471-4159.1989.tb09242.x. [DOI] [PubMed] [Google Scholar]
- 11.De Keyser J., Zeinstra E., Wilczak N. Astrocytic beta2-adrenergic receptors and multiple sclerosis. Neurobiol. Dis. 2004;15:331–339. doi: 10.1016/j.nbd.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Wang D., Xiang Y.K. beta-adrenergic receptor, amyloid beta-peptide, and Alzheimer's disease. Curr. Top. Membr. 2011;67:205–228. doi: 10.1016/B978-0-12-384921-2.00010-0. [DOI] [PubMed] [Google Scholar]
- 13.Yu J.T., Wang N.D., Ma T., Jiang H., Guan J., Tan L. Roles of beta-adrenergic receptors in Alzheimer's disease: implications for novel therapeutics. Brain Res. Bull. 2011;84:111–117. doi: 10.1016/j.brainresbull.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Kitazawa M., Medeiros R., Laferla F.M. Transgenic mouse models of Alzheimer disease: developing a better model as a tool for therapeutic interventions. Curr. Pharm. Des. 2012;18:1131–1147. doi: 10.2174/138161212799315786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshiyama Y., Higuchi M., Zhang B., Huang S.M., Iwata N., Saido T.C., Maeda J., Suhara T., Trojanowski J.Q., Lee V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 16.Klein S.M., Vykoukal J., Lechler P., Zeitler K., Gehmert S., Schreml S., Alt E., Bogdahn U., Prantl L. Noninvasive in vivo assessment of muscle impairment in the mdx mouse model—a comparison of two common wire hanging methods with two different results. J. Neurosci. Methods. 2012;203:292–297. doi: 10.1016/j.jneumeth.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Mandelkow E.M., Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012;2:a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zukerberg L.R., Patrick G.N., Nikolic M., Humbert S., Wu C.L., Lanier L.M., Gertler F.B., Vidal M., Van Etten R.A., Tsai L.H. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–646. doi: 10.1016/S0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 19.Cohen P., Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat. Rev. Drug. Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 20.Ksiezak-Reding H., Pyo H.K., Feinstein B., Pasinetti G.M. Akt/PKB kinase phosphorylates separately Thr212 and Ser214 of tau protein in vitro. Biochim. Biophys. Acta. 2003;1639:159–168. doi: 10.1016/j.bbadis.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Zheng M., Zhu W., Han Q., Xiao R.P. Emerging concepts and therapeutic implications of beta-adrenergic receptor subtype signaling. Pharmacol. Ther. 2005;108:257–268. doi: 10.1016/j.pharmthera.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Alessi D.R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshida H., Hastie C.J., McLauchlan H., Cohen P., Goedert M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK) J. Neurochem. 2004;90:352–358. doi: 10.1111/j.1471-4159.2004.02479.x. [DOI] [PubMed] [Google Scholar]
- 24.Jicha G.A., Weaver C., Lane E., Vianna C., Kress Y., Rockwood J., Davies P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer's disease. J. Neurosci. 1999;19:7486–7494. doi: 10.1523/JNEUROSCI.19-17-07486.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu B., Zhang L., Creighton J., Alexeyev M., Strada S.J., Stevens T. Protein kinase A phosphorylation of tau-serine 214 reorganizes microtubules and disrupts the endothelial cell barrier. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010;299:L493–L501. doi: 10.1152/ajplung.00431.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor S.S., Zhang P., Steichen J.M., Keshwani M.M., Kornev A.P. PKA: lessons learned after twenty years. Biochim. Biophys. Acta. 2013;1834:1271–1278. doi: 10.1016/j.bbapap.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalaria R.N., Harik S.I. Increased alpha 2- and beta 2-adrenergic receptors in cerebral microvessels in Alzheimer disease. Neurosci. Lett. 1989;106:233–238. doi: 10.1016/0304-3940(89)90231-0. [DOI] [PubMed] [Google Scholar]
- 28.Khachaturian A.S., Zandi P.P., Lyketsos C.G., Hayden K.M., Skoog I., Norton M.C., Tschanz J.T., Mayer L.S., Welsh-Bohmer K.A., Breitner J.C. Antihypertensive medication use and incident Alzheimer disease: the Cache County Study. Arch. Neurol. 2006;63:686–692. doi: 10.1001/archneur.63.5.noc60013. [DOI] [PubMed] [Google Scholar]
- 29.Green S.A., Liggett S.B. A proline-rich region of the third intracellular loop imparts phenotypic beta 1-versus beta 2-adrenergic receptor coupling and sequestration. J. Biol. Chem. 1994;269:26215–26219. [PubMed] [Google Scholar]
- 30.Green S.A., Turki J., Hall I.P., Liggett S.B. Implications of genetic variability of human beta 2-adrenergic receptor structure. Pulm. Pharmacol. 1995;8:1–10. doi: 10.1006/pulp.1995.1001. [DOI] [PubMed] [Google Scholar]
- 31.Yasar S., Xia J., Yao W., Furberg C.D., Xue Q.L., Mercado C.I., Fitzpatrick A.L., Fried L.P., Kawas C.H., Sink K.M., et al. Antihypertensive drugs decrease risk of Alzheimer disease: Ginkgo Evaluation of Memory Study. Neurology. 2013;81:896–903. doi: 10.1212/WNL.0b013e3182a35228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marien M.R., Colpaert F.C., Rosenquist A.C. Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res. Brain Res. Rev. 2004;45:38–78. doi: 10.1016/j.brainresrev.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 33.Szot P., White S.S., Greenup J.L., Leverenz J.B., Peskind E.R., Raskind M.A. Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer's disease and dementia with Lewy bodies. J. Neurosci. 2006;26:467–478. doi: 10.1523/JNEUROSCI.4265-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang D., Govindaiah G., Liu R., De Arcangelis V., Cox C.L., Xiang Y.K. Binding of amyloid beta peptide to beta2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. FASEB J. 2010;24:3511–3521. doi: 10.1096/fj.10-156661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang D., Fu Q., Zhou Y., Xu B., Shi Q., Igwe B., Matt L., Hell J.W., Wisely E.V., Oddo S., et al. beta2 adrenergic receptor, protein kinase A (PKA) and c-Jun N-terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J. Biol. Chem. 2013;288:10298–10307. doi: 10.1074/jbc.M112.415141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oddo S., Caccamo A., Tseng B., Cheng D., Vasilevko V., Cribbs D.H., LaFerla F.M. Blocking Abeta42 accumulation delays the onset and progression of tau pathology via the C terminus of heat shock protein70-interacting protein: a mechanistic link between Abeta and tau pathology. J. Neurosci. 2008;28:12163–12175. doi: 10.1523/JNEUROSCI.2464-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oddo S., Vasilevko V., Caccamo A., Kitazawa M., Cribbs D.H., LaFerla F.M. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J. Biol. Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 38.Oddo S., Caccamo A., Shepherd J.D., Murphy M.P., Golde T.E., Kayed R., Metherate R., Mattson M.P., Akbari Y., LaFerla F.M. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 39.Gotz J., Chen F., van Dorpe J., Nitsch R.M. Formation of neurofibrillary tangles in P301 l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 40.Bierhaus A., Wolf J., Andrassy M., Rohleder N., Humpert P.M., Petrov D., Ferstl R., von Eynatten M., Wendt T., Rudofsky G., et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc. Natl Acad. Sci. USA. 2003;100:1920–1925. doi: 10.1073/pnas.0438019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yin D., Tuthill D., Mufson R.A., Shi Y. Chronic restraint stress promotes lymphocyte apoptosis by modulating CD95 expression. J. Exp. Med. 2000;191:1423–1428. doi: 10.1084/jem.191.8.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caccamo A., Medina D.X., Oddo S. Glucocorticoids exacerbate cognitive deficits in TDP-25 transgenic mice via a glutathione-mediated mechanism: implications for aging, stress and TDP-43 proteinopathies. J. Neurosci. 2013;33:906–913. doi: 10.1523/JNEUROSCI.3314-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothman S.M., Mattson M.P. Adverse stress, hippocampal networks, and Alzheimer's disease. Neuromol. Med. 2010;12:56–70. doi: 10.1007/s12017-009-8107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott C.W., Spreen R.C., Herman J.L., Chow F.P., Davison M.D., Young J., Caputo C.B. Phosphorylation of recombinant tau by cAMP-dependent protein kinase. Identification of phosphorylation sites and effect on microtubule assembly. J. Biol. Chem. 1993;268:1166–1173. [PubMed] [Google Scholar]
- 45.Brandt R., Lee G., Teplow D.B., Shalloway D., Abdel-Ghany M. Differential effect of phosphorylation and substrate modulation on tau's ability to promote microtubule growth and nucleation. J. Biol. Chem. 1994;269:11776–11782. [PubMed] [Google Scholar]
- 46.Leger J., Kempf M., Lee G., Brandt R. Conversion of serine to aspartate imitates phosphorylation-induced changes in the structure and function of microtubule-associated protein tau. J. Biol. Chem. 1997;272:8441–8446. doi: 10.1074/jbc.272.13.8441. [DOI] [PubMed] [Google Scholar]
- 47.Illenberger S., Zheng-Fischhofer Q., Preuss U., Stamer K., Baumann K., Trinczek B., Biernat J., Godemann R., Mandelkow E.M., Mandelkow E. The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: implications for Alzheimer's disease. Mol. Biol. Cell. 1998;9:1495–1512. doi: 10.1091/mbc.9.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jicha G.A., O'Donnell A., Weaver C., Angeletti R., Davies P. Hierarchical phosphorylation of recombinant tau by the paired-helical filament-associated protein kinase is dependent on cyclic AMP-dependent protein kinase. J. Neurochem. 1999;72:214–224. doi: 10.1046/j.1471-4159.1999.0720214.x. [DOI] [PubMed] [Google Scholar]
- 49.Li S., Jin M., Zhang D., Yang T., Koeglsperger T., Fu H., Selkoe D.J. Environmental novelty activates beta2-adrenergic signaling to prevent the impairment of hippocampal LTP by Abeta oligomers. Neuron. 2013;77:929–941. doi: 10.1016/j.neuron.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGaugh J.L., Cahill L., Roozendaal B. Involvement of the amygdala in memory storage: interaction with other brain systems. Proc. Natl Acad. Sci. USA. 1996;93:13508–13514. doi: 10.1073/pnas.93.24.13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou H.C., Sun Y.Y., Cai W., He X.T., Yi F., Li B.M., Zhang X.H. Activation of beta2-adrenoceptor enhances synaptic potentiation and behavioral memory via cAMP-PKA signaling in the medial prefrontal cortex of rats. Learn. Mem. 2013;20:274–284. doi: 10.1101/lm.030411.113. [DOI] [PubMed] [Google Scholar]
- 52.Schutsky K., Ouyang M., Castelino C.B., Zhang L., Thomas S.A. Stress and glucocorticoids impair memory retrieval via beta2-adrenergic, Gi/o-coupled suppression of cAMP signaling. J. Neurosci. 2011;31:14172–14181. doi: 10.1523/JNEUROSCI.2122-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Medina D.X., Orr M.E., Oddo S. Accumulation of C-terminal fragments of transactive response DNA-binding protein 43 leads to synaptic loss and cognitive deficits in human TDP-43 transgenic mice. Neurobiol. Aging. 2014;35:79–87. doi: 10.1016/j.neurobiolaging.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 54.Majumder S., Caccamo A., Medina D.X., Benavides A.D., Javors M.A., Kraig E., Strong R., Richardson A., Oddo S. Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancing NMDA signaling. Aging Cell. 2012;11:326–335. doi: 10.1111/j.1474-9726.2011.00791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Majumder S., Richardson A., Strong R., Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE. 2011;6:e25416. doi: 10.1371/journal.pone.0025416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.