

Abstract
Cholera toxin (CT), the principal virulence factor secreted by Vibrio cholerae, is an A-B5 type exotoxin that binds to host cell GM1-gangliosides and is responsible for cholera diarrhoea. We tested the hypothesis that the cyclic hexasaccharide α-cyclodextrin (α-CD), but not the cyclic heptasaccharides methyl-β-cyclodextrin (MD-β-CD) and hydroxypropyl-β-cyclodextrin (HP-β-CD) inhibit binding of CT to GM1-gangliosides. We report that α-CD decreases CT binding to GM1-ganglioside-coated microtitre plate wells and on the surface of fixed HeLa cells in a concentration-dependent manner, suggesting that this may be a promising lead for the development of compounds with therapeutic properties.
Introduction
Cyclodextrins (CDs) make up a family of cyclic oligosaccharides, composed of five, six, seven or eight α-d-glucopyranoside units linked 1→4, as they are in amylose. Specifically, α-CDs are six-sugar ring molecules, β-CDs are circular molecules made up of seven monosaccharides and γ-CD rings consist of eight sugars (Bender & Komiyama, 1978). α-CD derivatives have shown efficacy in blocking toxin-receptor interactions of bacterial pathogens, including Bacillus anthracis toxins (Karginov et al., 2005), the lethal toxin, Staphylococcus aureus α-haemolysin (Karginov et al., 2007), Clostridium botulinum C2 toxin and Clostridium perfringens iota toxin (Nestorovich et al., 2011).
Cholera toxin (CT) likely remains the most significant diarrhoeagenic virulence factor expressed by Vibrio cholerae (Herrington et al., 1988), despite other known contributing virulence factors and recently discovered toxin-delivery systems (Dziejman et al., 2005; Pukatzki et al., 2006). CT’s host receptors are pentameric GM1-gangliosides (GM1) that are bound by pentameric CT-B but not dissociated B subunits (De Wolf et al., 1981), even though structural studies show that each receptor-binding site is derived from the residues of a single B subunit (Merritt et al., 1994), suggesting the requirement for a defined geometric or architectonic motif for the interaction.
Much like the protective antigen (PA) of B. anthracis, CT is an A-B type exotoxin with receptor-binding B subunits and catalytic A subunits. Anthrax PA and CT’s B subunits are cell-binding proteins that share a repetitive cyclic structure consisting of seven and five homopeptides, respectively (Merritt et al., 1994; Nassi et al., 2002). A report by Karginov et al. (2005) demonstrated high-affinity blocking of PA by per-6-(3-aminopropylthio)-β-CD, a chemically modified β-CD, in part due to the shape and size of the cyclic heptasaccharide.
Upon examination of the size relationship between PA and CT-B toxins and cyclodextrins, we observed that α-CD, a cyclic hexasaccharide, shares a very close CD backbone-to-CT-B pore ratio (0.78) as the one reported for the heptasaccharide β-CD and PA toxin (0.75) by Karginov et al. (2005). In view of these considerations, we set out to test the hypothesis that α-CD, but not β-CD derivatives, inhibits CT binding to GM1 by ELISA.
Methods
Because β-CD could not be tested due to limited solubility in aqueous solution, we chose methyl-β-CD (M-β-CD) and hydroxypropyl-β-CD (HP-β-CD). The ELISA protocol employed before to quantify CT (Provenzano & Klose, 2000) was modified to accommodate a pre-incubation with CDs. Briefly, 200 μl titrated CDs (α-CD, M-β-CD or HP-β-CD from Sigma−Aldrich) suspended in PBS was added to microtitre plates (Thermo Scientific) with each well coated with 2 μg GM1 (Sigma−Aldrich). After 30 min incubation at room temperature, the CD-PBS solution was discarded, and 0.2 μg purified CT in 200 μl PBS with 2 mg BSA ml−1 was added to each well and incubated at room temperature for 30 min. After three PBS with 2 mg BSA ml−1 washes, a 1 : 2000 dilution of CT rabbit antiserum was added to each well, and the plates were incubated at room temperature for 30 min. After three additional PBS with 2 mg BSA ml−1 washes, the wells were incubated with a 1 : 4000 dilution of alkaline phosphatase-conjugated secondary antibody (Life Technologies). The plates were incubated once more at room temperature for 30 min, washed three times in PBS with 2 mg BSA ml−1, developed with 2 mg p-nitrophenylphosphate (Sigma−Aldrich) ml−1 and read in a microplate reader (Bio-Rad) at OD405. Positive controls for all ELISA experiments consisted of microtitre wells treated identically with 150 mM glucose/PBS or PBS alone for maximal CT binding values. Microtitre wells treated identically but without CT were utilized as negative controls and to subtract background. All experiments were performed in triplicate and replicated three times.
HeLa cells were grown to 60–70 % confluency in Dulbecco’s modified Eagle medium (DMEM) supplemented with 5 % fetal bovine serum in 50 ml tissue culture flasks. Live cells were fixed in 3.7 % formalin/PBS as previously described (Provenzano & Klose, 2000). After fixation, 50 mM α-CD or M-β-CD in 2 mg BSA ml−1 in PBS or 2 mg BSA ml−1 in PBS as negative and positive controls, respectively, were added to the tissue cultures and incubated for 1 h at room temperature. Next, cells were gently rinsed three times with 2 mg BSA ml−1 in PBS, and 1 μg CT ml−1 in 2 mg BSA ml−1 in PBS was added to the flasks, and the cells were incubated for 1 h at room temperature. After three 2 mg BSA ml−1 in PBS rinses, a 1 : 5000 dilution of rabbit CT antisera was added to the cells and incubated for 1 h at room temperature. Following three 2 mg BSA ml−1 in PBS rinses, the cells were incubated for 1 h at room temperature with a 1 : 10 000 dilution of fluorescein-labelled secondary antibody (Life Technologies). After three final 2 mg BSA ml−1 in PBS rinses, cells were examined by fluorescent microscopy, and four fields were photographed at 40× magnification. Normalized images of identical size, exposure and comparable cell density were quantified utilizing the pixel analysis feature of Adobe Photoshop (Adobe Corporation) from three independent replicates, and statistical analyses were carried out with SigmaPlot (Jandel Scientific).
Results and Discussion
We compared α-CD to M-β-CD and HP-β-CD to determine whether the ring size alone of the cyclic oligosaccharides could be a determinant in binding inhibition of CT to GM1 by ELISA. As shown in Fig. 1(a), 50 mM α-CD inhibited CT binding to GM1 by 48 % compared to M-β-CD and HP-β-CD, which afforded 1.8 % and 5.3 % binding inhibition, respectively. When CD concentrations were doubled to 100 mM, CT binding was inhibited by 75 %, whereas M-β-CD and HP-β-CD binding inhibition remained at 5.8 % and 8.2 %, respectively (Fig. 1a). Taken together, these results suggest that the size of the oligosaccharide ring is critical for inhibiting CT binding to GM1. We therefore utilized M-β-CD as a negative control in subsequent experiments and abandoned HP-β-CD.
Fig. 1.
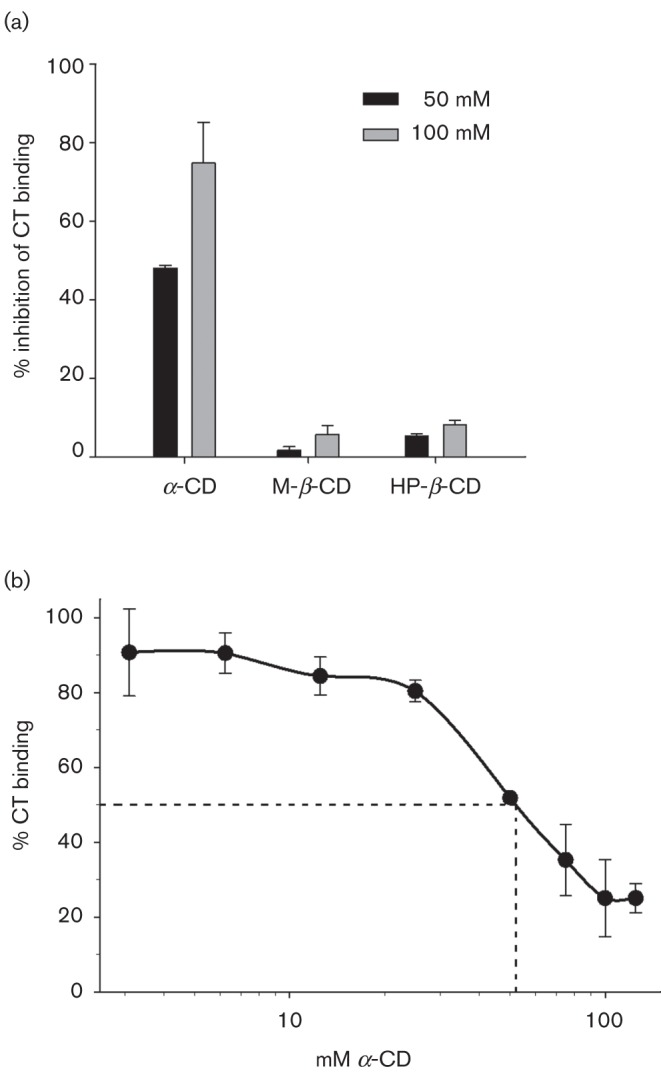
CT binding to GM1-coated microtitre wells was decreased in a concentration-dependent manner by α-CD but not β-CD derivatives. In (a), GM1 was preincubated with 50 or 100 mM α-CD, β-M-CD or β-HP-CD for 30 min prior to addition of CT, which was then quantified by ELISA. The amount of binding inhibition was plotted as the mean percentage from experiments conducted in triplicate, and the bars represent se. (b) The percentage CT binding to GM1-coated microtitre wells pretreated with 3.125, 6.25, 12.5, 25, 50, 75, 100 or 125 mM α-CD is shown. The dotted line highlights α-CD’s IC50 of 52 mM from experiments conducted in triplicate, and the bars represent se.
Next, we tested a range of α-CD concentrations to estimate binding inhibition range, efficacy and determine IC50. Fig. 1(b) shows a composite graph of CT binding to GM1 in the presence of 3.125, 6.25, 12.5, 25, 50, 75, 100 and 125 mM α-CD from one representative experiment performed in triplicate. Blocking of CT binding to GM1 by α-CD was concentration dependent and showed saturation kinetics above 100 mM. Concentrations of α-CD below 12.5 mM failed to inhibit CT binding to GM1 above 10 %. The dotted line highlights the 52 mM α-CD concentration required to block binding of CT to GM1 by 50 %, i.e. half the maximal inhibitory concentration (IC50) under the described experimental conditions.
GM1 randomly coats the bottom of microtitre plate wells and is not oriented in the same position as they are on the surface of eukaryotic cells. In subsequent experiments, we set out to determine whether α-CD blocking of CT binding is affected by GM1 orientation on the surface of fixed Hea cells compared to what we observed by ELISA. We chose to test 50 mM α-CD because at this concentration, M-β-CD did not significantly inhibit CT binding to GM1, but α-CD does. Fig. 2(a) shows three representative fluorescent micrographs of fixed HeLa cells incubated with 1 μg CT ml−1 alone (left panel), 50 mM M-β-CD (center panel) followed by 1 μg CT ml−1, or 50 mM α-CD (right panel) followed by 1 μg CT ml−1 and then probed with CT antiserum and fluorescein-labelled secondary antibody.
Fig. 2.

CT binding to GM1 on fixed Hea cells is significantly decreased by α-CD but not M-β-CD. (a, b) Three representative photomicrographs from independent replicates (a) utilized to generate the graph (b), which shows percentage CT binding to fixed HeLa cells pretreated with either 50 mM α-CD or M-β-CD quantified by fluorescence from three independent replicates. The bars represent sd, where P for the difference of means between the CT and CT+α-CD groups and the CT+M-β-CD and CT+α-CD groups is <0.05.
In Fig. 2(b) we quantified image fluorescence as shown in Fig. 1(a) from experiments carried out four times in triplicate. These efforts revealed that 50 mM α-CD reduced CT binding to fixed HeLa cells by 85.8 % (sd, 27.8 %), suggesting that GM1 orientation is important to binding inhibition. Surprisingly, binding inhibition of CT to HeLa cells by M-β-CD was also moderately improved at 22.3 % (sd, 33.8 %) compared to what we recorded in microtitre plates; however, it was not significantly different than CT binding to Hea cells alone (sd, 22.1 %), Student’s t-test P = 0.323. Student’s t-test analysis also confirmed that the difference between the means of the CT and CT+α-CD groups was P = 0.003 and between the CT+M-β-CD and CT+α-CD groups was P = 0.014, deeming the protection afforded by α-CD towards CT binding statistically significant compared to the control groups. Finally, we tested whether α-CD could block the CT-GM1 interaction when the cyclic hexasaccharide was first co-incubated with CT prior to exposure to the receptor and found that binding inhibition was reduced to background levels as those observed for M-β-CD (data not shown). Taken together, our results indicate that binding inhibition depends on α-CD interaction with GM1 rather than with CT.
In these initial studies, we wanted to ascertain whether the size of the cyclic hexasaccharide α-CD was sufficient to interfere with CT binding to GM1. Aware of the potentially cytopathic effect of α-CDs on viable cells described by Ohtani et al. (1989), we avoided working with live tissue cultures, especially at the high concentrations employed in these experiments. Our hypothesis was formulated in view of an elegant report by Karginov et al. (2005) showing PA toxin-receptor binding inhibition by β-CD derivatives. CT-B’s pentameric structure and size made it an attractive target for α-CD-mediated receptor interference. However, in contrast to the results described by Karginov et al. (2005), our data suggest that α-CD’s CT-blocking activity is mediated by binding to GM1 rather than to the toxin. CT binding to GM1-coated wells was blocked by ~50 % at 50 mM α-CD, yet the same concentration blocked CT binding to GM1 on fixed HeLa cells by 85.8 %, further lowering α-CD’s CT-binding IC50. Although the results of these experiments showed effectiveness at concentrations of CDs that exert cytopathic effects on live cells (Ohtani et al., 1989), they suggest that α-CD may be a promising lead for the development of compounds with therapeutic properties.
Acknowledgements
We thank Angel Olguin and Alisande Cardenas for technical support with ELISAs and reagent preparation. We are also grateful to Dr Jim Bina at the University of Pittsburg School of Medicine for the kind gift of CT antiserum and Dr Charles J. Gauntt, formerly at the University of Texas Health Science Center San Antonio, for the kind gift of HeLa cells. D. P. was supported by National Institutes of Health grants MD001091-01 and GMD068855-02.
Abbreviations:
- CT
cholera toxin
- α-CD
α-cyclodextrin
- β-CD
β-cyclodextrin
- GM1
GM1-gangliosides
- HP-β-CD
hydroxypropyl-β-cyclodextrin
- IC50
half inhibitory concentration
- M-β-CD
methyl-β-cyclodextrin
- PA
protective antigen
References
- Bender M. L., Komiyama M. (1978). Cyclodextrin Chemistry. Berlin: Springer-Verlag; 10.1007/978-3-642-66842-5 [DOI] [Google Scholar]
- De Wolf M. J. S., Fridkin M., Kohn L. D. (1981). Tryptophan residues of cholera toxin and its A and B protomers. Intrinsic fluorescence and solute quenching upon interacting with the ganglioside GM1, oligo-GM1, or dansylated oligo-GM1. J Biol Chem 256, 5489–5496 [PubMed] [Google Scholar]
- Dziejman M., Serruto D., Tam V. C., Sturtevant D., Diraphat P., Faruque S. M., Rahman M. H., Heidelberg J. F., Decker J., et al. (2005). Genomic characterization of non-O1, non-O139 Vibrio cholerae reveals genes for a type III secretion system. Proc Natl Acad Sci U S A 102, 3465–3470 10.1073/pnas.0409918102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington D. A., Hall R. H., Losonsky G., Mekalanos J. J., Taylor R. K., Levine M. M. (1988). Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168, 1487–1492 10.1084/jem.168.4.1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov V. A., Nestorovich E. M., Moayeri M., Leppla S. H., Bezrukov S. M. (2005). Blocking anthrax lethal toxin at the protective antigen channel by using structure-inspired drug design. Proc Natl Acad Sci U S A 102, 15075–15080 10.1073/pnas.0507488102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov V. A., Nestorovich E. M., Schmidtmann F., Robinson T. M., Yohannes A., Fahmi N. E., Bezrukov S. M., Hecht S. M. (2007). Inhibition of S. aureus alpha-hemolysin and B. anthracis lethal toxin by beta-cyclodextrin derivatives. Bioorg Med Chem 15, 5424–5431 10.1016/j.bmc.2007.05.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt E. A., Sarfaty S., van den Akker F., L’Hoir C., Martial J. A., Hol W. G. J. (1994). Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Protein Sci 3, 166–175 10.1002/pro.5560030202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassi S., Collier R. J., Finkelstein A. (2002). PA63 channel of anthrax toxin: an extended beta-barrel. Biochemistry 41, 1445–1450 10.1021/bi0119518 [DOI] [PubMed] [Google Scholar]
- Nestorovich E. M., Karginov V. A., Popoff M. R., Bezrukov S. M., Barth H. (2011). Tailored β-cyclodextrin blocks the translocation pores of binary exotoxins from C. botulinum and C. perfringens and protects cells from intoxication. PLoS ONE 6, e23927 10.1371/journal.pone.0023927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani Y., Irie T., Uekama K., Fukunaga K., Pitha J. (1989). Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur J Biochem 186, 17–22 10.1111/j.1432-1033.1989.tb15171.x [DOI] [PubMed] [Google Scholar]
- Provenzano D., Klose K. E. (2000). Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc Natl Acad Sci U S A 97, 10220–10224 10.1073/pnas.170219997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukatzki S., Ma A. T., Sturtevant D., Krastins B., Sarracino D., Nelson W. C., Heidelberg J. F., Mekalanos J. J. (2006). Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A 103, 1528–1533 10.1073/pnas.0510322103 [DOI] [PMC free article] [PubMed] [Google Scholar]