

Abstract

A metabolic system is composed of inherently interconnected metabolic precursors, intermediates, and products. The analysis of untargeted metabolomics data has conventionally been performed through the use of comparative statistics or multivariate statistical analysis-based approaches; however, each falls short in representing the related nature of metabolic perturbations. Herein, we describe a complementary method for the analysis of large metabolite inventories using a data-driven approach based upon a self-organizing map algorithm. This workflow allows for the unsupervised clustering, and subsequent prioritization of, correlated features through Gestalt comparisons of metabolic heat maps. We describe this methodology in detail, including a comparison to conventional metabolomics approaches, and demonstrate the application of this method to the analysis of the metabolic repercussions of prolonged cocaine exposure in rat sera profiles.
Genomic and transcriptomic measurements provide information that describes the capacity of a biological system to support specific phenotypes and functions, or the biological potential. Complementary, comprehensive end point molecular analyses, e.g., metabolomics and proteomics, provide information regarding the actual phenotype or functions of the system.1 Both classes of measurements can reveal the underlying complex nonlinear and time-dependent interactions upon which biological systems depend. Accepted multivariate statistical techniques such as principal component analysis (PCA) and orthogonal partial least-squares-discriminant analysis (OPLS-DA) can be used to identify statistical correlations in data, but they are as yet inadequate to elucidate complex interactions in high-dimensional data sets. We demonstrate here the utility of self-organizing maps (SOM) when applied to untargeted metabolomics studies, and how these maps relate to other common analysis techniques. The strength of SOM strategies is the ability to “[convert] complex, nonlinear statistical relationships between high-dimensional data into simple geometric relationships...”.2 Essentially, these techniques distill multivariate data into an accessible, visually interpretable format, while capturing inherent relationships among variables. Self-organizing maps have been used in diverse fields; these include the analysis of meteorological climate change,3 document text clustering,4 cattle management,5 crowd dynamics,6 and gene expression dynamics.7
Research endeavors in metabolomics seek to interrogate the global metabolite profile of a biological system of interest, with the intent of gaining insight into the system phenotype, or alternatively, how the system is interacting with the surrounding environment.8 In metabolomics studies, the complexity of metabolic profiles is often simplified to a fundamental comparison of large inventories of key, phenotypically descriptive small molecules that distinguish between differing physiological states of interest at a single time point (e.g., “normal” vs “diseased”). Differences in the relative concentrations in these metabolite inventories are then used to infer metabolic perturbations. Multivariate statistical analysis (MVSA) methods are used frequently to reduce large dimensional datasets which commonly result from mass spectrometry (MS)-based metabolomics experiments into relevant features (i.e., up- and down-regulated metabolites),9 with a feature defined as any detected monoisotopic molecular species with a discrete retention time and mass-to-charge ratio.10 The widespread application of MVSA to metabolomic studies provides a means of visualizing sample groupings and determining significant metabolite contributions through loadings plot interpretation. Clustering-based approaches deliver data organization based upon internal correlations, though interpretability of large cluster trees is time-consuming. Other metabolomic-based workflows utilize a univariate statistical approach and consider fold-change differences in molecular features.11 Thus, the interrelated nature of the metabolic fluctuations and underlying feature patterns arising from differing physiological states are often difficult to discern or are ignored in conventional feature prioritization workflows.
In this report, we demonstrate a SOM workflow to visualize metabolic phenotype and feature patterns in sera from rat models of cocaine addiction. This advances conventional metabolomics approaches by using SOM techniques, previously developed for gene expression analyses,7 and adapts them for untargeted metabolic profiling. The present workflow uses SOM-based methods to cluster and prioritize analytes of interest by similar expression profiles, in addition to data visualization as previously demonstrated.12,13 Our SOM approach groups features that are annotated by both retention time and mass-to-charge ratio based upon similarities in signal intensity profiles across biological sample sets. The grouping procedure is performed in an iterative manner for a defined training period and metabolites are clustered based upon underlying trends in the metabolic inventories to create the SOM. These maps are then averaged across experimental groups and compared to provide a heat map of up- and down-regulated metabolites as a function of experimental group. This added dimension of feature–feature correlation provides valuable insight into recognizing experimental subpopulations and relevant biomolecules, as well as allowing for the removal of biologically insignificant background features.
We have termed this workflow, which consists of studying MS-based metabolic profiles using SOMs, Molecular Expression Dynamics Investigator (MEDI). This manuscript describes the MEDI workflow method in detail, and subsequently applies MEDI to determine the effects of long-term cocaine exposure upon the serum metabolic profile of rat populations. Currently, we display the application of SOMs to a mass spectrometry-based metabolomic analysis of sera samples from cocaine-naïve rats and behaviorally distinct cocaine-exposed rat models (behaviorally “nonaddicted” and “addicted”).14 Although cocaine and cocaine metabolites are cleared from the animal in 2–4 days, the present workflow using MEDI can distinguish between the metabolite inventories from sera harvested from cocaine-naïve and cocaine-experienced rats two weeks after the last administration of cocaine.
Experimental Section
An overview of the rat cocaine addiction behavioral protocol, data acquisition, and processing methods can be found in Figure 1, with representative data shown in Figure 2. These will be described in greater detail in the Results and Discussion section.
Figure 1.

Self-administration experimental design and data acquisition, processing, and interpretation workflow. (a) The experimental design for self-administration is shown. All rats are first subjected to a behavioral conditioning phase, during which they are trained to self-administer cocaine through operant conditioning. An extinction phase follows, with the intent of extinguishing cocaine-seeking behavior. Subsequently a reinstatement injection is given, and the drug-seeking behavior of the rat then classifies the level of addiction. Time scales are shown below. (b) The complete analytical process for data acquisition, processing, and interrogation is shown. Each step is described in detail in the text.
Figure 2.
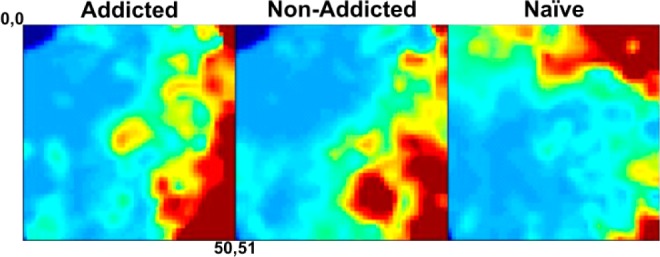
Representative MEDI heat maps indicating relative analyte intensity. For each of the two behavioral groups of cocaine use (addicted and nonaddicted) and cocaine-naïve rat sera metabolomes, corresponding average MEDI heat maps are presented. The static metabolite phenotypes displayed through self-organizing maps indicate gross differences between each group. (Maps represent averages of technical triplicates for three naïve biological samples, four nonaddicted biological samples, and two addicted biological samples, respectively.)
Rat Cocaine Addiction Behavioral Model
Addiction models were prepared using a long-access self-administration protocol.14−16 Briefly, rats were trained to press a lever to receive an infusion of cocaine (0.8 mg/kg/infusion), receiving one drug infusion for each lever press. They were placed in the operant chambers and allowed to self-administer cocaine for 6 h per day, or until they received 50 infusions per day, whichever came first. This behavior was subsequently extinguished through the replacement of cocaine injections with saline injections. Extinction was performed for 8 days, at which point the animals performed minimal lever presses. Addiction classification was determined using a drug-induced reinstatement test. Briefly, a priming injection of 10 mg/kg cocaine was delivered intraperitoneally, and the rats were allowed to again lever press for saline. Rats that obtained 50 saline injections after the cocaine injection were labeled “addicted,” while those that performed less than 50 injections (most of which performed less than 20 injections) were labeled “nonaddicted.” Upon completion of the behavioral study, rats were sacrificed and their blood frozen at −80 °C before being used for analyses. Aliquots were transported on dry ice from Duke University (Durham, NC) to Vanderbilt University (Nashville, TN) and subsequently stored at −80 °C prior to mass spectrometry analysis. In total, three naïve biological samples, four nonaddicted biological samples, and two addicted biological samples were produced for subsequent analyses.
Rat Serum Sample Preparation for Analysis
Frozen whole blood samples from addicted, nonaddicted, and naïve rats were thawed at 4 °C, and then centrifuged at 14 000 rpm for 2 min. 60 μL of serum was removed and metabolites were isolated by precipitating the proteins with 3:1 v:v cold methanol kept on dry ice. Samples were vortexed for 10 s and centrifuged at 14 000 rpm for 10 min at 4 °C. 150 uL of supernatant was extracted and dried down in a SpeedVac, after which the samples were reconstituted in 100 uL of mobile phase A.
Liquid Chromatography-Ion Mobility-Mass Spectrometry Analysis
UPLC-IM-MS and UPLC-IM-MSE analyses were performed on a SYNAPT G2 HDMS (Waters, Milford, MA) mass spectrometer equipped with a nanoAcquity UPLC and autosampler (Waters, Milford, MA). Metabolites were separated on a 75 μm × 100 mm HSS C18 (1.7 μm particle size) column and 180 μm × 20 mm HSS C18 (5 μm particle size) trap column. Column temperature was maintained at 45 °C to minimize chromatographic drift, and the autoinjector sample tray held at 4 °C to minimize sample degradation. A double-loop injection volume of 10 μL was injected in a 5 μL loop. Chromatographic separations were performed by using a 20 min method at a flow rate of 450 nL/min using a gradient mixer of 0.1% formic acid in H2O (mobile phase A) and 0.1% formic acid in ACN (mobile phase B). Briefly, a 3 min wash period at a flow rate of 15 μL/min was performed, during which the eluent was diverted to waste prior to analytical separation. Following removal of residual salts and trapping of analytes on the trap column, flow was redirected to flow through the analytical column with an initial 99% mobile phase A for 0.5 min. Mobile phase B was increased to 60% over 6.5 min and up to 99% in 4 min, and then held at 99% for 3 min. The column was re-equilibrated to 99% mobile phase A over 0.5 min and held for 5.5 min after each run. All analytes were analyzed using positive mode nanoelectrospray ionization. Typical parameters include a capillary voltage of 3.5 kV, sampling cone setting of 25.0 and extraction cone setting of 4.0, source temperature of 80 °C, desolvation gas (N2) flow of 600 L/h, and a cone gas flow of 20 L/h. Data were acquired in MSE mode, which acquires both a low-energy spectrum and a high-energy spectrum. Collision-induced dissociation (CID) was performed post mobility separation with a ramped energy profile from 20 V to 60 V in the high CID acquisition. Traveling wave velocity was held constant at 550 m/s and a height of 40.0 V. Data were acquired at a sampling rate of 2 Hz over the mass range 50–1400 m/z. Sodium formate (10 μg/mL) in 90:10 propan-2-ol:water (v:v) was used to calibrate over this range with <1 ppm mass accuracy. Leucine enkephalin in 50:50 H2O:ACN with 0.1% formic acid (v:v) was used as a lock mass compound (accurate mass 556.2771 Da) at a flow rate of 0.6 μL/min and a concentration of 2 ng/mL every 10 s. Data acquisition was performed from 0 to 20 min of the liquid chromatography separation. Triplicate technical analysis was performed in a randomized fashion, with quality control samples analyzed every five injections. Quality control samples contained equal volume aliquots of each sample mixed together.
Data Processing and Multivariate Statistical Analysis
Data were mass-corrected post-acquisition and centroided. Peaks were deisotoped and normalized using MarkerLynx data processing software (Waters, Milford, MA). Peak-picking using chromatographic profiles was also performed using MarkerLynx. Peak detection was performed on low-energy data across the mass range of 50–2000 Da with retention times between 1 min and 20 min with peak widths ≤30 s (no applied smoothing), intensity threshold of 1000, mass window of 0.03 Da and retention time window of 0.1 min. Data were deisotoped and areas normalized to 10 000 counts per sample.
Multivariate statistical analyses were performed using Umetrics extended statistics software EZinfo version 2.0.0.0 (Waters, Milford, MA). Principal component analysis (PCA) and orthogonal partial least-squares-discriminate analysis (OPLS-DA) were performed on all data acquired and pareto scaled.
Molecular Expression Dynamics Investigator Parameters
After initial peak filtering, detecting, aligning, and normalizing, features were exported in a tab-delimited file congruent with GEDI (format may be found at http://apps.childrenshospital.org/clinical/research/ingber/GEDI/gedihome.htm). The minimum system requirements necessary include a video display capable of displaying at least 1024 × 768 pixels and the latest version of Java (Sun Microsystems, Inc.). A 50 × 51 grid was defined and trained using 80 first-phase and 160 second-phase iterations. A neighborhood radius of 4.0 was used during the first phase, and 1.0 during the second phase. The learning factor was 0.5 for the first phase and 0.05 for the second phase. The neighborhood block sizes applied for the first and second phases were 4 and 2, respectively. A conscience of 3.0 was used for both phases. A random seed value of 1 was used, and Euclidean distance was applied for the distance metric. A linear initialization method was applied. Samples were averaged across technical and biological replicates, and subtracted across experimental groups within the Gene Expression Dynamics Inspector (GEDI) software. GEDI maps were exported through the software, in addition to “Gene Assignment Lists,” which indicate node location of features, and “Map Centroids,” which were used for the generation of intensity values for regions of interest.
Statistical Analysis of Regions in Molecular Expression Dynamics Investigator Heat Maps
The cocaine-experienced MEDI self-organized heat map (Figure 3c) shows the summed ion signal intensities for specific regions across sample types (cocaine-naïve, cocaine-addicted, and cocaine-nonaddicted). This MEDI heat map has allowed us to quantify and determine the statistically significant regions, labeled a-f, using a one-way ANOVA test among sample types. A one-way ANOVA compares the effect of cocaine use using the grouped “neighborhoods” of signal ion intensities for nonaddicted, addicted, experienced (average of nonaddicted and addicted samples) and naïve rats. We observed a significant effect (p < 0.01) based on cocaine exposure for all regions, and for those regions with unequal variance between groups, a Kruskal–Wallis one-way ANOVA was conducted (regions a, d, e, and f) [a: H(3,37 ) = 22.03, p < 0.0001; b: F(3,37) = 4.99, p = 0.0052; c: H(3,37) = 21.83, p < 0.0001; d: F(3, 37) = 7.24, p = 0.0006; e: H(3,37) = 18.9, p = 0.0003; f: H(3,37) = 25.6, p < 0.0001], indicating that post hoc comparisons were appropriate. Post hoc comparisons using a Bonferroni–Holm’s test show statistically significant differences (p ≤ 0.01) for all regions (a–f) when comparing cocaine-experienced, cocaine-addicted, cocaine-nonaddicted, and cocaine-naïve sample types. In addition, region f showed a statistically significant difference (p < 0.01) between addicted and nonaddicted rat models (see f in Figure 4). Region c, however, does not show a statistically significant difference (p = 0.06) between nonaddicted and addicted sample groups, which indicates that there are neighborhoods that further discriminate cocaine-nonaddicted and cocaine-addicted models.
Figure 3.

Rat sera metabolome depictions for cocaine-experienced versus naïve classes. (a) Principal component analysis (PCA) of cocaine-experienced (block markers) rat sera metabolomes plotted with cocaine-naïve (diamond markers). Behavioral subclasses are indicated by color. (b) S-plot comparing cocaine-experienced (−1) to cocaine-naïve (+1) metabolomes. (Marker color corresponds to the boxes in panel c (inset shows a magnified subimage).) (c) Differential MEDI heat map of average cocaine-experienced metabolic profiles with average cocaine-naïve profiles subtracted. Boxed-in regions are then delineated in panel (d), which is an annotated representative UPLC-MS heat map marking feature location on the cocaine-experienced UPLC-MS plot. The colored dots correspond to the different feature islands in panel c [Box dimensions: a (39,38:50,51); b (46,33:50,37); c (28,34:38,46); d (39,28:45,36); e (36,20:42,27); f (46,20:50,32)]. (Analysis contains technical triplicates for three naïve biological samples, two addicted biological samples, and four nonaddicted biological samples.)
Figure 4.
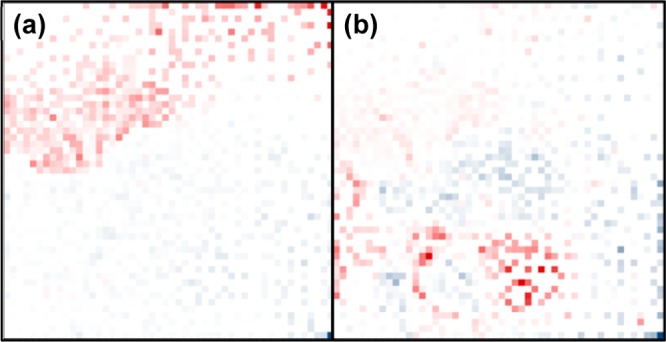
Loadings contribution of nodes to PCA. The contributions of each node to (a) the first principal component and (b) the second principal component are indicated by color intensity. Red indicates a negative contribution and blue indicates a positive contribution.
Feature Identification
Putative identifications were performed using the monoisotopic accurate mass and raw data to determine molecular ion type. Monoisotopic masses were searched against the Human Metabolome Database (HMDB),17,18 METLIN,19 and LIPID MAPS20 databases for putative identifications, with a mass tolerance of 0.01 Da. When possible, fragmentation data were used to support identifications, utilizing mobility separation prior to fragmentation to isolate parent ions in separations space. Data may be found in the Supporting Information regarding the putative identification assignments for prioritized metabolites.
Results and Discussion
Overview of the Data Analysis Approach, Molecular Expression Dynamics Inspection
For mass spectrometry-based metabolomic data analysis, MEDI incorporates Gene Expression Dynamics Inspector (GEDI) software. Figure 1b outlines the general workflow for the analysis of metabolomics data using a self-organizing map algorithm to sort detected features in an unsupervised, data-driven manner. This workflow facilitates the identification of unique expression profiles across samples based on feature patterns detected in the metabolomic analysis. Briefly, this workflow consist of six steps: (1) data acquisition, (2) data preprocessing which incorporates peak detection, alignment, and normalization, (3) generation of self-organizing maps, (4) differential analysis intensity maps generated in step (3) to determine relevant feature clusters, (5) interrogation of metabolite feature assignment maps to determine unique features/analytes of interest, and finally (6) identification of peaks of interest. The MEDI workflow is applicable across platforms, with the exception of peak identification, which will be technique/detector-dependent. The generalized MEDI workflow is now described in greater detail.
Data Acquisition and Preprocessing (MEDI Workflow: Steps 1 and 2)
The first step in the proposed workflow is the initial data acquisition. It is important to note that no one sample preparation or mass spectrometry analysis technique will give a global metabolomic view simply based on the chemical diversity associated with metabolites. It is therefore essential to plan experiments, extractions, and analysis techniques accordingly. It is beyond the scope of this report to discuss all metabolite extraction and analysis protocols, but it is important to understand the limitations of each sample preparation and analytical method. We suggest a few relevant reviews on this topic.9,21,22
After data acquisition, raw data must be described as discrete features. This involves centroiding and aligning retention time and mass spectral profiles, in addition to deisotoping data to ensure monoisotopic peak comparison. Subsequent normalization scales data to reduce the impact of technical variation. Publically available software (e.g., XCMS) can be used to filter, detect, and align peaks; therefore, this workflow is compatible across numerous mass spectrometry platforms (e.g., LC-MS/MS, LC-IM-MS/MS, GC-MS).11 Normalized metabolomics data are then appropriate for feature organization through the self-organizing map algorithm.
Feature Organization and Analysis (MEDI Workflow: Steps 3 and 4)
In these MEDI workflow steps, a SOM algorithm is used to assign detected features to a grid of user-defined dimensions. Specifically, the algorithm arranges features in an iterative manner based on similarity in intensity profiles across samples. Initially, the grid is populated with randomly generated intensity profiles. Randomly selected features from the input data are placed in the grid location, or node, which best matches the intensity profile of that feature. The profile of this node is then adjusted to more closely resemble the profile of the matched feature. Surrounding nodes are also adjusted to more closely resemble the matched feature, but to a lesser extent. This process is then performed again with a different, randomly selected input feature. Features that behave similarly across samples are assigned to a particular neighborhood, or in nodes adjacent to like features. The assignment of features to specific nodes is then iterated as node profiles evolve. After features are assigned to specific node coordinates, intensity maps are generated for each sample using the summed intensity for each node. These profile the metabolic phenotype for each sample, displaying all detected features in a heat map. The relative number of features that contribute to a neighborhood is shown in the feature density map. Subsequently, samples can be averaged and subtracted based on sample group and experimental specifics. Differential profile maps show clusters/regions of up- or down-regulated features, which are potential metabolites of interest. This specific analysis reveals regions (boxed regions), which should be prioritized and compared across samples. Importantly, the feature density map indicates that many of these regions of interest are resultant of a small number of metabolites.
Cluster Decryption (MEDI Workflow: Step 5)
To extract which metabolomic features contribute to neighborhoods of interest, clusters must be decrypted into constituent features. Each node has a finite number of associated features, which determine the resultant heat maps. Multiple features can contribute to a single coordinate intensity, thus images need to be “decrypted” by determining which features contribute to specific neighborhoods. The descriptive data for a feature indicate the chemical properties of the feature and the location in the raw data.
Feature Identification (MEDI Workflow: Step 6)
The final step in the proposed workflow includes the identification of significant features. These identifications will be platform-specific, thus the tools used for analyte identification will also be platform-specific. There are several tools available for fragmentation prediction and matching of spectra for metabolomic-based studies, as well as databases (e.g., Human Metabolome Database, Lipid Maps Structure Database, METLIN, Kyoto Encyclopedia of Genes and Genomes).17−20,23,24 The metabolites detected provide the basis for inferences on metabolic perturbations between sample groups. Guidelines for metabolite identification confidence have been outlined by the Metabolomics Standards Initiative.25
Applying MEDI to Explore the Effects of Long-Term Cocaine Exposure
To display the utility of this workflow, we applied the MEDI process to liquid chromatography–mass spectral (LC-MS) sera profiles from cocaine-naïve rats and behaviorally distinct cocaine-addicted and cocaine-nonaddicted rats. In these data, ∼2266 unique features (RT – m/z pairs) were detected across all samples after peak picking, alignment, and normalization using MarkerLynx software (Waters Corporation, Milford, MA). Detected features were self-organized using the GEDI software, and average group heat maps are seen in Figure 2. This function simply averages node intensities across a selected group of samples, which, in this case, is determined by behavioral class. For comparison of the generalized effects of cocaine exposure, the two behavioral classes were pooled and are classified “cocaine-experienced.” These individual self-organizing heat maps clearly show distinct differences among groups. The density of metabolites for given nodes can be seen in Figure S1 in the Supporting Information.
Visual inspection of the metabolite heat maps demonstrates that the sera profiles for the three groups have both shared and distinct characteristics. This displays a significant advantage to using MEDI for metabolic phenotype investigation. By virtue of the feature organization process, the groupings that result from the self-organizing algorithm are driven by hierarchical specificity. As a result, if experimental groups have significant differences, groups will emerge that concentrate these differences. In addition, background signals will be organized together and essentially eliminated from the analysis. Features that are specific to a particular sample will then occupy separate regions. To gain perspective on how the MEDI process compares to multivariate statistical analysis methods, we performed principal component analysis and orthogonal partial least squares-discriminant analysis on detected features.
Multivariate Statistical Analysis
The use of multivariate statistical analysis methods is a common informatics approach for metabolomics data. This enables researchers to determine significant features in complex datasets, interrogate sample grouping, investigate data acquisition reproducibility, and classify unknown samples based on example training sets. Figure 3a is a principal component analysis (PCA) of the three sample groups analyzed. In PCA, the intensity of each feature (considered a dimension) is used to describe a given sample. Briefly, PCA determines the largest eigenvalue eigenvector of the covariance matrix of the data, which is the first principal component. This eigenvector describes the largest differences in the samples. The second principal component is orthogonal to the first principal component and describes the next largest differences in the data. The result is sample grouping based on similarity, and separation based on the largest global feature differences in the first principal component, and the next largest differences in the second principal component. The first and second principal components are plotted in Figure 3a as the abscissa and ordinate, respectively. This displays the samples in maximally distinguishing two-dimensional space, based on feature intensity. Subsequent principal components are all orthogonal and describe progressively less variation (i.e., lower eigenvalue eigenvectors of the covariance matrix). The PCA plot in Figure 3a illustrates the ability for MS-based metabolic profiles from cocaine-naïve and cocaine-experienced rat sera to separate in the first principal component (see x-axis in Figure 3a). This also shows a separation in the second principal component (see y-axis); cocaine-experienced rats further separate into two main groups. The top group (quadrant I) consists of data from both behaviorally addicted (black) and nonaddicted rats (red), while the other group (quadrant IV) comprises data generated from biological duplicates of behaviorally nonaddicted rats (multiple points due to technical replicates). The grouping consistency in the biological duplicates indicates biological variation from the other cocaine-exposed rats. It is unclear if this secondary separation is a result of behavioral class, or simply specific to the rats. Importantly, the first principal component separates metabolomic profiles based on cocaine history, independent of behavioral class. In these experiments, rat serum was obtained two weeks after cocaine administration; therefore, cocaine and its metabolites were not detected in these analyses. Although the PCA plot indicates separation based on exposure, it describes only 24% of the variation with the first two principal components. For the purposes of subsequent binary comparisons, the samples are grouped as either cocaine-naïve or cocaine-experienced.
Orthogonal partial least-squares-discriminant analysis (OPLS-DA) can also be used to compare cocaine-naïve to cocaine-experienced rat sera profiles (see Figure S2 in the Supporting Information).26 OPLS-DA, in this case, finds the relationship between the UPLC-MS data and cocaine history as a supervised method. OPLS-DA orients the model such that the abscissa is the predictive component, or between-group variation. The orthogonal ordinate then describes intragroup variation. Specifically, this model explains 98% of the data variation between groups. Figure 3b shows a corresponding S-plot used to determine metabolites of significance. This S-plot graphs features based on group specificity or correlation (ordinate) and covariance (abscissa). Features with a high group correlation, or specific to either cocaine-exposed or cocaine-naïve condition, in this case, have a large magnitude in the y-dimension. Features with a large loadings contribution to the predictive component possess large magnitudes in the x-dimension. In this manner, we are able to determine the features that are specific to long-term cocaine exposure.
MEDI Heat Map Interpretation
The SOM approach to feature organization places features with similar sample intensity profiles proximal in the coordinate grid, as mentioned above. This generates regions of features that are up- and down-regulated (e.g., Figure 3c, red and blue, respectively) consistently across samples, in addition to regions that are specific to a subset of samples. This provides additional flexibility to data organization beyond the conventional dimensions of loadings analyses of MVSA methods. Shown in Figure 3c is an average MEDI heat map of cocaine-experienced serum profiles (i.e., both behavioral classes) with the average naïve heat map subtracted to display metabolites that are either up-regulated (yellow to red), or down-regulated (blue to dark blue) as a general result of prolonged cocaine exposure. This differential analysis subtracts the average node intensities of cocaine-naïve rat sera profiles from the averaged experimental group. Although many islands exist, the more intense regions of up-regulation are outlined for comparative purposes. These regions are both annotated and outlined with a colored box in Figures 3c. The colored boxes are present for comparative purposes and indicate the feature location in other data representations (Figures 3b and 3c) [Box coordinates: a (39,38:50,51); b (46,33:50,37); c (28,34:38,46); d (39,28:45,36); e (36,20:42,27); f (46,20:50,32)]. It should be noted that these boxes are consistent across sample groups and determined by group perimeter. Thresholding and feature recognition software is being implemented for future applications.
Correlating the regions of interest in Figure 3c with a raw LC-MS plot (Figure 3d) illustrates the concept that grouped features display a large range of chemical properties, as they occupy different regions of separations space. The marker color in Figure 3d corresponds to the location in the heat map in Figure 3c. The S-plot in Figure 3b, described above as a common method to extract meaningful features from OPLS-DA binary analyses, has been modified so the color of the markers corresponds to the regions of interest in Figure 3c. All the features that would be prioritized through OPLS-DA are encapsuled in the regions of interest. These features occupy dispersed regions of the S-plot.
Loadings Contributions of MEDI Coordinates
Correlations between the MEDI feature assignment location and the loadings contributions to PCA are seen in Figures 4a and 4b. The loadings contribution of a feature to a particular principal component indicates the weight of that feature regarding sample magnitude in that dimension. In other words, a feature with a large negative loading value in the first principal component will influence a sample containing that feature to have a negative value in that component. Representing the loadings in this medium provides insight into neighborhood formation. The static nature of these samples means any clustering that occurs is sample-group-specific, assuming technical reproducibility. As such, what occurs in all cocaine-experienced sera profiles will group, and subsequent neighborhoods will form based on sample specificity. Figures 4a and 4b should be considered with both Figures 3a and 3c, as the loadings contributions link the MEDI heat map to the PCA scores plot. The largest trends are resultant of cocaine experience and have been organized into two main regions of the map accordingly. The upper portion of the map corresponds to features that are down-regulated, generally, in the cocaine-experienced. This is indicated by the dominantly red upper portion. Features initially partition based on the global group differences. Considering the second principal component loadings, there is very little contribution from the features that are found in elevated intensities in the cocaine-naïve samples, which is seen by the relatively small contribution of the red region in Figure 4a. The second principal component loadings map offers more insight into the formation of feature islands in the cocaine-experienced group. The significant feature loadings are those describing intraexperienced separations, which are indicated by the scores plot. The nodes that contribute greatly to the second principal component form regions in the MEDI plot. This demonstrates the finer clustering effects of the feature sorting algorithm. Subpopulations of samples, such as the biologically distinct cocaine-exposed rats, produce regions representing features that are unique to that subgroup. The grouping in principal component analysis, in addition to individual sample MEDI heat map investigation, provides insight into the interpretation of these underlying features.
Specifically, coordinate (50,51), or the extreme bottom right node, has the greatest contribution to a positive loading in the first and second principal components. This feature group, consisting of putatively identified deoxyuridine, and three other features, contains the most distinguishing features to cocaine experience in PCA. This group is also prioritized during the MEDI process. Other contributing signals are listed based on their group occupation, feature descriptors, and, when applicable, a putative identification in Table 1. The greatest negative contributors to principal component two are found within region “c,” in addition to more peripheral regions. These features are up-regulated in the distinct cocaine-exposed rat sera. As a result, these features provide insight into the difference between this rat and other cocaine-exposed models. In addition, region “f” has a positive contribution to the second principal component, and thus is up-regulated in most cocaine-exposed rats. As a result, this region may provide alternative insight into the intragroup separation.
Table 1. Putative Metabolite Identificationa.
| retention time (min) | m/z | MEDI region | putative identification |
|---|---|---|---|
| 11.76 | 130.066 | a | 3-methylene-indolenine |
| 9.92 | 182.192 | a | no database match |
| 10.82 | 185.080 | a | (3-methoxy-4-hydroxyphenyl) ethylene glycol |
| 11.78 | 190.048 | a | kynurenic acid |
| 9.85 | 229.091 | a | deoxyuridine |
| 10.53 | 229.142 | a | no database match |
| 10.77 | 243.095 | a | thymidine |
| 10.34 | 273.175 | a | estradiol |
| 10.19 | 308.091 | a | glutathione |
| 9.49 | 338.087 | a | 3-indole carboxylic acid glucuronide |
| 9.74 | 340.104 | a | 5-hydroxy-6-methoxyindole glucuronide |
| 10.55 | 361.138 | a | dityrosine |
| 14.09 | 500.277 | a | LPE(20:5) |
| 9.72 | 520.336 | a | LPC(18:2) |
| 9.09 | 201.068 | b | bilirubin oxidation product |
| 12.62 | 225.088 | c | 3-hydroxykynurenine |
| 9.22 | 305.159 | c | sodiated steroid-like molecule |
| 12.18 | 430.296 | c | glycocholic acid −2H2O |
| 12.18 | 448.306 | c | glycocholic acid-H2O |
| 12.19 | 466.318 | c | glycocholic acid |
| 10.19 | 162.056 | d | 4,6-dihydroxyquinoline |
| 9.55 | 269.132 | d | 3-carboxy-4-methyl-5-pentyl-2-furanpropionic acid |
| 9.48 | 371.227 | d | 6-keto-prostaglandin F1a |
| 9.54 | 399.625 | e | [M+2H]2+ |
| 1.20 | 203.054 | f | succinyl acetoacetate |
| 11.94 | 223.066 | f | no database match |
| 14.11 | 542.327 | f | LPC(20:5) |
Metabolites were given preliminary identifications based upon accurate mass, ion type, and database searching.
Region Interpretation and Comparison
Further heat map interpretation prioritizes regions of interest, which consist of grouped features. Figure 5 compares the summed node intensities for outlined regions of the MEDI heat map. This provides insight into the significance of regions, in addition to the intragroup differences that occur. The summed intensities corroborate the loadings analysis, as the regions selected have significant differences in intensities between cocaine-experienced and cocaine-naïve groups, and each have positive contributions to the first principal component. The regions that show differences within the experienced group (i.e., regions c and f) have varied intensities between behavioral classes, and also hold negative and positive contributions to the second principal component, respectively. The significance of the displayed differences was subsequently determined.
Figure 5.
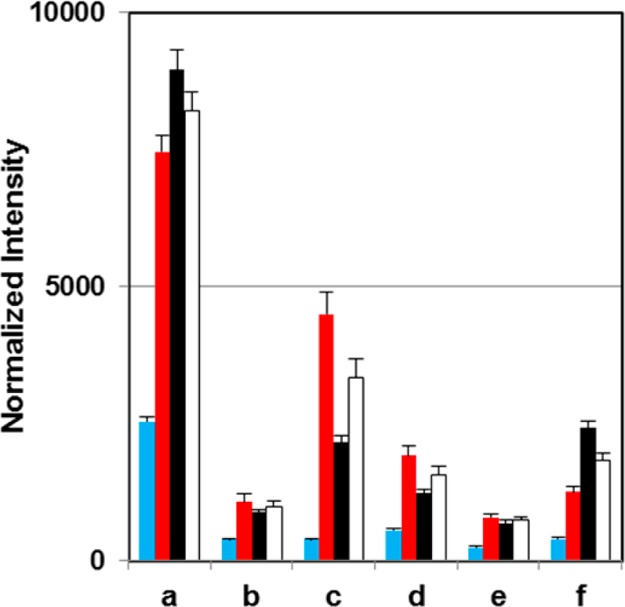
Histogram depicting the intensity integrated over six of the enclosed regions of MEDI heat map in Figure 3b. Rats with a history of cocaine exposure (white) had mean intensities significantly higher than cocaine-naïve rats (blue) in all regions tested (p < 0.05). In addition, the mean intensity of region “f” was significantly higher in cocaine-addicted rats (black) than nonaddicted (red) (p < 0.01).
Determination of Contributing Features
Extracting relevant biological information is paramount in metabolomic experiments. Following regional analysis of MEDI heat maps, the extraction of contributing features is performed through the interface of GEDI by selecting a node, or through an exported “Gene Assignments List.” Table 1 lists relevant feature identifiers, region located, and, when available, putative identifications. The organization of features is a function of whether the feature is present in the same permutation of samples at relatively similar intensities. As a result, the subpopulations that arise indicate biological subtleties that exist among groups. The putatively identified features in disparate regions provide insight into these biological differences.
Regions a–f all show distinct up-regulation as a result of prolonged cocaine exposure, as seen in both Figure 5 and the loadings representation in Figure 4a. Region “a” contains metabolites that occur in a majority of the cocaine-exposed group. These include metabolites indicative of compensatory mechanisms of the biochemical effects of prolonged cocaine exposure. A largely up-regulated metabolite found in this region is (3-methoxy-4-hydroxyphenyl)ethylene glycol, which is the primary serum metabolite of norepinephrine and has been found to be dysregulated following cocaine withdrawal.27 This metabolite is downstream of dopamine (and more generally catecholamine) synthesis.28 Kynurenic acid is a naturally occurring metabolite resulting from tryptophan metabolism, and it has displayed protective effects against cocaine toxicity.29 The inhibitory effects of cocaine exposure on glial cell growth have also been shown, significantly decreasing incorporation of thymidine in DNA synthesis, which inhibits growth.30 The marked increase in serum thymidine concentrations is perhaps a compensatory result of cocaine exposure, or perhaps a result of metabolite pooling resulting from lack of incorporation. This could be the reason for up-regulation of deoxyuridine as well. Alterations in estradiol concentrations have been seen in response to cocaine exposure, although why up-regulation occurs in male rats following cocaine extinction is unclear. The presence of 3-carboxy-4-methyl-5-pentyl-2-furanpropionic acid could speak to renal health as a result of the prolonged cocaine exposure.31,32
Region “c” concentrates the biological differences that exist within the cocaine-exposed group. The increased 3-hydroxykynurenine that appears to exist implicates further tryptophan perturbations and is indicative of neural inflammation.33 This metabolite has been associated with oxidative stress and neuronal cell death. Unidentified features that exist in this region may ultimately provide more insight into the biological variation that exists.
The other distinct region of difference within the cocaine-experienced group is region “f.” The presence of succinylacetoacetate is a unique metabolite resulting from tyrosinemia, which has been shown to result from long-term cocaine exposure.34 The differences in lysophosphocholines observed should also be noted.
The current study focuses entirely on the up-regulated features found in the cocaine-experienced group. There is a wealth of information that is present in the regions of down-regulation. However, considering the pedagogical nature of this data set, we have chosen to demonstrate the relevance of the MEDI method using a subset of the prioritized features. For definitive assignments, putative identifications should be validated against standards with retention time and MS/MS matching.
Conclusions
We have described the MEDI workflow and applied this method to a static set of sera samples from behaviorally conditioned cocaine addiction rat models. We have demonstrated the utility of SOM to distinguish underlying feature motifs. Features that contributed to these regions were putatively identified and metabolic connections that have been well-described as consequential of cocaine exposure were established.
Although this method was applied to data acquired using mass spectrometry-based detection, it is easily applied to other metabolomics platforms (e.g., NMR), or to additional dimensions of separation combined with MS. We have also indicated the application of the MEDI workflow only on static samples, although the GEDI core software is ideal to determine underlying temporal dynamics. MEDI is a method that provides a medium to express metabolic phenotype and prioritize features based upon underlying sample patterns.
It should be noted that the dataset investigated in this study was for proof of principle, and caution should be observed in interpreting these results regarding cocaine classification, as a small sample set was investigated and absolute identification of analytes necessitates further studies. To enhance confidence in metabolites indicative of behavioral classification, a larger cohort is required. However, for the purposes of this report, we consider these data to be an instructive application of the MEDI process.
Although we apply this method to end point analyses, this method is also well-suited for comparison of temporal dynamic samples, as the name implies. For the purpose of this work, however, we endeavor to describe this method on temporally pooled samples. Specifically, the MEDI workflow begins with data acquisition and ends with the identification of biologically significant up- and down-regulated mass spectrometry signals.
Acknowledgments
We thank Dr. Amina S. Woods (National Institutes of Health-National Institute on Drug Abuse, Baltimore, MD) for initial discussions on the cocaine model investigated. This work was supported in part by the National Institutes of Health (NIH Grant RC2DA028981), the U.S. Defense Threat Reduction Agency (Grant HDTRA1-09-1-0013), the Vanderbilt Institute for Integrative Biosystems Research and Education, the Vanderbilt Institute of Chemical Biology, and the Systems Biology and Bioengineering Undergraduate Research Experience (funded by Gideon Searle at Vanderbilt University). We thank Nolan Smith for his technical assistance, and Allison Price for her editorial assistance.
Supporting Information Available
Additional information as noted in the text is presented in the Supporting Information, including molecular species information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Patti G. J.; Yanes O.; Siuzdak G. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohonen T.; Oja E.; Simula O.; Visa A.; Kangas J. Proc. IEEE 1996, 84, 1358–1384. [Google Scholar]
- Skific N.; Francis J. A.; Cassano J. J. J. Climate 2009, 22, 4135–4153. [Google Scholar]
- Kohonen T.; Niklasson L.; Bodén M.; Ziemke T. In Proceedings of ICANN98, The 8th International Conference on Artificial Neural Networks; Springer, 1998; Vol. 1, pp 65–74. [Google Scholar]
- Correia Baptista Soares de Mello J. C.; Goncalves Gomes E.; Angulo Meza L.; Biondi Neto L.; Gomes Pinto de Abreu U.; de Carvalho T. B.; de Zen S. In Applications of Self-Organizing Maps, 1st Edition; Johnsson M., Ed.; InTech Publishers: Midlothian, TX, 2012; Chapter 4, pp 67–88. [Google Scholar]
- Owens J.; Hunter A. In Proceedings of IEEE International Workshop on Visual Surveillance, Dublin, Ireland, July 1, 2000; pp 77–83. [Google Scholar]
- Eichler G. S.; Huang S.; Ingber D. E. Bioinformatics 2003, 19, 2321–2322. [DOI] [PubMed] [Google Scholar]
- Rochfort S. J. Nat. Prod. 2005, 68, 1813–1820. [DOI] [PubMed] [Google Scholar]
- Goodacre R.; Vaidyanathan S.; Dunn W. B.; Harrigan G. G.; Kell D. B. Trends Biotechnol. 2004, 22, 245–252. [DOI] [PubMed] [Google Scholar]
- Nordstrom A.; O’Maille G.; Qin C.; Siuzdak G. Anal. Chem. 2006, 78, 3289–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A.; Elizabeth J.; O’Maille G.; Abagyan R.; Siuzdak G. Anal. Chem. 2006, 78, 779–787. [DOI] [PubMed] [Google Scholar]
- Patterson A. D.; Li H.; Eichler G. S.; Krausz K. W.; Weinstein J. N.; Fornace A. J. Jr; Gonzalez F. J.; Jeffrey R. Anal. Chem. 2008, 80, 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyburski J. B.; Patterson A. D.; Krausz K. W.; Slavik J.; Fornace A. J. Jr; Gonzalez F. J.; Idle J. R. Radiat. Res. 2008, 170, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm-Sapyta N. L.; Olsen C. M.; Winder D. G. Neuropsychopharmacology 2005, 31, 1444–1451. [DOI] [PubMed] [Google Scholar]
- Schramm-Sapyta N. L.; Cauley M. C.; Stangl D. K.; Glowacz S.; Stepp K. A.; Levin E. D.; Kuhn C. M. Psychopharmacology 2011, 215, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley J. W.; Fryer T. D.; Brichard L.; Robinson E. S. J.; Theobald D. E. H.; Laane K.; Pena Y.; Murphy E. R.; Shah Y.; Probst K. Science 2007, 315, 1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Knox C.; Guo A. C.; Eisner R.; Young N.; Gautam B.; Hau D. D.; Psychogios N.; Dong E.; Bouatra S. Nucleic Acids Res. 2009, 37, D603–D610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Tzur D.; Knox C.; Eisner R.; Guo A. C.; Young N.; Cheng D.; Jewell K.; Arndt D.; Sawhney S. Nucleic Acids Res. 2007, 35, D521–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A.; Maille G. O.; Want E. J.; Qin C.; Trauger S. A.; Brandon T. R.; Custodio D. E.; Abagyan R.; Siuzdak G. Ther. Drug Monit. 2005, 27, 747. [DOI] [PubMed] [Google Scholar]
- Sud M.; Fahy E.; Cotter D.; Brown A.; Dennis E. A.; Glass C. K.; Merrill A. H. Jr; Murphy R. C.; Raetz C. R. H.; Russell D. W. Nucleic Acids Res. 2007, 35, D527–D532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakova L.; Vlckova H. Anal. Chim. Acta 2009, 656, 8–35. [DOI] [PubMed] [Google Scholar]
- Alvarez-Sanchez B.; Priego-Capote F.; Luque de Castro M. D. TrAC, Trends Anal. Chem. 2010, 29, 111–119. [Google Scholar]
- Ogata H.; Goto S.; Sato K.; Fujibuchi W.; Bono H.; Kanehisa M. Nucleic Acids Res. 1999, 27, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M.; Goto S.; Hattori M.; Aoki-Kinoshita K. F.; Itoh M.; Kawashima S.; Katayama T.; Araki M.; Hirakawa M. Nucleic Acids Res. 2006, 34, D354–D357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner L. W.; Amberg A.; Barrett D.; Beale M. H.; Beger R.; Daykin C. A.; Fan T. W. M.; Fiehn O.; Goodacre R.; Griffin J. L. Metabolomics 2007, 3, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiklund S.; Johansson E.; Sjstrm L.; Mellerowicz E. J.; Edlund U.; Shockcor J. P.; Gottfries J.; Moritz T.; Trygg J. Anal. Chem. 2008, 80, 115–122. [DOI] [PubMed] [Google Scholar]
- McDougle C. J.; Black J. E.; Malison R. T.; Zimmerman R. C.; Kosten T. R.; Heninger G. R.; Price L. R. Arch. Gen. Psychiatry 1994, 51, 713–719. [DOI] [PubMed] [Google Scholar]
- Lovenberg W.; Bruckwick E. A.; Hanbauer I. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockhold R. W.; Oden G.; Ho I. K.; Andrew M.; Farley J. M. Brain Res. Bull. 1991, 27, 721–723. [DOI] [PubMed] [Google Scholar]
- Garg U. C.; Turndorf H.; Bansinath M. Neuroscience 1993, 57, 467–472. [DOI] [PubMed] [Google Scholar]
- Niwa T.; Takeda N.; Maeda K.; Shibata M.; Tatematsu A. Clin. Chim. Acta 1988, 173, 127–138. [DOI] [PubMed] [Google Scholar]
- Costigan M. G.; Yaqoob M.; Lindup W. E. Nephrol. Dial. Transplant. 1996, 11, 803–807. [DOI] [PubMed] [Google Scholar]
- Okuda S.; Nishiyama N.; Saito H.; Katsuki H. J. Neurochem. 1998, 70, 299–307. [DOI] [PubMed] [Google Scholar]
- Fallstrom S.-P.; Lindblad B.; Steen G. Acta Paediatr. (Stockholm) 1981, 70, 315–320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.