

Abstract
We present a novel excitation-emission multiplexed fluorescence lifetime microscopy (FLIM) method that surpasses current FLIM techniques in multiplexing capability. The method employs Fourier multiplexing to simultaneously acquire confocal fluorescence lifetime images of multiple excitation wavelength and emission color combinations at 44,000 pixels/sec. The system is built with low-cost CW laser sources and standard PMTs with versatile spectral configuration, which can be implemented as an add-on to commercial confocal microscopes. The Fourier lifetime confocal method allows fast multiplexed FLIM imaging, which makes it possible to monitor multiple biological processes in live cells. The low cost and compatibility with commercial systems could also make multiplexed FLIM more accessible to biological research community.
OCIS codes: (170.2520) Fluorescence microscopy; (170.6280) Spectroscopy, fluorescence and luminescence
1. Introduction
Fluorescence lifetime microscopy (FLIM) is a powerful tool for functional imaging of biochemical environments and molecular interactions in biological specimens [1]. Existing FLIMs techniques can be grouped into two categories: full-frame 2D FLIM based on gated or modulated cameras, and point-scanning 3D FLIM.
Full-frame 2D FLIM techniques have faster frame rates than point-scanning FLIM, but the gated or modulated cameras carry a high instrument cost for full-frame FLIM systems. While recent development of CMOS based single-photon avalanche diodes [2, 3] or CCD/CMOS hybrid lock-in imagers [4] may provide a low-cost alternative for image detection in the future, currently the numbers of pixels reported in such detectors are limited compared to gated cameras. Furthermore because camera-based epi-detection does not have native depth-section ability, for 3D imaging of biological specimen, sophisticated designs such as structured illumination [5], spinning disk confocal microscopy [6] or select plane illumination [7] are needed with full-frame FLIM systems.
Point scanning FLIM is compatible with confocal or two photon microscopy that have native depth sectioning capabilities for 3D lifetime imaging. Both the frequency domain [8] and time domain [9] fluorescence lifetime methods have been widely used in point-scanning FLIM. Both methods have their pros and cons, and the choice of a FLIM method often has to compromise between speed, accuracy and cost:
-
(1)
The time-domain method needs less photon counts to achieve the same lifetime accuracy [10], however, it has a higher cost due to the picosecond pulsed laser source and time-correlated single photon counting (TCSPC) detection system needed. The TCSPC system has a limited count rate, which becomes the bottleneck for increasing imaging speed. Multi-channel TCSPC can alleviate the count-rate limit at the expense of higher instrument cost.
-
(2)
The frequency-domain method is highly versatile in hardware and can be the lowest-cost system in all FLIM approaches [11, 12]. The excitation source of the frequency domain method can be either low-cost modulated CW source or pulsed laser/LED source. The detector can be in either the single photon counting mode or analog mode, which does not suffer from the count-rate bottleneck in TCSPC. However, due to limitations in light source and RF hardware, existing frequency domain FLIM systems cannot easily perform fast measurements at multiple modulation frequencies and typically only operate at one or two modulation frequencies. Single or dual-frequency FLIM can only detect trends of lifetime change [13, 14] and are not sufficient for analyzing complex decay models. While multi-frequency measurements can be performed with pulsed excitation utilizing higher order harmonics of the fundamental pulse frequency [10, 15], such systems require picosecond pulsed light source similar in cost to those used in time-domain systems.
In recent years, FLIM has seen significant improvement in instrumentations including light sources [16, 17], detectors [3, 18, 19] and experimental and data analysis schemes [12, 15, 20]. However one limitation persists in FLIM, which is the inability to perform paralleled detection on multiple excitation wavelengths. Fluorescence imaging at multiples excitation wavelengths, FLIM or not, are generally carried out in sequential excitation multiplexing, where different excitation wavelengths are switched on one at a time and the sample is imaged sequentially. As the result, the total image acquisition speed is decreased. As the imaging speed of FLIM is already slower than intensity-only imaging method because it needs higher photon counts to extract quantitative information on the fluorescence decay, further slowing down the imaging speed with sequential excitation multiplexing may affect FLIM’s ability to image live samples. Thus, although emission-multiplexed FLIM systems are frequently reported in literature, only few excitation-multiplexed FLIM systems exist [17, 21].
Parallel excitation multiplexing FLIM had been previously demonstrated with multiple electro-optical modulators, each modulating an excitation laser at a unique frequency [22, 23]. The parallel multiplexing is achieved by the frequency multiplexing principle similar to Frequency Division Multiple Access (FDMA). However, in previous systems only one modulation frequency was measured per excitation line, which is not sufficient for complex decay analysis.
Our previous work [24] demonstrated that the Fourier transform fluorescence lifetime spectroscopy allows rapid parallel excitation-multiplexed lifetime measurements at continuous frequency points by modulating multiple laser lines with a fast frequency-sweeping Fourier transform interferometer. The parallel excitation-multiplexing is achieved by modulating multiple laser lines via interference, which create wavelength-encoded modulation frequency . The velocity of the interferometer delay line v changes linearly, creating wavelength-encoded modulations sweeping from 0 to >150 MHz in 23 μs, allowing nanosecond lifetime measurements with continuous frequency sampling at multiple excitation wavelengths simultaneously at 44,000 sample/s.
A Fourier lifetime confocal microscope based on the Fourier transform fluorescence lifetime technique would be able to perform raster-scanning lifetime measurements at a high pixel rate of 44,000 pixels/sec, suitable for volumetric confocal laser-scanning imaging in live specimens. With multiple emission detectors, the system would be able to simultaneously image all possible excitation-emission channels. The approach would dramatically increase FLIM’s spectral multiplexing capability. Furthermore, the Fourier lifetime confocal microscope would use low-cost CW lasers and regular analog PMTs commonly used in confocal microscopes, so that it could potentially be implemented as an add-on to existing confocal microscopes with minimal additional cost. The continuous multi-frequency measurements on fluorescence lifetime responses could facilitate analysis of complex fluorescence decay behaviors [25].
However, the previous Fourier lifetime fluorometer is not compatible with confocal imaging due to the following reasons: (1) The fluorometer acquires lifetime data via multichannel GHz digitalization. The data rate is unsustainable and impractical in imaging mode at multiple excitation-emission wavelengths combinations; (2) Small beam walk-off exists in the laser excitation of the fluorometer, which is caused by the single-pass polygon optical delay line used by the interferometer. The unwanted beam walk-off leads to lifetime and intensity artifacts in confocal images of heterogeneous samples.
Furthermore, as FLIM needs thousands of photons per pixel to accurately measure lifetimes, the imaging speed of FLIM is often limited by the brightness of fluorescent biological samples, not the hardware acquisition speed. Theoretical analysis and experimental tests are needed to prove that the full hardware speed of a lifetime spectroscopic method can be realized in live imaging of biological samples.
The purpose of this paper is to address these challenges and questions, and to report a Fourier lifetime confocal microscopy instrument as a novel, accurate and low-cost solution to excitation-emission multiplexed 3D FLIM.
2. Microscope system
2.1 System scheme: eliminate beam walk-off and perform confocal laser scanning imaging
The schematic of the microscope is shown in Fig. 1(a). Similar to the fluorometer we previously reported, the system has four excitation laser lines (405, 488, 561 and 640 nm) to perform parallel multi-excitation lifetime imaging. All lasers are directed into a Michelson interferometer with a scanning optical delay line, which produces frequency sweeping interferometric modulations on all laser lines with modulation frequencies inversely proportional to laser wavelengths (Figs. 1(b) and 1(c)).
Fig. 1.
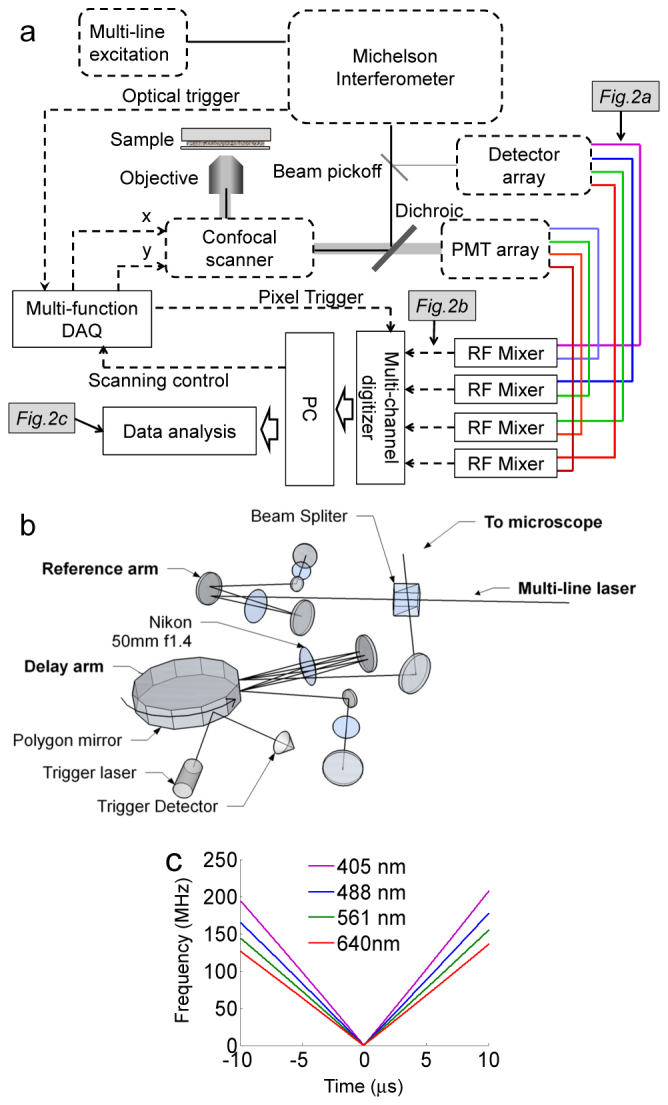
Schematics of Fourier confocal FLIM. (a) Schematic of the optical and signal acquisition system. (b) Fourier transform interferometer with double passed delay line. The interferometer has a 23 μs scan repetition time and no beam walk-off. (c) Wavelength-dependent interference modulation sweep twice between 0 to 150 MHz within 23 μs.
To eliminate beam walk-off, the delay line in the confocal microscope employs a double-passed geometry (Fig. 1(b)) [26] instead of the single-pass geometry used previously. The change also removes beam spot movements caused by mechanical vibrations of the spinning polygon mirror. A 48-facet 55,000 rpm polygon mirror scanner (23 μs per facet, Lincoln Lasers) is used in the double-pass optical delay line. An IR laser at 780 nm wavelength is placed at the side of the polygon mirror scanner in order to provide an optical trigger signal of the facet spinning.
Modulation frequencies of visible lasers sweep from 0~150 MHz twice within 23 μs (Fig. 1(c)). The modulation frequency is highly repeatable due to the small motor speed instability (0.03%), which corresponds to frequency variations less than 45 kHz during the sweep. The frequency instability does not cause detectable lifetime artifacts because: (1) the frequency instability is small in comparison to the typical nanosecond lifetime and the 1 MHz detection bandwidth, and (2) the emission signal is demodulated in reference to the real-time laser modulation signal in a way similar to lock-in detection.
The modulated laser output from the interferometer is split by a beam pick-off. A small portion of the output is sent to a series of amplified photodetectors to monitor the interference modulations of individual excitation lasers. The majority of the output is directed to a confocal microscope built on an inverted microscope frame (Olympus IX71). The excitation beam passes through a pinhole and is focused onto the sample through a 60X NA1.4 oil objective lens. A pair of close-loop x-y galvanometer mirrors (Thorlabs) scans the laser focus across the sample. Confocal scanning is performed in the one-directional scan mode with fast pull back. Typically a 400-by-400 pixel scan at 44,000 pixel/s takes a total time of 4 seconds. The typical excitation power used for live cell imaging is 10 to 200 μW per laser line. Fluorescence emission is spatially filtered by the same pinhole and separated from the excitation by a quad-band dichroic mirror (Semrock Di01-R405/488/561/635). The emission is further split into multiple spectral bands and detected by multiple photomultiplier tube detectors (PMTs, Hamamatsu H7422). The typical PMT gain setting is 500 to 600 Volts (maximum gain setting is 1000 Volts).
2.2 Analog-digital hybrid signal processing enable sustainable 4-by-4 lifetime EEM imaging
The Fourier lifetime fluorometer we previously developed records high frequency excitation and fluorescence modulations at their native frequencies simultaneously with a GHz digitizer, creating an unsustainable data rate and an extreme large volume of data that needs to be processed digitally. To apply the Fourier lifetime method in a continuous-imaging mode, an analog-digital hybrid signal processing and lifetime analysis method was developed for Fourier lifetime confocal imaging. The new method decreases the data rate per channel and allows simultaneous acquisition of lifetime imaging in up to 4-by-4 excitation-emission wavelength combinations, which form a lifetime excitation-emission matrix (EEM) image.
To process a lifetime information in the ijth Ex-Em channel, the signal of the jth emission spectral channel (Fig. 2(a)) is heterodyne down-mixed with the delayed reference modulation signal of the ith laser line (Fig. 2(a)) by an RF mixer (Mini-Circuits ZX05-1L-S + ) (Fig. 2(b)). Because the modualtion of ith laser modulation is linearly sweeping, the time delay between excitation and emission signals corresponds to a constant frequency shift . The mixed signal is
| (1) |
The signal in the jth emission channel is excited by multiple excitation lines
| (2) |
where and are the modulation and phase of the fluorescence lifetime frequency response of the kjth Ex-Em channel, and is the brightness of the Ex-Em channel. The signal after mixer is
| (3) |
Because when , after low-pass filtering, only emission signal that is excited by the ith laser line remains
| (4) |
The mixing and low-pass filtering procedure allows all combinations of Ex-Em channels to be acquired independently in parallel.
Fig. 2.
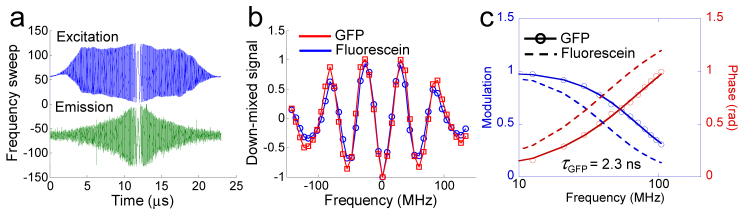
Analog-digital hybrid data processing of Fourier confocal FLIM. (a) Frequency-sweeping laser excitation and fluorescence emission signals from the sample. (b) Down-mixed emission signals. (c) Measured modulation and phase signal of GFP in comparison to those of fluorescein.
The carrier frequency of the down-mixed signal is set at 200~300 kHz by adjusting the time delay between the emission and reference signals. An increase in the differential delay between the emission signal and the reference signal will increase the down-mixed frequency, and vice versa.
The down-mixing process allows the digitalization rate to drop from the previous 750 MHz per Ex-Em channel to 2 MHz, which is sustainable with a typical PC. In our system, up to 4-by-4 Ex-Em channels are digitalized by a multi-channel high-speed digitizer (NI 5752&7962) at 2 MHz. 40 frequency points are sampled during each round trip frequency sweep and treated as a single pixel multi-frequency measurement. The confocal scanning is controlled by a multi-function DAQ (NI 6115). Confocal scanning and data acquisition are synchronized by the optical trigger signal from the polygon scanner.
The RF response of each Ex-Em channels are corrected by measuring established frequency-domain lifetime standards [14, 27]. Coumarin 6 (2.5 ns), fluorescein (3.9 ns) and Rhodamin B (1.8 ns) are used for Ex-Em channels with 405 nm, 488 and 561 nm excitations respectively [28]. The correction factor is calculated by
| (5) |
where refers to the Hilbert transform, and are modulation and phase responses of the lifetime standard calculated from its known lifetime. The correction factor needs to be re-measured if the optical system is realigned or any electronic component is changed.
3. Multi-label 3D lifetime imaging
3.1 Lifetime analysis method
The modulation and phase response of an Ex-Em channel m(ω), ϕ(ω) is recovered by correcting the down-mixed signal at each pixel with the prerecorded correction factor.
| (6) |
where is the complex phasor [29]. The intensity-weighted phasor response of each pixel is fitted with appropriate frequency domain fluorescence lifetime models [24, 25]. Typically the intensity weighed phasors at 40 frequency points, measured within a 20 μs frequency-sweep, is fitted with a single-exponential complex model to extract the lifetime at each pixel (Fig. 2(c)). The emission intensity image is calculated as the root square power of the down-mixed signal for each pixel. All images presented in this paper are acquired at full pixel rate (44,000 pixels/sec) without averaging.
3.2 3D confocal lifetime image of triple stained tissue: observe FRET in co-localized labels
To demonstrate the multiplexed 3D lifetime imaging capability of the microscope, cryostat section of mouse kidney stained with Alexa Fluor 488 wheat germ agglutinin, Alexa Fluor 568 phalloidin and DAPI (FluoCells prepared slide #3, Invitrogen) was scanned in 0.2-μm z-step with three lasers at 405, 488 and 561 nm simultaneously. Blue, green and red emissions are detected by three PMTs and down-mixed with three laser reference signals, respectively.
Figure 3 shows a confocal image slice (800x800 pixels, 0.15 μm per pixel) of 3D triple-channel confocal lifetime images of the above mentioned mouse kidney slide. Full 3D image set is provides in Media 1 (2.1MB, MP4) . Triple-channel fluorescence intensity (Fig. 3(a)) and lifetime images (Figs. 3(b)–3(d), false-colored in a 0 ns to 3 ns color scale) were acquired within a single scan. DAPI (blue in Fig. 3(a)) labels all nuclei. Alexa488-WGA (green in Fig. 3(a)) specifically stains the apical and basal area of proximal tubules, and basement membrane side of distal tubules [30]. Alexa568 phalloidin (red in Fig. 3(a)) stains actin filament of all cells. DAPI (blue in Fig. 3(a)) changes its lifetimes depending on its binding state to DNA, thus a highly heterogeneous lifetime distribution was observed (Fig. 3(b)). Alexa488 lifetimes (Fig. 3(c)) in the basal area of proximal tubules are shorter than lifetimes in the apical area of proximal tubules and basement membrane side of distal tubules. The lifetime shortening is likely due to co-localization of Alexa488 WGA and Alexa568 phalloidin, which generates Föster resonant energy transfer (FRET). The co-localization and FRET effect are more evident in Fig. 3(e), which show a cross section of the 3D confocal images. The lifetime of Alexa568 is uniform across the sample (Fig. 3(d)).
Fig. 3.

3D confocal lifetime image of triple-labeled mouse kidney slice. (a) False color intensity image of an x-y plane slice. (b) Lifetime x-y slice image of DAPI. (c) Lifetime x-y slice image of Alexa488 WGA. (d) Lifetime x-y slice image of Alexa568 phalloidin. (e) Sample x-z section. Top row: false color intensity image, showing co-localization of Alexa488 WGA and Alexa568 phalloidin at the basal area of proximal tubules. 2nd row: lifetime image of DAPI. 3rd row: lifetime image of Alexa488 WGA, showing decreased lifetime in the basal area of proximal tubules due to FRET between Alexa488 and Alexa568. Bottom row: lifetime image of Alexa568 phalloidin. The image size is 800-by-800 pixels with 0.15 μm pixel size, acquired at the full 44,000 pixel/s speed and processed without average. The full 3D data set is shown in Media 1 (2.1MB, MP4) . Scale bar 20 μm.
4. Sensitivity and accuracy analysis
Lifetime imaging of live cells with fluorescent protein labels is much more challenging than imaging stained tissue. On one hand, live cell imaging methods need to be fast in order to detect dynamic changes and avoid artifacts due to cell movements. On the other hand, the image speed is limited by two factors: (1) genetically labeled live cells are much dimmer than stained tissue, and (2) fluorescence proteins in general are less photostable than dye stains and are prone to photobleaching. As FLIM require a minimum number of photons to accurately resolve lifetime, the speed of live cell FLIM is often not limited by the hardware but by the photon budget available from the sample.
The imaging speed of a FLIM method is affected by two parameters: (1) the minimum number of photons needed to accurately measure the lifetime, which can be theoretically calculated, and (2) the available fluorescence photon flux at the detector, which depends on many factors, including the excitation power without excess photobleaching, the maximum count rate of the detector, and the detection duty cycle.
Our analysis shows that Fourier lifetime confocal imaging method has a typical photon number requirement similar to all frequency domain lifetime method using modulated CW sources, and the photon flux under typical confocal live cell imaging condition is higher than the minimal requirement.
4.1 F-number analysis of minimal photon counts for lifetime measurements
Following the methods in Ref [10], we calculated that in an ideal shot-noise limited situation, the 0-150 MHz multi-frequency heterodyne lifetime measurement and the complex phasor lifetime fitting used in our approach requires a similar number of photons as the tradition single-frequency homodyne frequency domain FLIM to achieve the same accuracy level.
Figure 4 plots the F-factor of lifetime fitting for the two methods. The figure of merit F of a lifetime method is defined as
| (7) |
where τ is the lifetime, Δτ is the lifetime accuracy, N is the number of detected photons and ΔN is the photon RMS noise. The F-factor is independent to the number of photons collected. With the multi-frequency heterodyne frequency-domain lifetime method, the photon efficiency is similar to the conventional single frequency homodyne frequency domain lifetime method, as shown in Fig. 4. The complex phasor fitting requires slightly less photons than modulation or phase-only fittings because both modulation and phase information are used. For fluorescence lifetimes in the range of 1~4 ns, approximately 5,000~10,000 photons are needed to resolve fluorescence lifetimes within 10% accuracy.
Fig. 4.
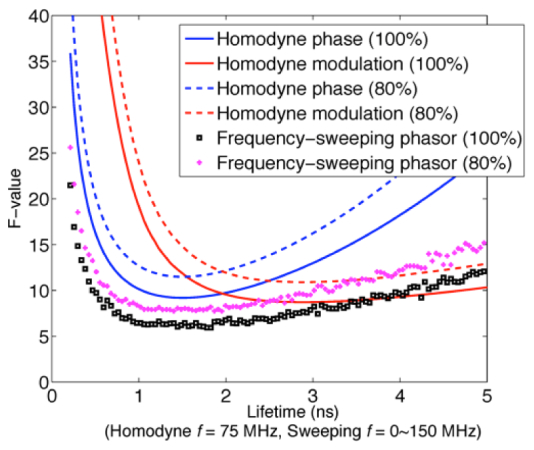
Monte Carlo simulation of F factor for heterodyne Fourier confocal FLIM compared to single frequency homodyne frequency domain lifetime method, at 100% and 80% excitation modulation depth. For 1~4 ns lifetimes Fourier confocal FLIM requires 5,000~1,0000 photons to achieve 10% lifetime accuracy. Homodyne lifetime F-factor curves are recalculated with same parameters as in Ref [10].
FLIM imaging speed is often limited by the detected photon flux not by hardware speed. In time-domain FLIM, the photon flux is likely limited by the power and repetition rate of the pulsed excitation, the TCSPC count rate or loss of photons outside of the detection time-gate.
In comparison to time-domain lifetime methods whose F-factors are lower, frequency domain methods in general require more photons to resolve lifetime to the same accuracy. Thus the photon flux requirement is higher for frequency domain FLIM operating at the same imaging speed.
Fortunately, frequency-domain FLIM using modulated CW excitation and analog detectors can detect a higher photon flux than time-domain FLIM. The higher photon flux is due to three factors: (1) CW laser has a higher excitation duty-cycle than pulsed laser and can excite more photons without saturation, (2) analog PMTs can detect continuous photon flux without saturation, and (3) analog PMTs operate under constant gain and high detection duty-cycle.
With the short-pulsed excitation, the emission photon flux is limited by excitation saturation. An 80 MHz pulse excitation can produce at most a photon flux of 80 MHz from a single fluorophore, which is about 12~32% of the CW saturation photon flux, estimated to be 250 MHz to 1GHz considering the typical 1-4 ns lifetime. With a typical 50 μW excitation power, the CW excitation intensity in our system is ~5% of the saturation intensity. Experimentally less than 1% of GFP bleaching was observed per scan under 50 μW excitation power. The typical detected photon flux in our system is about 109 photons/sec. The experimentally measured photon flux matches well with an photon flux estimation, assuming cells expressing GFP at a typical concentration of 10 μM (approximately 10 times brighter than cell autofluorescence [31] and can be easily achieved with a good promoter [32]), under typical confocal experimental conditions (50 μW excitation power, 1.4 NA objective, 10% total photon collection efficiency and 0.4 PMT quantum efficiency). The photon flux, according to Fig. 4, is sufficient for accurately resolving lifetime within 20 μs data acquisition.
In Fourier confocal lifetime imaging, the high photon flux is detected without saturation by setting the PMT gain voltage at 60% or less of its full range. In comparison, in TCSPC based fluorescence lifetime methods, the photon rate per channel has to be limited to 1% of the laser repetition rate to avoid pulse pile up [33]. Under an 80-MHz pulsed laser, a high-cost 16-channel TCSPC has a maximum count rate of 12.8 MHz, much less than the photon flux typical in our system.
Many FLIM system use gain-gated or gain-modulated detection, which rejects fluorescence photons outside of the detection time-gate and causes photon loss. In Fourier confocal lifetime imaging, PMTs are running under constant gain with 90% detection duty cycle. The only photon loss happens at the two ends of a facet scan where the edges of a facet rotate across the incident laser beam, which accounts for a 10% detection duty cycle loss.
It is worth noting that the above photon flux advantages apply to all frequency-domain FLIM methods that use modulated CW excitation and analog PMTs. However, in the past, due to limitations in optical modulators, such frequency-domain FLIM systems suffer from the inability to perform fast multi-frequency measurements. Single or dual-frequency FLIM, although can detect trends of lifetime change [13, 14], are not sufficient for analyzing complex decay models. The Fourier confocal FLIM breaks this hardware barrier by using an all-optical method to modulate laser intensity into a fast frequency sweep. The lifetime measurement is inherently multi-frequency, allowing the analysis of complex decay models such as multi-exponential decay and excited state reaction [25].
4.2 Live cell imaging experiments: shot-noise limited performance
Equation (7) predicts that if a FLIM system is photon shot-noise limited, the lifetime accuracy should be inversely proportional to the square root of the emission photon number or the square root of the excitation power. Experimental study was performed to prove that the Fourier confocal lifetime microscope is indeed shot-noise limited. Live HeLa cells expressing GFP were imaged repeatedly with increasing laser excitation power. As shown in Fig. 5, the width of GFP lifetime pixel histogram decreases with the increasing laser power (Fig. 5(a)). The standard deviation of measured GFP fluorescence lifetime is indeed inversely proportional to the square root of excitation laser power in Fig. 5(b), demonstrating that the microscope system and the lifetime analysis are limited by photon shot noise. GFP lifetime images at 200 μW and 30 μW are shown in Figs. 5(c) and 5(d), with the low excitation power image showing more pronounced pixel lifetime variations. The system achieved 10% lifetime accuracy in live imaging of GFP with 50 μW excitation power. Based on the average output level from the PMT, we estimated that the average photon count is 10,000 per pixel under this condition, consistent with previous numerical estimation. Considering the lifetime data analysis uses a 2-by-2 pixel data smoothing, the experimental lifetime accuracy matches the theoretically estimated lifetime accuracy within a factor of 2. The discrepancy in signal-to-noise is likely introduced by the reference laser modulation signal in the down mixing process, because reference signals are detected laser interference signal, which contain additional shot noise.
Fig. 5.
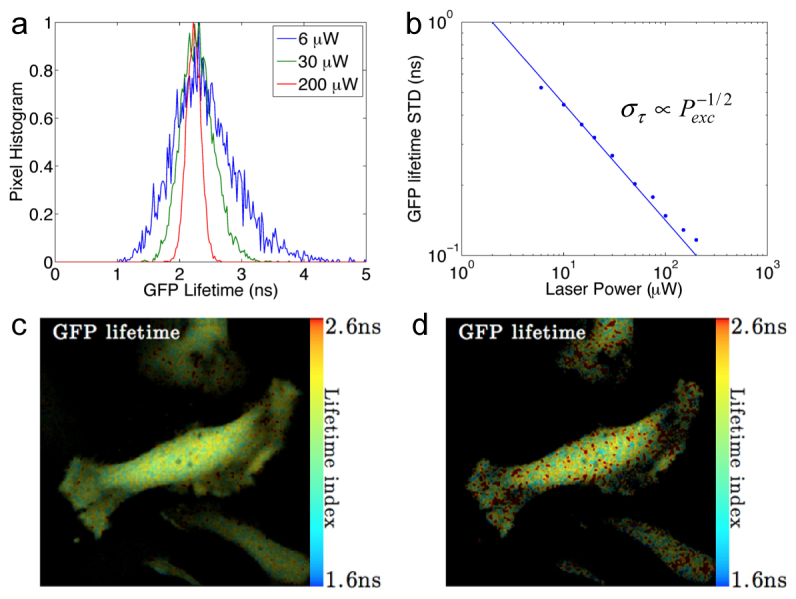
GFP lifetime accuracy in live cell imaging under different laser excitation power. (a) Histograms of GFP lifetime distribution narrow with increasing excitation power. (b) The standard deviation of GFP lifetime is inversely proportional to the square root of excitation power. (c) GFP lifetime image of HeLa cells at 200 μW excitation power. (d) GFP lifetime image of HeLa cells at 30 μW excitation power. GFP fluorescence lifetime was measured at 2.25 ± 0.16 ns at 200 μW excitation power and 2.3 ± 0.3 ns at 30 μW excitation power. Image size is 400-by-400 pixels with 0.3 μm pixel size. The image set was acquired at full 44,000 pixel/s speed. Lifetimes were fitted from data smoothed by a 2-by-2 pixel wide Gaussian window. The intensity image was processed at the original resolution without smoothing.
5. Discussion
The Fourier lifetime confocal imaging method provides a low cost solution to implement excitation-emission multiplexed FLIM as an add-on to confocal microscopes that are widely available in most biological research facilities. The multi-line CW laser sources in a standard confocal microscope can be converted to modulated light sources by the addition of the interferometer. Analog PMTs in standard confocal microscopes can be used without modification. The analog-digital data processing system uses readily available inexpensive off-the-shelf components. We estimate the cost different between our multi-channel FLIM confocal system and a standard multi-channel confocal microscope is about $30,000, which enables a confocal microscope to perform FLIM in 4-by-4 multiplexed excitation-emission channels simultaneously.
The Fourier lifetime method is universally versatile in spectral configuration. Figure 6 shows lifetime images of TagBFP and mCherry expressing HeLa cells, demonstrating that the system can achieve satisfactory lifetime accuracies on the full spectrum of fluorescence proteins. Our multiplexed FLIM system had successfully being applied to FRET study of protein conformation changes [34].
Fig. 6.

Lifetime images of HeLa cells expressing fluorescence proteins of blue and red colors. (a) TagBFP, 50 μW 405 nm excitation, 457 ± 20 nm emission. The excitation power and lifetime accuracy are lower because TagBFP is less photostable than GFP and mCherry; (b) mCherry, 200 μW 561 nm excitation, 593 ± 20 nm emission; (c) Lifetime histograms of three FPs; (d) Bar chart showing lifetime accuracy of three FPs. TagBFP: 2.79 ± 0.25 ns; GFP: 2.25 ± 0.16 ns; mCherry 1.80 ± 0.09 ns. All lifetimes match with literature values [35–37]. All images were acquired at the full 44,000 pixel/s speed. Lifetimes were fitted from data smoothed by a 2-by-2 pixel wide Gaussian window. The intensity images were processed at the original resolution. Scale bar 20 μm.
In addition to its fast-speed, low cost and spectral versatility, the Fourier lifetime confocal imaging method offers the unique advantage of excitation-emission spectral multiplexing. It allows application of multi-line excitation onto the sample simultaneously while still being able to differentiate between the excitation contributions from each line, which is not possible with any other fluorescence microscopy methods. With our method, imaging of multiple Ex-Em channels, FLIM or not, can be performed as fast and accurate as single Ex-Em channel imaging.
With its low-cost, multiplexed, depth-sectioned live fluorescence lifetime imaging capability, the Fourier lifetime confocal imaging method could promote the use of multiplexed imaging and FLIM in the study of biomolecular interactions inside living cells and tissue.
Acknowledgments
The authors thank Guang Yao for helping with cell cultures and transient transfection, and Weibin Zhou for helping interpreting the kidney slice lifetime images. This research was supported by NIH grants R00EB008737 and R01EB015481 to L. P.
References and links
- 1.A. Periasamy and R. M. Clegg, eds., FLIM Microscopy in Biology and Medicine (CRC, 2009). [Google Scholar]
- 2.Li D. U., Arlt J., Richardson J., Walker R., Buts A., Stoppa D., Charbon E., Henderson R., “Real-time fluorescence lifetime imaging system with a 32 × 32 013μm CMOS low dark-count single-photon avalanche diode array,” Opt. Express 18(10), 10257–10269 (2010). 10.1364/OE.18.010257 [DOI] [PubMed] [Google Scholar]
- 3.Li D. D. U., Arlt J., Tyndall D., Walker R., Richardson J., Stoppa D., Charbon E., Henderson R. K., “Video-rate fluorescence lifetime imaging camera with CMOS single-photon avalanche diode arrays and high-speed imaging algorithm,” J. Biomed. Opt. 16(9), 096012 (2011). 10.1117/1.3625288 [DOI] [PubMed] [Google Scholar]
- 4.Esposito A., Gerritsen H. C., Oggier T., Lustenberger F., Wouters F. S., “Innovating lifetime microscopy: a compact and simple tool for life sciences, screening, and diagnostics,” J. Biomed. Opt. 11(3), 034016 (2006). 10.1117/1.2208999 [DOI] [PubMed] [Google Scholar]
- 5.Cole M. J., Siegel J., Webb S. E. D., Jones R., Dowling K., French P. M. W., Lever M. J., Sucharov L. O. D., Neil M. A. A., Juskaitis R., Wilson T., “Whole-field optically sectioned fluorescence lifetime imaging,” Opt. Lett. 25(18), 1361–1363 (2000). 10.1364/OL.25.001361 [DOI] [PubMed] [Google Scholar]
- 6.Grant D. M., McGinty J., McGhee E. J., Bunney T. D., Owen D. M., Talbot C. B., Zhang W., Kumar S., Munro I., Lanigan P. M. P., Kennedy G. T., Dunsby C., Magee A. I., Courtney P., Katan M., Neil M. A. A., French P. M. W., “High speed optically sectioned fluorescence lifetime imaging permits study of live cell signaling events,” Opt. Express 15(24), 15656–15673 (2007). 10.1364/OE.15.015656 [DOI] [PubMed] [Google Scholar]
- 7.Greger K., Neetz M. J., Reynaud E. G., Stelzer E. H. K., “Three-dimensional fluorescence lifetime imaging with a single plane illumination microscope provides an improved signal to noise ratio,” Opt. Express 19(21), 20743–20750 (2011). 10.1364/OE.19.020743 [DOI] [PubMed] [Google Scholar]
- 8.So P. T. C., French T., Yu W. M., Berland K. M., Dong C. Y., Gratton E., “Time-resolved fluorescence microscopy using two-photon excitation,” Bioimaging 3(2), 49–63 (1995). [DOI] [Google Scholar]
- 9.Becker W., Bergmann A., Hink M. A., König K., Benndorf K., Biskup C., “Fluorescence lifetime imaging by time-correlated single-photon counting,” Microsc. Res. Tech. 63(1), 58–66 (2004). 10.1002/jemt.10421 [DOI] [PubMed] [Google Scholar]
- 10.Elder A., Schlachter S., Kaminski C. F., “Theoretical investigation of the photon efficiency in frequency-domain fluorescence lifetime imaging microscopy,” J. Opt. Soc. Am. A 25(2), 452–462 (2008). 10.1364/JOSAA.25.000452 [DOI] [PubMed] [Google Scholar]
- 11.Booth M. J., Wilson T., “Low-cost, frequency-domain, fluorescence lifetime confocal microscopy,” J. Microsc. 214(1), 36–42 (2004). 10.1111/j.0022-2720.2004.01316.x [DOI] [PubMed] [Google Scholar]
- 12.Colyer R. A., Lee C., Gratton E., “A novel fluorescence lifetime imaging system that optimizes photon efficiency,” Microsc. Res. Tech. 71(3), 201–213 (2008). 10.1002/jemt.20540 [DOI] [PubMed] [Google Scholar]
- 13.Gadella T. W. J., Jr, Jovin T. M., Clegg R. M., “Fluorescengce lifetime imaging microscopy (FLIM) - spatial resolution of microstructures on the nanosecond time-scale,” Biophys. Chem. 48(2), 221–239 (1993). 10.1016/0301-4622(93)85012-7 [DOI] [Google Scholar]
- 14.Hanley Q. S., Subramaniam V., Arndt-Jovin D. J., Jovin T. M., “Fluorescence lifetime imaging: multi-point calibration, minimum resolvable differences, and artifact suppression,” Cytometry 43(4), 248–260 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Chen H. T., Gratton E., “A practical implementation of multifrequency widefield frequency-domain fluorescence lifetime imaging microscopy,” Microsc. Res. Tech. 76(3), 282–289 (2013). 10.1002/jemt.22165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herman P., Maliwal B. P., Lin H. J., Lakowicz J. R., “Frequency-domain fluorescence microscopy with the LED as a light source,” J. Microsc. 203(2), 176–181 (2001). 10.1046/j.1365-2818.2001.00943.x [DOI] [PubMed] [Google Scholar]
- 17.Owen D. M., Auksorius E., Manning H. B., Talbot C. B., de Beule P. A. A., Dunsby C., Neil M. A. A., French P. M. W., “Excitation-resolved hyperspectral fluorescence lifetime imaging using a UV-extended supercontinuum source,” Opt. Lett. 32(23), 3408–3410 (2007). 10.1364/OL.32.003408 [DOI] [PubMed] [Google Scholar]
- 18.Elson D. S., Munro I., Requejo-Isidro J., McGinty J., Dunsby C., Galletly N., Stamp G. W., Neil M. A. A., Lever M. J., Kellett P. A., Dymoke-Bradshaw A., Hares J., French P. M. W., “Real-time time-domain fluorescence lifetime imaging including single-shot acquisition with a segmented optical image intensifier,” New J. Phys. 6, 180 (2004). 10.1088/1367-2630/6/1/180 [DOI] [Google Scholar]
- 19.Fereidouni F., Reitsma K., Gerritsen H. C., “High speed multispectral fluorescence lifetime imaging,” Opt. Express 21(10), 11769–11782 (2013). 10.1364/OE.21.011769 [DOI] [PubMed] [Google Scholar]
- 20.Colyer R. A., Siegmund O. H. W., Tremsin A. S., Vallerga J. V., Weiss S., Michalet X., “Phasor imaging with a widefield photon-counting detector,” J. Biomed. Opt. 17(1), 016008 (2012). 10.1117/1.JBO.17.1.016008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laurence T. A., Kong X. X., Jäger M., Weiss S., “Probing structural heterogeneities and fluctuations of nucleic acids and denatured proteins,” Proc. Natl. Acad. Sci. U.S.A. 102(48), 17348–17353 (2005). 10.1073/pnas.0508584102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carlsson K., Liljeborg A., “Confocal fluorescence microscopy using spectral and lifetime information to simultaneously record four fluorophores with high channel separation,” J. Microsc. 185(1), 37–46 (1997). 10.1046/j.1365-2818.1997.1450705.x [DOI] [Google Scholar]
- 23.Carlsson K., Liljeborg A., “Simultaneous confocal lifetime imaging of multiple fluorophores using the intensity-modulated multiple-wavelength scanning (IMS) technique,” J. Microsc. 191(2), 119–127 (1998). 10.1046/j.1365-2818.1998.00362.x [DOI] [PubMed] [Google Scholar]
- 24.Zhao M., Peng L., “Multiplexed fluorescence lifetime measurements by frequency-sweeping Fourier spectroscopy,” Opt. Lett. 35(17), 2910–2912 (2010). 10.1364/OL.35.002910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao M., Huang R., Peng L. L., “Quantitative multi-color FRET measurements by Fourier lifetime excitation-emission matrix spectroscopy,” Opt. Express 20(24), 26806–26827 (2012). 10.1364/OE.20.026806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oldenburg A. L., Reynolds J. J., Marks D. L., Boppart S. A., “Fast-Fourier-domain delay line for in vivo optical coherence tomography with a polygonal scanner,” Appl. Opt. 42(22), 4606–4611 (2003). 10.1364/AO.42.004606 [DOI] [PubMed] [Google Scholar]
- 27.J. R. Lakowicz, “Color effects and background fluorescence,” in Principles of Fluorescence Spectroscopy, 3rd ed. (Springer, 2006). [Google Scholar]
- 28.I. S. S. Inc, “Lifetime data of selected fluorophores”, retrieved http://www.iss.com/resources/reference/data_tables/LifetimeDataFluorophores.html
- 29.Digman M. A., Caiolfa V. R., Zamai M., Gratton E., “The phasor approach to fluorescence lifetime imaging analysis,” Biophys. J. 94(2), L14–L16 (2008). 10.1529/biophysj.107.120154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holthöfer H., “Lectin binding sites in kidney. a comparative study of 14 animal species,” J. Histochem. Cytochem. 31(4), 531–537 (1983). 10.1177/31.4.6827083 [DOI] [PubMed] [Google Scholar]
- 31.Niswender K. D., Blackman S. M., Rohde L., Magnuson M. A., Piston D. W., “Quantitative imaging of Green Fluorescent Protein in cultured cells: comparison of microscopic techniques, use in fusion proteins and detection limits,” J. Microsc. 180(2), 109–116 (1995). [DOI] [PubMed] [Google Scholar]
- 32.Furtado A., Henry R., “Measurement of green fluorescent protein concentration in single cells by image analysis,” Anal. Biochem. 310(1), 84–92 (2002). 10.1016/S0003-2697(02)00281-6 [DOI] [PubMed] [Google Scholar]
- 33.J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd ed. (Springer, 2006). [Google Scholar]
- 34.Kim S.-E., Huang H., Zhao M., Zhang X., Zhang A., Semonov M. V., MacDonald B. T., Zhang X., Abreu J. G., Peng L., He X., “Wnt stabilization of β-catenin reveals principles for morphogen receptor-scaffold assemblies,” Science 340(6134), 867–870 (2013). 10.1126/science.1232389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subach O. M., Gundorov I. S., Yoshimura M., Subach F. V., Zhang J., Grüenwald D., Souslova E. A., Chudakov D. M., Verkhusha V. V., “Conversion of red fluorescent protein into a bright blue probe,” Chem. Biol. 15(10), 1116–1124 (2008). 10.1016/j.chembiol.2008.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Treanor B., Lanigan P. M. P., Suhling K., Schreiber T., Munro I., Neil M. A. A., Phillips D., Davis D. M., French P. M. W., “Imaging fluorescence lifetime heterogeneity applied to GFP-tagged MHC protein at an immunological synapse,” J. Microsc. 217(1), 36–43 (2005). 10.1111/j.0022-2720.2005.01430.x [DOI] [PubMed] [Google Scholar]
- 37.Visser A. J. W. G., Laptenok S. P., Visser N. V., van Hoek A., Birch D. J. S., Brochon J.-C., Borst J. W., “Time-resolved FRET fluorescence spectroscopy of visible fluorescent protein pairs,” Eur. Biophys. J. 39(2), 241–253 (2010). 10.1007/s00249-009-0528-8 [DOI] [PubMed] [Google Scholar]