

Summary
Regulatory T (Treg) cells play a vital role in the prevention of autoimmunity and the maintenance of self-tolerance but also have an active role in inhibiting immune responses during viral, bacterial and parasitic infections. Whereas excessive Treg activity can lead to immunodeficiency, chronic infection and cancer, too little Treg activity results in autoimmunity and immunopathology, and impairs the quality of pathogen-specific responses. Recent studies have helped define the homeostatic mechanisms that support the diverse pool of peripheral Treg cells under steady-state conditions, and delineate how the abundance and function of Treg cells changes during inflammation. These findings are highly relevant for developing effective strategies to manipulate Treg cell activity to promote allograft tolerance and treat autoimmunity, chronic infection and cancer.
Keywords: regulatory T cells, homeostasis, autoimmunity, inflammation
Introduction
Immune homeostasis ensures that the number, diversity and functional competency of lymphocytes is maintained, both during periods with and without overt inflammation. The term homeostasis was derived from the Greek word “stasis”, meaning “standing still”, although this seemingly restful condition is the result of mechanisms that actively maintain the various populations of leukocytes and preserve their functional specialization. When functioning properly, homeostatic mechanisms act to maintain the appropriate balance between effector and regulatory cells, thereby ensuring efficient pathogen control while preventing unwanted immunopathology and autoimmune disease. Therapeutic manipulation of the immune homeostasis can be used to restore this balance in patients with chronic infection or autoimmune disease and to prevent graft rejection or promote immune responses directed against tumors.
The homeostatic equilibrium is accomplished through a combination of soluble cytokine signals and direct interactions between different cell types, which largely occur in a tissue- and microenvironment-specific fashion. Moreover, different populations of cells are placed under unique homeostatic constraints, such that competition for limited resources is minimized via compartmentalization. Among the various immune cell types, control of T cell homeostasis is probably the best understood. Indeed, pioneering work over the last two decades has demonstrated that T cells alter the homeostatic requirements as they emigrate from the thymus as naïve T cells that recirculate through secondary lymphoid tissues, differentiate into effector T cells after antigen stimulation, and persist as long-lived memory cells (1).
Similar to conventional CD4+ T cells, most CD4+Foxp3+ regulatory T (Treg) cells are produced in the thymus, although they display a distinct T cell receptor (TCR) repertoire that is skewed for the high-affinity recognition of self-peptides (2). Treg cells have been shown to play a dominant role in maintaining tolerance and immune homeostasis, as evidenced by the widespread lymphoproliferative disease and autoimmunity that develops in Foxp3-deficient IPEX patients and scurfy mice, and decreased Treg cell activity has also been implicated in development of a number of more common autoimmune and inflammatory diseases, including type-1 diabetes, rheumatoid arthritis, multiple sclerosis and systemic lupus erythematosus. Treg cells have been shown to suppress conventional T cells through multiple mechanisms, including the generation of immunosuppressive cytokines such as tissue growth factor-β (TGF-β) or interleukin (IL)-10 and via direct contact with effector T cells or antigen-presenting cells (APCs), and these have been reviewed at length elsewhere (3,4). Moreover, many parallels have been drawn between Treg cells and conventional CD4+ T cells in terms of their ability to co-opt similar transcriptional and activation profiles to respond to specific types of inflammation (5). However, unlike the detailed understanding of conventional T cell homeostasis, the homeostatic mechanisms that maintain the complex and functionally diverse Treg cell pool in different tissue sites remain poorly understood. In this review, we focus on the cytokine-, cell type- and tissue-specific factors governing Treg cell maintenance, discuss how these mechanisms differ from those governing conventional CD4+ T cells, and how these systems evolve during periods of inflammation.
Part I: Homeostasis of Treg cells in the steady state
The role of IL-2 in peripheral Treg cell homeostasis
IL-2 was originally characterized as a potent T cell growth factor, promoting the expansion of antigen-activated T cells in an autocrine manner. This cytokine is produced mainly by activated CD4+ and CD8+ T cells in secondary lymphoid tissues, where it is consumed primarily by cells expressing the high-affinity form of the IL-2 receptor (6). High-affinity signaling is made possible by the association of CD25 (also known as IL-2Rα), which does not directly participate in signaling but rather increases the affinity of the IL-2R for ligand by 10–100 fold, with dimers of CD122 (IL-2Rβ) and CD132 (the γc chain). Signal transduction occurs via activation of the Janus kinase (Jak)/signal transducer and activator of transcription (Stat) pathway (primarily via dimers of phosphorylated Stat5), as well as mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways activated via phosphorylation of the signaling adaptor Shc (7). Other γc chain cytokines such as IL-7 and IL-15 are capable of transducing similar signals, and the IL-15R also uses CD122 and CD132 as its primary signal transduction chains. IL-7 and IL-15 have been shown to play important roles in the development and peripheral homeostasis of conventional CD4+ and CD8+ T cells (8). However, although thymic development of Treg cells requires T cell-intrinsic Stat5 signaling, this function is mediated primarily by IL-2, with minimal roles for IL-7 and IL-15 that only partially compensate for the loss of IL-2 (9,10), and the role of IL-2 in the thymic generation of Treg cells has been expertly reviewed elsewhere (11).
The main consequences of IL-2 signaling include cell cycle progression and the expression of anti-apoptotic proteins such as Bcl-2 and Mcl-1 (12,13). Many immune cell types, including CD4+ T cells, CD8+ T cells and NK cells, can upregulate CD25 expression upon activation. However, Foxp3 directly promotes CD25 expression (14,15), and as a consequence, Treg cells are unique in that they constitutively express the high-affinity IL-2 receptor. Additionally, IL-2 signaling further promotes CD25 expression via activated Stat5 (16). However, because Runx1 cooperates with Foxp3 and NFAT to bind to the IL-2 promoter and halt its transcription, Treg cells do not themselves produce IL-2 and are instead reliant on paracrine IL-2 produced by other activated T cells (17,18). Thus, the influence of IL-2 on Treg cell homeostasis is dependent on both the rate of IL-2 production and the rate of IL-2 consumption in the steady state.
The key finding showing the important role for IL-2 in Treg cell development and homeostasis came from the surprising discovery of the autoimmune manifestations that occur in mice deficient for IL-2 or CD25. Instead of immunodeficiency, which was expected to result from the loss of an important T cell growth factor, animals lacking IL-2 signaling demonstrate over-active T cell responses, massive lymphoproliferation, lymphadenopathy and splenomegaly and succumb to inflammatory disease within 6–8 weeks of life (19,20). Because Treg cells were first identified by their high-level expression of CD25, the apparent lack of Treg cells in mice deficient for IL-2 or its receptor led to the conclusion that these autoimmune manifestations were the result of decreased Treg cells and their failure to control autoreactive, inflammatory responses. However, the subsequent identification of Treg cells based on Foxp3, as well as the development of mice in which Foxp3 expression could be directly identified using fluorescent proteins such as GFP, revealed that CD25-deficient mice contain near-normal frequencies and numbers of Treg cells in both the thymus and periphery (9,21,22). Furthermore, mice in which CD122 is only expressed during thymic development, resulting in T cell unresponsiveness to IL-2 in the periphery, fail to demonstrate the lethal autoimmunity observed in full CD122-deficient animals (23). Moreover, in contrast to scurfy mice, which lack all Treg cells due to mutation in Foxp3, IL-2- and CD25-deficient mice exhibit less severe autoimmune disease and their lifespan that is approximately twice as long as that of scurfy mice, indicating that at least some Treg cell function is preserved in the absence of IL-2 signaling. Thus, the role of IL-2 in controlling Treg cell abundance and activity is more complicated than previously appreciated, and although IL-2 is clearly required to maintain Treg-mediated tolerance in vivo, these findings indicate that the peripheral homeostasis of Treg cells is at least partially IL-2 independent.
The precise role for IL-2 in Treg cell survival versus function remains debated. Indeed, because Treg cell numbers have been directly linked to their ability to control the size of effector T cell populations, there is likely considerable overlap between survival and function. Evidence in support of a role for IL-2 in Treg cell function includes the observation that IL-2 neutralization in vivo results in the development of autoimmune gastritis on the Balb/c background and exacerbated diabetes on the NOD background (24). In fact, spontaneous autoimmunity in NOD mice is genetically dependent on alterations in the IL-2 gene that result in lower production of IL-2 and consequent autoimmune-mediated diabetes through the impaired regulation of Treg cells (25). Accordingly, IL-2 therapy using anti-IL-2-IL-2 immune complexes, which increases Treg cell numbers by selectively stimulating CD25+ cells (26), was shown to prevent the development of diabetes in NOD mice (25), as well as block induction of experimental autoimmune encephalomyelitis (EAE) and induce tolerance to major histocompatibility complex-incompatible pancreatic islet transplantation (27). In addition, Foxp3 function is closely linked to Treg cell fitness and establishment of the Treg cell transcriptional program, and deficiency in CD25 has been shown to result in lower amounts of Foxp3, especially in the thymus (21). In fact, forced expression of Foxp3 in IL-2Rβ-deficient mice restored Treg cell function and prevented autoimmunity, demonstrating that IL-2 signaling is functionally linked to the control of Foxp3 expression (22). In contrast, the in vitro suppressive function of IL-2- and CD25− deficient Treg cells is intact, and gene expression analysis suggested that the failure of these cells to control the development of autoimmunity was instead related to impaired metabolic fitness (21). To address the function of Treg cells independent of the pro-survival effects of IL-2, IL-2 knockout mice were crossed to Bim knockout mice. However, the enhanced survival of Treg cells via deletion of this pro-apoptotic factor failed to prevent autoimmune disease development, indicating that despite its ability to induce Treg survival, the principle effect of IL-2 on Treg cell function is qualitative rather than quantitative (28). However, due to their high rate of steady-state proliferation and the pro-apoptotic effects of Foxp3 itself (29), it is not surprising that Treg cells must coordinate survival and death signals to maintain stable numbers. In fact, a recent study linked IL-2-driven upregulation of the pro-survival Bcl-2 family member Mcl-1 to Treg cell survival and showed that deletion of Mcl-1 (with the corresponding unrestrained action of Bim) caused a rapid loss of Treg cells and the onset of fatal autoimmunity (12).
The aforementioned studies indicate that IL-2 has pleiotropic effects on Treg cell development, survival and function, and the specific requirement for IL-2-mediated signals likely differs under different conditions. Indeed, whereas previous research in this field has focused on IL-2 as a global survival or maturation factor, recent work from our laboratory has demonstrated that different subsets of Treg cells display distinct homeostatic behaviors and that IL-2 predominantly controls the homeostatic maintenance of a specific Treg cell subset: namely quiescent CCR7hiCD44loCD62Lhi cells that we have termed central Treg (or cTR) cells, which are primarily localized within the T cell zones of secondary lymphoid tissues (30). In this location, these cTR cells are correctly positioned to receive paracrine IL-2 from activated T cells and to inhibit APCs and autoreactive T cell priming (31). Indeed, we found that Stat5 phosphorylation is highly enriched in this Treg cell subset directly ex vivo, and we observed the selective loss of cTR cells in CD25-deficient and anti-IL-2-treated mice. By contrast, the maintenance and proliferation of highly proliferative CD44hiCD62LloCCR7lo effector Treg (eTR) cells is IL-2 independent, and these are the Treg cells remaining in mice with impaired IL-2 signaling. These eTR cells are the dominant Treg cell population found in non-lymphoid tissues, where they employ suppressive mechanisms such as IL-10 secretion to dampen the tissue inflammatory responses of effector T cells (32).
The notion that cytokine requirements fluctuate with the development of distinct T cell populations is a theme that has been extensively studied in conventional T cells. Naïve T cells primarily rely on IL-7 signals for survival, and in many ways, the role of IL-7 in naïve T cell homeostasis is analogous to the role of IL-2 in Treg homeostasis (specifically naïve-like cTR cells). For example, conventional T cells do not produce IL-7 and instead rely on paracrine IL-7 produced by stromal cells within the T cells zones of secondary lymphoid tissues (33). Like IL-2 in Treg cells, IL-7 signaling primarily regulates naïve T cell abundance by inducing the expression of key anti-apoptotic proteins, such as Bcl-2 and Mcl-1, while inhibiting the expression of pro-apoptotic factors such as Bid, Bim and Bad (34,35). Additionally, IL-7 has a minimal effect on naïve T cell proliferation unless it is present in vast excess, e.g. during lymphopenia-induced proliferation or upon administration of super-agonistic IL-7/anti-IL-7 immune complexes (36). Similarly, although we showed that IL-2 signaling is not associated with Treg cell proliferation in the steady state (30), IL-2 immune complexes potently induce Treg cell proliferation (26), and IL-2 can drive Treg cell proliferation during ‘niche-filling’ following ablation of half of the Treg cell compartment (12).. Moreover, effector and memory CD4+ and CD8+ T cell lose their strict dependence on IL-7 for their survival and instead rely on combined signals from the TCR as well as IL-15 for their maintenance, and this mirrors the loss of IL-2 dependence we observed in eTR cells. As a result, similar to IL-2- and CD25-deficient mice that selectively lack the CD44loCD62LhiCCR7hi Treg cell subset, mice lacking IL-7 or IL-7Rα have an almost complete deficiency in CD44loCD62LhiCCR7hi naïve T cells, whereas resitudal CD44hi effector/memory cell populations can be readily detected in these mice (unpublished observations). Based on these data, we feel that IL-2 is not a pan-Treg survival factor, but rather that it has a specific role in the maintenance of naïve-like cTR cells; moreover, the autoimmunity that results in IL-2- and CD25− deficient mice indicates that maintenance of normal immune tolerance requires the combined action of both cTR and eTR cells.
T cell receptor signaling and transcriptional control over Treg homeostasis
In addition to cytokines such as IL-2 and IL-7, TCR signaling also has a key function in the homeostatic maintenance and proliferation of T cells. As noted previously, although the fine specificity of Treg cells is still largely undetermined, several lines of evidence indicate that as a population they are enriched in self-reactive cells. First, thymic development of Treg cells appears to be driven by encounter with self-antigen (37). This has been recapitulated in several TCR transgenic models in which cognate antigen is also expressed in thymus (38–40) and was also recently demonstrated for a prostate-specific self-antigen in the endogenous Treg cell repertoire (41). Additionally, TCRs cloned from Treg cells drive a high-degree of homeostatic proliferation and autoimmune/inflammatory disease when ectopically expressed in T cells transferred to lymphopenic hosts (42), and Treg cells from transgenic mice expressing GFP from the TCR-responsive Nr4a1 (Nur77) locus demonstrated heightened and sustained TCR signaling in comparison to conventional T cells (43). The high degree of self-reactivity among Treg cells indicates the potential for frequent encounters with antigen in the periphery that could help control the size and composition of the Treg cell pool in different tissue locations (44). Indeed, antigen-presenting dendritic cells (DCs) are known to support the maintenance of peripheral Treg cells, and DC numbers are tightly linked to Treg numbers via a regulatory loop, whereby increased numbers of DCs leads to increased Treg cell division and accumulation via a mechanism requiring MHC II expression on DCs (45). To evaluate the requirement for continued TCR signaling on Treg cell function and homeostasis, a recent study inactivated p56Lck in Treg cells using Ox40-cre and a conditional null allele of the Lck gene and observed impaired turnover, loss of suppressive function and dramatic changes in Treg gene expression (46).
Although TCR signaling clearly helps regulate Treg cell homeostasis in vivo, the signaling pathways engaged and responsible for this differ somewhat from those associated with activation of conventional Foxp3− T cells. For instance, immediately downstream of the TCR complex, the tyrosine kinase Zap70 is recruited to phosphorylated immunoreceptor tyrosine-based activation motifs (ITAMs), where it is phosphorylated by Lck and becomes catalytically active. Because Zap70 is critically required for thymic T cell development, its role in peripheral T cell homeostasis was not evaluated until recently. However, by generating mice expressing a Zap70 mutant whose catalytic activity could be selectively blocked with a small-molecule inhibitor, Au-Yeung et al. showed that although the catalytic activity of Zap70 was required for activation of conventional naïve, effector and memory T cells, the Zap70 kinase-independent pathway was sufficient for Treg cell function in vitro (47). Additionally, activation of the PI3K-Akt pathway is essential for proper activation of conventional T cells but inhibitory to Treg cell differentiation and expansion. Accordingly, reduced levels of active Akt caused by a kinase-inactive form of PI3K p110δ or deficiency in sphingosine 1-phosphate receptor (S1P1) were shown to favor Treg cell development (48), whereas overexpression of constitutively active Akt or S1P1 led to reduced Treg cell frequencies (49,50). Another example of the negative regulation of Treg cells by Akt signaling is demonstrated by the expansion of selective expansion of Treg cells when activated in the presence of rapamycin, an mTOR (mammalian target of rapamycin) inhibitor. mTOR signaling (resulting from the distinct mTORC1 and mTORC2 complexes) integrates diverse immune signals (TCR signaling, co-stimulation, IL-2R signaling, etc.) with metabolic cues (such as glucose transport) to direct T cell fate decisions. In fact, mTOR is required for the differentiation of effector CD4+ T cells but is generally regarded as a negative regulator of Treg cell differentiation (51). However, the impact of mTORC signaling in Treg cells is more complicated than previously appreciated. For instance, Treg-specific deletion of the mTORC1 component Raptor led to a profound loss of Treg cell suppressive activity in vivo and the development of fatal inflammation (52). Thus, although conventional T cells and Treg cells possess similar molecular machinery to respond to TCR signals, the orchestration of these signals differs dramatically between cell types, and this has significant consequences for the control of Treg cell abundance and function in vivo.
The role of co-stimulation in Treg homeostasis
For full activation, naive T cells require two signals for productive activation: ‘signal 1’ delivered through the TCR and ‘signal 2’ provided by co-stimulatory molecules. CD28 was the first costimulatory molecule discovered, and the interaction between CD28 and its ligands CD80 (B7-1) and CD86 (B7-2) remains the best-characterized co-stimulatory pathway and the subject of active clinical research. The importance of CD28 signaling in Treg cell development and homeostasis was first appreciated nearly a decade ago by the seemingly paradoxical finding that rather than being protected, the development of diabetes in NOD mice deficient for either CD28 or its ligands was actually accelerated, and this could be prevented by the transfer wild-type Treg cells (53). Indeed, like most T cells, Treg cells constitutively express CD28, and removal of CD28 co-stimulation results in decreased numbers of thymic and peripheral Treg cells (54). Although initial reports were hindered by potential trans effects of loss of CD28-mediated co-stimulation on IL-2 production by effector T cells, a recent study generated CD28-conditional knockout mice and reported the cell-intrinsic requirement for CD28 in Treg cell survival and function, as these mice developed severe autoimmunity with increased numbers of activated effector cells (55). Consistent with their potent antigen-presenting and costimulatory function, Bar-On et al. used mixed chimeras to demonstrate that DCs were the primary cell type responsible for providing CD80/86 co-stimulation to Treg cells (56).
Similar to CD28, inducible costimulator (ICOS) is another member of the immunoglobulin (Ig)-like family of costimulatory receptors that helps control the development and function of nearly all CD4+ T cell subsets. Expression of ICOS is rapidly induced on T cells following activation and is influenced by both TCR and CD28 signaling; however, unlike CD28 that is expressed by nearly all T cells, ICOS is expressed at higher levels in the steady state by Treg cells compared to conventional T cells. Dendritic cells and B cells express high levels of ICOS-ligand (ICOSL), and ICOS has also been shown to control regulatory T cell abundance and function in vivo. Treg cell numbers are reduced in ICOS-deficient mice, and, in contrast to CD28-deficient animals, this seems to be solely the result of defective peripheral maintenance, as normal thymic Treg populations were reported in these animals (57). Moreover, blockade of ICOS was shown to promote the development of diabetes in BDC2.5 TCR-transgenic mice, as a result of dysregulation among ICOS+IL-10-producing Treg cells (58). Because IL-10 production has been correlated with eTR phenotype cells, we analyzed the specific requirement for ICOS in cTR vs. eTR cells and found that blockade of ICOS signaling resulted in the unique loss of eTR cells without any effect on cTR cell abundance. Despite its well-established costimulatory function, the remaining eTR cells in ICOSL-blocked mice demonstrated normal proliferation, and our data are most consistent with a role for ICOS in promoting eTR survival rather than proliferation (30).
Although the largest frequencies of ICOS+ eTR cells are found in peripheral tissue sites such as the intestines, a specific population of CD44hiICOS+Bcl6+CXCR5+ Treg cells that localize to germinal centers within lymphoid tissues, termed T follicular regulatory or Tfr cells, was recently described. These Tfr cells are specialized to suppress the generation of germinal center B cells, the affinity maturation of antibodies and the differentiation of plasma cells (59,60). Interestingly, the generation and maintenance of these cells requires CD28 and ICOS, whereas this cell subset is negatively regulated via signals from PD-1 and PD-L1 (61). It is likely that follicular B cells expressing ICOSL may provide homeostatic signals to ICOS-expressing Tfr cells, although additional work is required to address this hypothesis.
The potential for redundancy between CD28− and IL-2-mediated homeostatic signals in Treg cells has been proposed, and indeed, IL-2−/−CD28−/− double knockout mice demonstrate a more significant decline in peripheral Treg cell frequencies compared to single knockouts alone, indicating an additive role for these homeostatic pathways in Treg cell maintenance (62). However, an integrated understanding of how IL-2 and TCR/co-stimulatory signals combine to control the homeostasis of different Treg cell populations in distinct tissue locations is lacking. For instance, one possibility may be that IL-2 simply potentiates antigen-driven proliferation/selection of highly self-reactive Treg cells in the periphery. Alternatively, IL-2 and TCR/costimulatory signals could drive ‘parallel’ pathways of Treg cell homeostasis (63). To distinguish between these possibilities, we analyzed the requirement for CD28 in the expansion of Treg cells induced by super-agonistic IL-2 immune complexes, or by increasing DC numbers following transplanting B16 melanoma cells overexpressing Flt3L (B16.Flt3L cells). We found that although the IL-2-induced Treg cell expansion was CD28− independent, the increase in Treg cells observed in wild-type B16.Flt3L recipients did not occur in the absence of CD28 (Figure 1). Conversely, we found that Treg cell numbers were increased to a similar extent when B16.Flt3L cell recipients were treated with blocking IL-2 antibodies (30). Moreover, increasing the number of DCs specifically expanded effector Treg (eTR) cells with the CCR7loCD44hiCD62Llo phenotype, without altering the number or frequency of IL-2-responsive cTR cells. Thus, our data indicate that rather than acting in the same pathway, TCR/co-stimulatory signals and IL-2 act in parallel to control the abundance of distinct Treg cell subsets (Figure 2).
Figure 1. DC-driven Treg cell expansion is CD28-dependent.

Total number (left) and frequency (right) of Foxp3+ Treg cells in the spleens of wild-type and Cd28−/− mice treated with B16.Flt3L tumors for two weeks or with IL-2 immune complexes for 7 days as indicated.
Figure 2. Separate homeostatic pathways for different Treg cell populations.
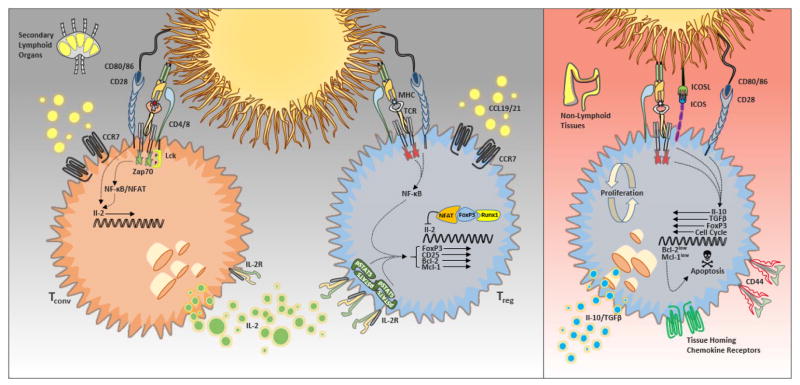
(Left) Paracrine IL-2 maintains quiescent CD44loCCR7hi ‘central’ Treg cells in secondary lymphoid tissues by induction of pro-survival factors such as Mcl-1. (Right) ‘Effector’ Treg cells in non-lymphoid tissues balance rapid proliferation and cell death, and this balance depends in part TCR/co-stimulatory receptor signaling.
The molecular mechanisms by which CD28 and ICOS control Treg cell development and homeostasis remain poorly understood. However, as the induction and maintenance of Foxp3 expression relies in part on activation of NF-κB, the ability of CD28 to dramatically enhance TCR-driven NF-κB activity may underlie much of its effect on Treg cells. Interestingly, both CD28 and ICOS are potent activators of the PI3K and Akt signaling cascade, which is somewhat paradoxical given the antagonistic effect PI3K/Akt activation has on Treg cell differentiation and function. However, eTR cells that lack survival signals via IL-2 may rely on of ICOS-dependent PI3K activation to properly balance their high rate of cell proliferation with their rate of cell death to maintain stable numbers, and this would help explain why blocking ICOS signaling leads to a decrease in the number of eTR cells without impacting their proliferation.
How anatomical location dictates Treg homeostasis
CCR7 provides Treg cell access to IL-2 in secondary lymphoid tissues
Lymphoid tissues are complex structures where numerous cell types with distinct functions are organized into specific compartments to facilitate the cellular communication necessary for the generation of an appropriate immune response. Migration to and within specific compartments of lymphoid tissues is particularly important for the modulation of antigen-specific responses, and integrin and chemokine signals play a key role in this process. Under steady-state conditions, the chemokines CCL19 and CCL21 are constitutively expressed by stromal cells in the T cell areas of the secondary lymphoid tissues, where they act as the sole ligands for the chemokine receptor CCR7. By directing the migration of naïve T cells and activated dendritic cells to secondary lymphoid tissues, CCR7 and its ligands coordinate the key cellular interaction needed for T cell-mediated immunosurvailance. As a result, CCR7-deficient mice display grossly altered lymphoid tissue architecture associated with impaired T cell priming (64). However, although delayed, mice deficient in CCR7 or its ligands often show exacerbated T cell responses compared with wild type animals (65,66), and older Ccr7−/− mice even develop multi-organ autoimmunity, suggesting that immunoregulatory mechanisms are impaired in the absence of CCR7 (67). Indeed, although dispensable for their in vitro activity, CCR7 is essential for the in vivo function of Treg cells. Based on the high level of CCR7 expression on IL-2-dependnet cTR cells, we hypothesized that one way in which CCR7 controls Treg cell function is by allowing cTR cells to access paracrine IL-2 in the T cell zones of secondary lymphoid tissues. Indeed, we found that Treg cells in Ccr7−/− mice showed decreased phosphorylation of pStat5 directly ex vivo and were predominantly eTR phenotype cells. Moreover, cell-intrinsic expression of CCR7 was required for Treg cells to receive IL-2 signals and phosphorylate Stat5 within the T cell zones of the spleen (30). Thus, CCR7 gives cTR cells preferential access to IL-2 and is required to maintain the balance between cTR and eTR cells, and our results provide additional mechanistic insight into the link between CCR7, IL-2, Treg cell function and the development of autoimmunity.
Although CCR7 facilitates the microenvironmental acquisition of paracrine IL-2 by Treg cells, this IL-2 ‘niche’ remains poorly defined. The T cell zones of secondary lymphoid tissues are not homogenous and instead can be further divided into a number of specialized domains that differ in cellular composition and function. This organization is dictated largely by non-hematopoietic stromal cells, which have recently been identified as key accessory cells that help promote immune function and tolerance (68). In fact, stromal cells highly express CCL19 and CCL21 (69), raising the possibility that these cells could act as cellular scaffolds to bring CCR7+Treg cells into proximity with the appropriate DCs and IL-2-producing T cells. However, additional work is required to determine how loss of CCR7 ligand expression in different cellular compartments impacts paracrine IL-2 signaling and Treg cell homeostasis. During activation, the T cell-APC contact is termed the immunological synapse, and the integrin LFA-1 supports these contacts around TCR clusters. Upon examining T cell-T cell clustering during activation, a recent study demonstrated that paracrine IL-2 signaling between adjacent activated T cells occurs via a synaptic mechanism involving homotypic LFA-1 interactions (70). However, unpublished data from our lab indicate that Itgb2−/− Treg cells lacking LFA-1 expression can enter the white pulp and phosphorylate Stat5 normally upon transfer into WT recipients (Figure 3). Thus, tolerogenic IL-2 cross-talk between memory T cells and Treg cells may be mechanistically distinct from pro-inflammatory IL-2 signaling between effector T cells, and it is tempting to speculate that synaptic IL-2 signaling between effector T cells during strong inflammatory responses could act to ‘hide’ IL-2 from nearby Treg cells.
Figure 3. CD18 is not required for Treg cell access to IL-2 in secondary lymphoid tissues.
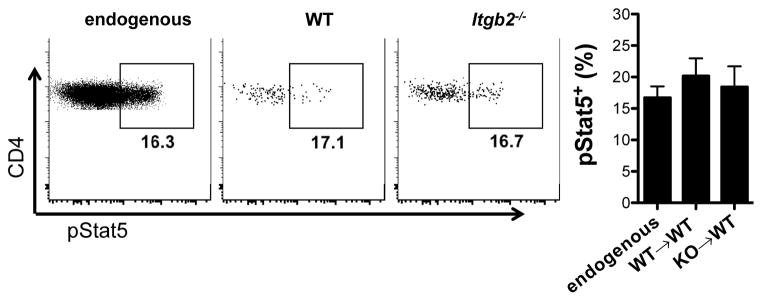
CD4+CD25+ Treg cells were isolated from WT and Itgb2−/− (CD18-knockout) mice and adoptively transferred into congenically marked WT recipient mice. At 36 hours after transfer, pStat5 staining was performed directly ex vivo to determine the ability of endogenous Treg cells (left), recovered WT Treg cells (middle) and recovered Itgb2−/− Treg cells (right) to access paracrine IL-2 in the spleen. The average pStat5+ frequencies for these Treg cell populations are shown in the bar graph at the far right (n=3 data points per group). The CD18 integrin constitutes the beta subunit of LFA-1 (when paired with CD11a). Whereas LFA-1-dependent cellular synapses were shown to facilitate IL-2 signaling in effector T cells (see reference, these data suggest that Treg cells utilize a mechanism distinct from LFA-1 integrin interactions to access paracrine IL-2 in the T cell zones of the spleen.
Non-lymphoid tissues
Treg cells can be found in virtually all non-lymphoid tissues, including the skin and hair follicles, lung, liver, intestines, adipose tissue, brain and spinal cord, placenta, tumors and atherosclerotic plaques (71). Although their frequency at these sites is increased during periods of inflammation, eTR cells express a wide array of homing receptors during the steady-state that enable their continued migration and persistence in peripheral tissue sites (72). Previous work from our group and others has demonstrated that homing to non-lymphoid tissues is critical for controlling Treg cell homeostasis and function at these sites. For instance, mice whose Treg cells cannot access the skin due to lack of the chemokine receptor CCR4 or the α-1,3-fucosyltransferase IV and VII (FuT4/7) enzymes (thereby lacking functional ligands for the P- and E-selectin adhesion molecules) develop spontaneous inflammatory disease specifically in the skin and show impaired immune responses during cutaneous infection with vaccinia virus (73–75). Mechanistically, the transcription factors Blimp-1 and IRF-4 were recently shown to be required for effector Treg cell IL-10 production, ICOS expression and homeostasis in tissues such as the lung and intestines (76). This helps explain the immunopathology observed in Blimp-1-deficient mice and mice with Treg-cell specific IL-10 deficiency (77,78).
The self-reactivity of Treg cells also contributes to their localization in specific non-lymphoid tissues, presumably to prevent autoimmune responses and collateral damage to self during periods of strong inflammation. The recent generation of elegant TCR-transgenic mouse models has significantly contributed to our understanding of how TCR specificity dictates Treg migration, homeostasis and function. For instance, a recent study from our lab showed that Treg cells from mice engineered to express an autoreactive TCR isolated from a scurfy mouse displayed an activated phenotype with the expression of skin-homing molecules such as CD103, and E/P-selectin ligands (79). Similarly, by cloning the TCR of tumor-infiltrating Treg cells in a mouse model of prostate tumor development, Malchow et al. demonstrated that prostate-specific Treg cells were generated in the thymus and accumulated in peripheral tissues on the basis of antigen recognition (41). To address the continued requirement for antigen signaling by Treg cells in peripheral tissues, Rosenblum et al. developed an ovalbumin (OVA)-specific TCR-transgenic model in which skin-specific expression of OVA could be experimentally induced (80). With this system, these authors demonstrated that expression of an antigen in a peripheral tissue could promote the accumulation of antigen-specific Treg cells required for the resolution of tissue-specific autoimmunity. Moreover, these skin-resident OVA-specific Treg cells were termed memory Treg cells because they could survive in this nonlymphoid site for extended periods without antigen expression but were ‘primed’ to control subsequent inflammation upon forced re-expression of OVA. However, unlike the IL-2-dependent cTR cells and ICOS-dependent eTR cells, these memory Treg cells are dependent on IL-7 for maintenance in peripheral tissues following removal of antigen (81). Thus, Treg cells generated against foreign antigens, such as commensal and pathogenic micro-organisms, or paternal alloantigens likely can persist long-term in the absence of TCR stimulation and influence the outcome of future antigen encounters.
Part II: Treg cells and inflammation
In addition to preventing autoimmunity and maintaining self-tolerance, Treg cells can also inhibit immune responses during viral, bacterial, and parasitic infections (82). A major challenge Treg cells face during infection is how to maintain self-tolerance while allowing protective pathogen-specific immune responses to occur. Whereas excessive Treg cell activity can lead to chronic infection and cancer, too little Treg cell activity during infection can result in collateral autoimmunity and immunopathology and can even impair the quality of pathogen-specific response (83–85). These studies highlight the need for Treg cells to integrate environmental cues and precisely tailor their activity to different immune contexts accordingly (Figure 4).
Figure 4.

Control of Treg cell activity is critical for proper resolution of inflammation.
Treg cells are sensitive to a number of external cues in the immune environment that are induced during inflammation. Many of these are factors that already regulate Treg cell survival and activity during steady-state conditions, such as IL-2 and TCR co-stimulatory molecules, but whose expression is altered upon during inflammation. Other factors, like pro-inflammatory cytokines and detection of pathogen- or danger-associated molecular patterns (PAMPs and DAMPs), are specifically induced during infection and can modulate Treg cell activity both directly and indirectly. Understanding the balance between these factors is critical to our understanding of how Treg cells tailor their activity to meet the demands of different inflammatory settings. In the following section, we will review how changes in Treg cell abundance and function influence the development of chronic infection, autoimmunity, and cancer, and then examine how changes in specific cytokine and co-stimulatory signals may control Treg cell activity during inflammation.
Treg Cells during Infection, Autoimmunity, and Cancer
Infection
During an infection, the immune system must recognize and eliminate the invading pathogen while limiting collateral damage to self-tissue that can result from uncontrolled effector T cell responses. While Treg cell-mediated suppression can impair clearance of harmful pathogens, this activity can actually be beneficial to the host in other instances. Treg cell activity, thus, must be tightly regulated in different infectious settings, as both too little and too much Treg cell activity can harm the quality of pathogen-specific effector immune responses.
Some inhibition of Treg activity early during infection is critical for the development of effective pathogen-specific T cell responses. Considering the abundance of Treg cell-activating factors present during infection, like IL-2 and activated DCs, the host must employ counter-regulatory mechanisms to circumvent Treg cell activity and ensure pathogen control. Indeed, a number of acute infections induce a transient decline in Treg cell numbers that correlates with the expansion of effector T cells (86). This inhibition of Treg cell activity can be accomplished indirectly in some instances: for example, activation of APCs by Toll-like receptor (TLR) or CD40 stimulation protects them from Treg cell-mediated suppression (87–89), and pro-inflammatory cytokines such as IL-1β and IL-6 can render effector T cells resistant to Treg cell-mediated inhibition (90,91). Alternatively, proinflammatory cytokines like TNFα can directly inhibit Treg cell activity (92), and recent work in our lab demonstrates that type I interferons (IFNs) directly repress Treg cell activity during viral infection to allow optimal anti-viral T cell responses and efficient viral clearance (Srivastava et al., Submitted).
Failure to limit Treg activity by such mechanisms can impair effector T cell responses, promoting pathogen persistence and chronic infection. Elevated Treg cell numbers, for example, are associated with higher viral burden and exaggerated liver pathology following infection with hepatitis C virus (93,94), and with lung pathology and active disease following Mycobacterium tuberculosis (Mtb) infection (95). In fact, recent studies have demonstrated that thymically-derived Treg cells specific for Mtb antigen expand early during Mtb infection, delay the priming of CD4+ and CD8+ T cells in draining lymph nodes, and block their migration to infected lungs, resulting in higher bacterial burden (96,97). In such infectious settings where excessive Treg cell activity is detrimental to the host, Treg cell depletion can restore protective immune responses. For example, Treg cell depletion enhances bacterial clearance following Mtb infection (98), and protects mice infected with Plasmodium yoelii from death by restoring anti-parasite effector responses (99).
Although Treg cells are known for their ability to interfere with effector T cell responses, several recent studies highlight an underappreciated role for Treg cells in shaping the quality of pathogen-specific responses. In a ground-breaking study, Lund et al. demonstrated that Treg cells are actually essential for the development of appropriate anti-viral T cell responses during murine infection with herpes simplex virus-2 (HSV-2) (84). In this study, depletion of Treg cells following intravaginal infection with HSV-2 accentuated T cell priming and proliferation in the draining lymph node, but uncontrolled T cell activation prevented effector T cell mobilization from the lymph node to the vaginal epithelium, resulting in uncontrolled viral replication and death. Interestingly, Treg cell depletion can also impair the avidity of CD8+ T cell responses during Listeria monocytogenes infection (LM) (85). In this model, Treg cells promote high-avidity CD8+ T cell responses to LM infection by destabilizing low-affinity T cell-DC interactions. Treg cells can also indirectly promote memory responses: following infection with the intracellular parasite Leishmania major, Treg cells block the sterile eradication of the pathogen, providing a source of persistent antigen that is essential for the maintenance of protective memory responses (100). In other instances, the ability of Treg cells to limit effector T cell activity can be beneficial to the host by limiting immunopathology in the presence of persistent antigen. Patients with chronic HCV infection have higher numbers of Treg cells in blood and liver biopsies than uninfected patients, with Treg number inversely correlated with histological severity (94,101). However, although depletion of Treg cells enhances HCV-specific CD8+ T cell function in vitro, sustained CD8+ T cell responses correlate strongly with immunopathology and liver damage (102). Likewise, Treg depletion increases damage to the liver in mice chronically infected with Schistosoma mansoni, and Treg cells protect against inflammatory lesions in a mouse model of HSV infection (103,104). Treg cells, thus, can play a protective role during infection by regulating both the quality and quantity of effector responses to pathogens.
Taken together, these studies underscore the need for Treg cells to integrate cues from the immune environment in order to appropriately modulate their activity depending on the immune context. Too much Treg cell activity can result in immunosuppression and impaired pathogen clearance, whereas too little Treg cell activity can impair effector T cell mobilization and avidity during infection and unleash potentially fatal inflammatory and autoimmune diseases. Identifying the cellular and molecular signals that control Treg cell homeostasis and function is essential for understanding how Treg cells influence the outcome of normal and pathological immune responses.
Autoimmunity
Thought to be largely specific for self-antigens, Treg cells are classically known for their role in preventing the activation of self-specific effector T cells and autoimmunity. The importance of Treg cells in maintaining self-tolerance is highlighted by the severe multi-organ inflammation that develops in Foxp3-deficient scurfy mice and IPEX patients carrying mutations in Foxp3. Though Treg suppressive activity is clearly impaired in autoimmune disorders, what factors lead to this dysfunction are still being elucidated. Treg and effector T cell-intrinsic defects may contribute to loss of tolerance, while overexpression of inflammatory mediators can activate APCs and effector T cells, making both cell types refractory to Treg cell-mediated suppression. Interestingly, circulating numbers of Treg cells appear to be mostly normal in patients with autoimmunity. Although the data from these studies is complicated by the lack of precise markers used to identify Treg cells in humans, most studies have demonstrated no difference in the numbers of CD4+Foxp3+ Treg cells in the peripheral blood of patients with type I diabetes (T1D) and multiple sclerosis (MS) compared with healthy controls (105,106). On the other hand most studies of patients with systemic lupus erythematosus (SLE) detected a decrease in circulating numbers of Treg cells that correlated with disease activity (107,108). Of course, these findings are limited only to peripheral blood of patients and do not necessarily reflect changes in Treg abundance or activity at sites of on-going inflammation. In studies where sites of inflammation were also analyzed, Treg numbers were actually increased in the synovial fluid of patients with rheumatoid arthritis (RA) and in the inflamed lamina propria of patients with inflammatory bowel disease (IBD) (109,110). These increases in Treg abundance are likely secondary to Treg cell-activating signals abundant in inflamed tissues. However, in many cases Treg cells from autoimmune patients show functional defects in in vitro suppression assays. In some cases, dysfunction appears to be due to Treg cell-intrinsic defects. For example, Treg cells isolated from patients with RA were unable to suppress the activation of autologous effector T cells from healthy controls as well as control Treg cells (111,112). However, Treg cells from IBD or T1D patitents were able to suppress healthy control effector T cells to similar levels as control Treg cells (113,114). In these cases, other factors including local inflammatory cytokine production, APC activation, and effector T cell resistance to suppression may indirectly subvert Treg function in vivo.
The resistance of effector T cells to Treg-mediated suppression has been described both in patients and in mouse models of T1D, MS, and SLE. Effector T cells isolated from T1D patients, for example, could not be suppressed by either T1D or healthy control Treg cells (113,115). Likewise, Treg cells are able to migrate to the central nervous system (CNS) during experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, but are unable to suppress effector T cells during active disease due to the production of IL-6 and TNFα, both of which have been implicated in driving effector T cell activation and resistance to suppression (116). Other studies indicate that changes in APC activity and cytokine production can also subvert Treg suppressive function. Treg cells from SLE patients showed defective suppression in the presence of APCs from SLE patients, and this was linked to their production of IFNα (117). Similarly, DCs and monocytes from the synovial fluid of RA patients have a more activated phenotype and can induce Th17 cell polarization better than those from peripheral blood (118), while DCs in the CNS promote Th17 maintenance via secretion of IL-23 during EAE (116). Thus, despite the presence of normal numbers of functional Treg cells, a number of factors in the inflammatory milieu may still circumvent Treg-mediated suppression and drive autoimmunity.
This indirect subversion of Treg cell function may make putative therapies targeted at Treg expansion difficult to implement. Adoptive Treg cell therapy trials, involving the ex vivo expansion of Treg cells and reinfusion into patients, are currently underway and will help determine whether transferring greater numbers of Treg cells is sufficient to overcome Treg cell dysfunction in patients with autoimmunity, or whether extrinsic factors in the inflammatory environment will still render these expanded Treg cells ineffective.
Cancer
The current model of cancer immunosurveillance argues that nascent tumors are continually being eliminated by the immune system, and only tumors that manage to escape immune detection eventually develop into malignancies. This model is validated in part by the dramatic increase in spontaneous and carcinogen-induced cancer found in immunocompromised patients and in immunodeficient mice (119). In line with this, recent studies indicate that Treg cells play an important role in modulating the anti-tumor effector T cell response and promoting tumor immune evasion (120). Not only do Treg cells dampen anti-tumor immune responses, but Facciabene et al. demonstrated that hypoxia induced CCL28 expression by tumor cells promoted the recruitment of CCR10+ Treg cells into the hypoxic tumor microenvironment, and that the newly recruited Treg cells produced VEGFA and promoted tumor angiogenesis (121). Other studies indicate that RANKL-expressing Treg cells promote tumor metastasis via their interactions with RANK-expressing tumor-associated DCs (122). Treg cells, thus, may promote tumorigenesis via their effects on both immune effector cells as well as on tumor metastasis and angiogenesis. It is important to note, however, that Treg cell number is also associated with improved prognosis for certain malignancies, like colorectal cancer and certain types of lymphoma(123–125). Several studies now indicate that chronic inflammation can promote cancer by creating a setting in which cell death, necrosis, and mutagenesis are increased (126), and as such, Treg cells may play a protective role in these contexts by dampening inflammation-induced cancer.
Treg cells may accumulate at tumor sites by a number of mechanisms. Tumor cells can secrete factors like TGFβ, IL-10, and prostaglandin E2 (PGE2) which block DC maturation and promote Treg cell expansion and conversion from naïve CD4+ T cells. In addition to promoting Treg conversion from naïve CD4+ T cells (127), tumor cells can also recruit circulating natural Treg cells to tumor sites, in part through secretion of specific chemokines (121,128,129). As discussed previously, Malchow et al. demonstrated that the Treg cells that accumulate in prostate tumors were specific for normal prostate antigens, not tumor antigens, and developed in the thymus even in the absence of cancer, suggesting that tumors co-opt pre-existing self-specific Treg cells for effective immunosuppression (41).
Given the association of Treg accumulation at tumor sites and poor prognosis for many cancers, many cancer therapies have been aimed at limiting excessive Treg activity. Depletion of Treg cells via anti-CD25 monoclonal antibodies in mouse models resulted in enhancement of tumor-specific effector T cell responses and tumor regression (130,131). However, clinical trials of anti-CD25 therapy in cancer patients had only limited success (120), most likely due to their lack of specificity, since activated CD4+ and CD8+ effector T cells also express CD25. Another approach has been to target CTLA-4, an inhibitory receptor highly expressed by Treg cells that can restrict anti-tumor immune responses. A CTLA-4-specific antibody (Ipilimumab) was recently approved for the treatment of advanced melanoma, with evidence for tumor regression in treated patients and improved anti-tumor responses in patients with other tumor types (132,133). Anti-CTLA-4 treatment increases the ratio of effector T cells to Treg cells specifically at the tumor site, in part through depletion of tumor-infiltrating CTLA-4+ Treg cells via FcγR-expressing macrophages (134). Anti-CTLA-4 antibody treatment, however, was associated with the development of severe autoimmune side effects in a number of patients, complicating its utility as an effective cancer treatment. Recently, the cell surface receptor neuropilin-1 was shown to help maintain Treg cell function within tumors, and this may prove an attractive target in anti-tumor immunotherapry (135).
Factors Modulating Treg Cells During Inflammation
Gamma-common (γc) chain cytokines
IL-2 is classically produced by pathogen-specific effector T cells upon activation in the periphery, and Treg cells are among the first cells to respond to IL-2 upon antigenic challenge in vivo (136,137). That Treg cells are stimulated by IL-2 early in an immune response when their activity may need to be curbed is puzzling. One possibility is that parallel activation of Treg cells with effector T cells may prevent the development of collateral autoimmunity. However, IL-2 expression varies widely in different infectious models, and reduced IL-2 production by effector T cells is thought to underlie a transient decrease in Treg cell number during infection with Toxoplasma gondii, Listeria monocytogenes, and vaccinia virus (86,138). IL-2 may also drive the proliferation of Treg cells late in infection, after the clearance of pathogen, in order to turn off the effector T cell response and limit immunopathology. For instance, we have shown that Treg cells peak in proliferation and number two weeks post-infection with LCMV, correlating with the contraction of CD8+ and CD4+ effector T cell responses. IL-2 expression in the spleen and STAT5 phosphorylation in Treg cells also peak at this time point, suggesting a role for IL-2 in driving Treg cell expansion late in infection (Srivastava et al., Submitted).
Like IL-2, IL-15 is a common gamma chain cytokine that can drive Treg cell proliferation both in vitro and in vivo (139–141), in addition to its role in DC, NK, and CD8+ memory T cell activation and survival. Depending on the balance of these different effects, IL-15 can both promote and inhibit Treg cell activity in different contexts. For example, IL-15 treatment promotes Treg expansion and Foxp3 expression and can protect against diabetes onset when coupled with NK cell depletion (141). Additionally, IL-15 can promote the ability of TGFβ to induce Foxp3 expression in naïve CD4+CD25− T cells (142). In other contexts, IL-15 can work to subvert Treg cell activity. IL-15 is induced early during viral infection and can render effector CD4+ and CD8+ T cells resistant to Treg-mediated suppression via activation of the PI3K signaling pathway (142). Interestingly, DePaulo et al. recently demonstrated that IL-15, which is upregulated in the intestines of patients with celiac disease, can modulate the tolerogenic effects of retinoic acid and promote inflammation rather than oral tolerance to gut antigens (143). Similarly, synovial fibroblasts in patients with RA constitutively express IL-15, and co-culture with RA fibroblasts induced proliferation of both Treg cells and effector− T cells with the net effect of increasing the production of pathogenic cytokines by effector T cells (144). Thus, the evidence suggests that during inflammation, IL-15’s pro-proliferative effects on Treg cells are usually outweighed by its activating effects on effector T cells and NK cells.
Co-stimulatory molecules
During inflammation, recognition of PAMPs or DAMPS drives DC activation and maturation, resulting in increased antigen presentation and expression of MHC class II and the co-stimulatory ligands CD80, CD86 and ICOSL. As discussed in the previous section, these molecules have a dramatic impact on the abundance and function of Treg cells, and this also holds true in infection and cancer as well as autoimmunity (145,146). Indeed, the expansion of ICOS+ Treg cells during therapeutic IL-2 administration is associated with poor clinical responses in melanoma patients (147). Among the other co-stimulatory molecules that help control inflammatory responses, OX40 is constitutively expressed on Treg cells, although it can also be expressed on activated effector T cells following TCR/CD28 co-stimulation. Like ICOSL, OX40L is expressed on activated DCs and B cells following exposure to pathogens, and its expression can be further enhanced by CD40 signaling. OX40 plays an important role in supporting Treg cell survival, proliferation and suppressive function. OX40 signaling induces Treg proliferation (148), and protects against experimental autoimmune thyroiditis and T1D in NOD mice (149). Moreover, OX40-deficient Treg cells are unable to control inflammatory colitis (150). Another study, however, showed OX40 induces proliferation of Treg cells, but these expanded Treg cells have reduced levels of Foxp3 and exhibit poor suppressive function that could be rescued by administration of IL-2 (151). In addition to its direct effects on Treg cells, OX40 can also activate effector T cells, making them resistant to Treg-mediated suppression (152), and can prevent Foxp3 induction in naïve conventional T cells (153). Consequently, intra-tumor injection of OX40 agonists activated effector T cells and resulted in tumor rejection (154). Interestingly, this study also identified a direct role for OX40 in inhibiting Treg cells and demonstrated that OX40 signaling in both Treg and effector T cells was required for tumor rejection. Thus, the effects of OX40 on both regulatory and effector T cells are complex, and the balance of these signals can result in either be pro- or anti-inflammatory outcomes.
Pro-inflammatory cytokines
A number of pro-inflammatory cytokines induced during infection have been shown to subvert Treg cell activity, both directly and indirectly. While these cytokines may play a protective role during acute infection by circumventing Treg cell activity, their overexpression is largely associated with autoimmune disorders and Treg cell dysfunction.
IL-1β and IL-6 are pro-inflammatory cytokines largely known for their ability to induce systemic inflammation and for their role in the pathology of autoimmune diseases like RA, type I diabetes, and MS. Treatments aimed at blocking both cytokines, via blocking antibodies (anti-IL-6R, tocilizumab) or soluble receptor antagonists (sIL-1RA, anakinra) have both been approved for rheumatoid arthritis and are being tested for the treatment of other autoimmune diseases. In addition to promoting Th17 cell differentiation, several studies have demonstrated that these cytokines can indirectly subvert Treg cell function by activating effector T cells and rendering them resistant to Treg cell-mediated suppression (90,91). In fact, IL-6Rα and STAT3 phosphorylation are increased in effector T cells from patients with relapsing-remitting multiple sclerosis (RRMS), and inhibition of STAT3 in these cells restored their ability to be suppressed by Treg cells (155). The direct effects of these cytokines on Treg cells, however, have not been clearly established. Treg cells express both IL-1R and IL-6R and can activate p38/JNK and STAT3 signaling in response to these cytokines, respectively (156,157). IL-1β may affect Treg cell stability, as IL-1R signaling has been shown to induce IL-17 production in both human and murine Treg cells and has been implicated in the conversion of induced Treg cells to Th17 cells (156,158). Similarly, IL-6R signaling also STAT3 phosphorylation and inhibits Treg cell function in a mouse model of allergic inflammation (157), and a recent study demonstrated that STAT3 signaling in both Treg cells and effector T cells contributes to IL-6-mediated Treg cell dysfunction. Thus, it appears that through both direct and indirect mechanisms, IL-1 and IL-6 subvert Treg cell function during inflammation.
Like IL-1 and IL-6, tumor necrosis factor alpha (TNF) is a pleiotropic cytokine promoting systemic inflammation through its induction of fever, the acute phase response, and activation of macrophages and neutrophils. Although TNF is critical for host defense against a variety of pathogens, it is strongly implicated in the pathogenesis of several inflammatory disorders like IBD, RA and psoriasis, and TNF inhibitors are highly efficacious in treating these diseases (159). How TNF modulates Treg cell activity, however, remains controversial. TNF can increase IL-2 induced STAT5 phosphorylation in Treg cells and can increase Treg suppressive activity in vitro, though this study did not distinguish between direct effects on Treg cells and effector T cells (160). A direct role for TNF signaling in maintaining Treg function in vivo has been demonstrated in a model of inflammatory colitis, as TNFR2−/− Treg cells expressed lower levels of Foxp3 and were unable to suppress disease as efficiently as wild-type Treg cells (161,162). However, a number of studies indicate that TNF actually impairs Treg suppressive activity both directly and indirectly. For instance, Wehrens et al. revealed that over-expression of TNF and IL-6 in the inflamed synovium of patients with JIA induces protein kinase B (PKB) hyperactivation in effector T cells, making them resistant to suppression (163). Moreover, Treg cells are dysfunctional in the inflamed joints of patients with RA, and several recent studies have pointed to TNF as the major mediator of this dysfunction (92,164,165). Thus, TNF seems to have opposing and context-dependent effects on Treg cell stability and function, and the mechanisms underlying this complexity remain poorly defined.
Like, TNF-α, the type I interferons (IFNs) are a family of pro-inflammatory cytokines that are essential for anti-viral immunity in both mice and humans but whose over-production is associated with a number of organ-specific and systemic autoimmune disorders. These cytokines signal through the ubiquitously expressed heterodimeric type I IFN receptor (IFNαR), leading to phosphorylation and activation of STAT1 and STAT2, and induction of hundreds of IFN-stimulated genes (ISGs). Type I IFNs can induce apoptosis, block translation, and inhibit cellular proliferation of many cell types, limiting viral spread and making type I IFNs clinically useful in the treatment of chronic viral infection and certain types of leukemia. Also similar to TNF-α, studies have provided conflicting results regarding the impact of type I IFNs on Treg cells (166–170), and have generally not used experimental systems to examine the direct effects of IFNs on Treg cell homeostasis and function. However, using experimental systems in which the direct effects of type-1 IFN on Treg cells can be isolated, we have shown that type I IFNs directly inhibit Treg cell activity in vitro and in vivo during acute infection with LCMV, and that this transient inhibition of Treg cells is necessary for the generation of optimal antiviral T cell responses (Srivastava et al., Submitted). However, the immunomodulatory effects of type I IFNs are incredibly complex, and recent studies have suggested that type I IFNs may exert opposing effects based on the timing and extent of their expression. For instance, during acute viral infection, IFNs are induced transiently and promote viral clearance, whereas during chronic infection, heightened and prolonged IFN expression promotes viral persistence and immunosuppression (171). It is therefore important to understand exactly how IFNs regulate Treg cell activity, both directly and indirectly, and further studies are required to determine how acute and prolonged IFN production impacts Treg cell function, and how this may in turn contribute to immune dysfunction in chronic infection and type I IFN-associated autoimmune diseases.
IFN-γ is a pleiotropic cytokine classically known for its role during type I inflammation, where it is required for the clearance of intracellular pathogens such as Mycobacterium tuberculosis, Leishmania major and Listeria monocytogenes. In addition to its activating effects on CD8+ cytotoxic T cells, NK cells, and macrophages, IFN-γ also polarizes naïve CD4+ T cells towards the Th1 lineage and potently inhibits the induction of Foxp3 expression during T cell activation in the periphery (172). In this, IFN-γ is similar to IL-27, and both cytokines signal in large part through activation of STAT1 (173,174). However, both IFNγ and IL-27 can potently induce T-bet expression in thymus-derived Treg cells, enabling them to better control Th1-mediated inflammatory responses (175,176). Moreover, delayed induction of IL12Rβ2 in IFN-γ-stimulated Treg cells prevents them from continuing IL-12/STAT4-dependent Th1 cell development and allows them to maintain their suppressive function (177). Interestingly, several studies have demonstrated that Treg cells can produce IFN-γ when stimulated in a strongly inflammatory Th1 environment. In some contexts, Treg cell-derived IFN-γ is protective and required for normal suppressive function. For example, Ifng−/− CD4+CD25+ Treg cells fail to protect against skin graft rejection and graft versus host disease in models of allogeneic transplantation (178). By contrast, IFN-γ production has also been associated with Treg cell dysfunction during infection with Toxoplasma gondii (138), and excessive STAT1 activation, via either loss of the STAT1-inhibitors mir146a or Socs1, can also result in IFNγ production by Treg cells, resulting in loss of suppressive function and Th1-mediated pathology (179). IFN-γ, thus, can both promote and subvert Treg suppressive activity in different settings, and the balance between these opposing functions likely depends on contextual factors such as the timing and extent of expression.
IL-10
In addition to being a potent anti-inflammatory cytokine produced by Treg cells, several recent studies have highlighted a direct role for IL-10 in the maintenance of Treg suppressive function during inflammation. Murai et al. demonstrated that IL-10 produced by macrophages in the lamina propria is required to maintain Foxp3 expression and suppressive function in Treg cells during inflammation (180). Treg cells deficient in the IL-10 receptor β chain lost expression of Foxp3 and were unable to control inflammatory disease as well as wild-type Treg cells in a transfer model of colitis. Interestingly, IL-10RB-deficient Treg cells did not lose Foxp3 expression when co-transferred with wild-type Treg cells that sufficiently protected against colitis, suggesting that loss of Foxp3 expression in these cells is due to the inflammatory environment. Similarly, Chaudhry et al. showed that IL-10 induces STAT3 phosphorylation in Treg cells, and that ablation of IL-10RB or STAT3 expression in Treg cells resulted in spontaneous dysregulation of Th17 cell responses and inflammatory colitis (181,182). Thus, in addition to its effects on T cells and APCs, IL-10 mediates its anti-inflammatory effects in part through its regulation of Treg stability and function both during steady-state conditions and during inflammation.
Pathogen recognition receptors
Detection of invading pathogens occurs through a number of different pathogen recognition receptors (PRRs), a class of receptors that recognizes pathogen-associated molecular patterns (PAMPs). Infected cells can directly sense the presence of pathogens via the cell-intrinsic RIG-I-like receptors (RLRs) or via stimulation of cell surface-localized TLRs, which signal via the signaling adaptor MyD88. Conversely, professional APCs can endocytose infected cells and detect pathogens via stimulation of endosomal TLRs (TLR3, 7/8, 9). As engagement of PRRs generally leads to production of pro-inflammatory cytokines, it is not surprising that these receptors can have a dramatic impact on Treg cell abundance and function. For instance, production of IL-1β and IL-6 following TLR7 and TLR9 activation in DCs potently blocks Treg suppressive function (91,183). Moreover, Tlr9−/− mice displayed elevated frequencies of Treg cells and reduced frequencies of IL-17- and IFNγ-producing effector T cells in the intestine, resulting in impaired responses to oral infection and vaccianation (184). Recent studies, however, have demonstrated that Treg cells express a number of TLRs that may directly regulate their activity. For instance, TLR8 is preferentially expressed on human Treg cells compared to effector T cells, and both synthetic and natural TLR8 agonists abrogate Treg suppressive function and dampen their tumor-promoting activity (185). Likewise, Treg cells treated with the synthetic TLR2 ligand Pam3Cys transiently reduce Foxp3 expression and lose their suppressive function in vitro and in vivo in models of colitis and Leishmania infection (186). By contrast, the endogenous TLR2 agonist heat-shock protein 60 (Hsp60) enhances human Treg cell activity, and Tlr2−/− mice have reduced numbers of Treg cells and enhanced resistance to Candida albicans (187). These divergent effects may be explained in part by the fact that TLR2 can heterodimerize with TLR1 and TLR6, and thus may signal differently in response to different TLR1/2 or TLR2/6 agonists. Other TLRs, like TLR4 and TLR5, have clearer activating roles on Treg cells. Direct engagement of TLR4 by LPS induces Treg proliferation and enhances suppressive function (188), while engagement of TLR5 by flagellin enhances Foxp3 expression and suppressive activity (189). Treg cells also express the cell-intrinsic nucleic acid receptors RIG-I and MDA5, which are critical for viral recognition and signal through the common signaling adaptor IPS-1. Indeed, Ips1−/− mice infected with West Nile virus show defective Treg cell expansion and dysregulated T cell activation, failing to control the infection (190). Whether this defect in Treg cell activity was due to cell-intrinsic loss of IPS-1 or was secondary to the elevated systemic proinflammatory cytokines observed in these mice is unclear. Activation of Treg-intrinsic MDA5 by infection with encephalomyocarditis virus, however, was shown to subvert Treg suppressive function in vitro in co-cultures, even in the absence of APCs or MDA5 expression in effector T cells (191), demonstrating that intracellular nucleic acid sensing can directly modulate Treg cell activity during infection.
CONCLUSION
As can be seen, maintenance of the diverse pool of peripheral Treg cells is a dynamically regulated process that allows the immune system to appropriately initiate and resolve immune responses to invading pathogens and tumors, while preventing autoimmunity and limiting collateral tissue damage and immunopathology. Treg homeostasis is governed by a number of factors, ranging from cytokine and TCR/co-stimulatory signaling to anatomical location, and interestingly, many of the same environmental signals are perceived differently by effector and regulatory T cells, allowing these functionally opposed populations to be differentially regulated in the same microenvironment. Treatments for autoimmunity, like low dose IL-2 therapy or TNF blockade, are exploiting these differences to enhance Treg cell function without increasing effector T cell function.
Manipulation of Treg cell numbers has been proposed as a promising new avenue for the treatment of chronic infection, autoimmunity, and cancer. These strategies, however, may prove challenging given the harmful effects that can arise from the presence of too many or too few Treg cell numbers, and thus targeted approaches to enhance or limit the function of particular subsets of Treg cells may prove more effective than global changes in Treg cell numbers, and further studies are required to determine how best to manipulate both the quality and quantity of Treg cell responses for the treatment of human disease.
References
- 1.Boyman O, Krieg C, Homann D, Sprent J. Homeostatic maintenance of T cells and natural killer cells. Cell Mol Life Sci. 2012;69:1597–1608. doi: 10.1007/s00018-012-0968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lio CW, Hsieh CS. Becoming self-aware: the thymic education of regulatory T cells. Curr Opin Immunol. 2010 doi: 10.1016/j.coi.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von BH. Mechanisms of suppression by suppressor T cells. Nat Immunol. 2005;6:338–344. doi: 10.1038/ni1180. [DOI] [PubMed] [Google Scholar]
- 5.Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3(+) regulatory T cells. Nat Rev Immunol. 2011;11:119–130. doi: 10.1038/nri2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–479. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 7.Gu H, Maeda H, Moon JJ, Lord JD, Yoakim M, Nelson BH, Neel BG. New role for Shc in activation of the phosphatidylinositol 3-kinase/Akt pathway. Mol Cell Biol. 2000;20:7109–7120. doi: 10.1128/mcb.20.19.7109-7120.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HR, Hwang KA, Park SH, Kang I. IL-7 and IL-15: biology and roles in T-Cell immunity in health and disease. Crit Rev Immunol. 2008;28:325–339. doi: 10.1615/critrevimmunol.v28.i4.40. [DOI] [PubMed] [Google Scholar]
- 9.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 10.Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol. 2008;181:3285–3290. doi: 10.4049/jimmunol.181.5.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahmud SA, Manlove LS, Farrar MA. Interleukin-2 and STAT5 in regulatory T cell development and function. JAKSTAT. 2013;2:e23154. doi: 10.4161/jkst.23154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, Humblet-Baron S, Schonefeldt S, Herold MJ, Hildeman D, Strasser A, Bouillet P, Lu LF, Matthys P, Freitas AA, Luther RJ, Weaver CT, Dooley J, Gray DH, Liston A. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3(+) regulatory T cells. Nat Immunol. 2013;14:959–965. doi: 10.1038/ni.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng G, Podack ER. Suppression of apoptosis in a cytotoxic T-cell line by interleukin 2-mediated gene transcription and deregulated expression of the protooncogene bcl-2. Proc Natl Acad Sci U S A. 1993;90:2189–2193. doi: 10.1073/pnas.90.6.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen C, Rowell EA, Thomas RM, Hancock WW, Wells AD. Transcriptional regulation by Foxp3 is associated with direct promoter occupancy and modulation of histone acetylation. J Biol Chem. 2006;281:36828–36834. doi: 10.1074/jbc.M608848200. [DOI] [PubMed] [Google Scholar]
- 15.Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 16.Kim HP, Kelly J, Leonard WJ. The basis for IL-2-induced IL-2 receptor alpha chain gene regulation: importance of two widely separated IL-2 response elements. Immunity. 2001;15:159–172. doi: 10.1016/s1074-7613(01)00167-4. [DOI] [PubMed] [Google Scholar]
- 17.Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- 18.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, Mathis D, Benoist C, Chen L, Rao A. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 19.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 20.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 21.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 22.Soper DM, Kasprowicz DJ, Ziegler SF. IL-2Rbeta links IL-2R signaling with Foxp3 expression. Eur J Immunol. 2007;37:1817–1826. doi: 10.1002/eji.200737101. [DOI] [PubMed] [Google Scholar]
- 23.Malek TR, Porter BO, Codias EK, Scibelli P, Yu A. Normal lymphoid homeostasis and lack of lethal autoimmunity in mice containing mature T cells with severely impaired IL-2 receptors. J Immunol. 2000;164:2905–2914. doi: 10.4049/jimmunol.164.6.2905. [DOI] [PubMed] [Google Scholar]
- 24.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, Bluestone JA. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- 27.Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, Grey ST, Sprent J. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med. 2009;206:751–760. doi: 10.1084/jem.20082824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O’Gorman WE, Abbas AK. Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J Immunol. 2010;185:6426–6430. doi: 10.4049/jimmunol.0903940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tai X, Erman B, Alag A, Mu J, Kimura M, Katz G, Guinter T, McCaughtry T, Etzensperger R, Feigenbaum L, Singer DS, Singer A. Foxp3 transcription factor is proapoptotic and lethal to developing regulatory T cells unless counterbalanced by cytokine survival signals. Immunity. 2013;38:1116–1128. doi: 10.1016/j.immuni.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smigiel KS, Richards E, Srivastava S, Thomas KN, Dudda JC, Klonowski KD, Campbell DJ. CCR7 provides localized access to IL-2 and defines homeostatically distinct regualtory T cell subsets. 2014 doi: 10.1084/jem.20131142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, Lafaille JJ, Dustin ML. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med. 2006;203:505–511. doi: 10.1084/jem.20050783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cretney E, Kallies A, Nutt SL. Differentiation and function of Foxp3(+) effector regulatory T cells. Trends Immunol. 2013;34:74–80. doi: 10.1016/j.it.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, Cyster JG, Luther SA. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. 2007;8:1255–1265. doi: 10.1038/ni1513. [DOI] [PubMed] [Google Scholar]
- 34.Wojciechowski S, Tripathi P, Bourdeau T, Acero L, Grimes HL, Katz JD, Finkelman FD, Hildeman DA. Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J Exp Med. 2007;204:1665–1675. doi: 10.1084/jem.20070618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 36.Boyman O, Ramsey C, Kim DM, Sprent J, Surh CD. IL-7/anti-IL-7 mAb complexes restore T cell development and induce homeostatic T Cell expansion without lymphopenia. J Immunol. 2008;180:7265–7275. doi: 10.4049/jimmunol.180.11.7265. [DOI] [PubMed] [Google Scholar]
- 37.Simons DM, Picca CC, Oh S, Perng OA, Aitken M, Erikson J, Caton AJ. How specificity for self-peptides shapes the development and function of regulatory T cells. J Leukoc Biol. 2010;88:1099–1107. doi: 10.1189/jlb.0310183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med. 2003;198:249–258. doi: 10.1084/jem.20030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat Immunol. 2002;3:756–763. doi: 10.1038/ni816. [DOI] [PubMed] [Google Scholar]
- 40.Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 41.Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, Amit AS, Kang C, Geddes JE, Allison JP, Socci ND, Savage PA. Aire-dependent thymic development of tumor-associated regulatory T cells. Science. 2013;339:1219–1224. doi: 10.1126/science.1233913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–277. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 43.Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, Hogquist KA. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lathrop SK, Santacruz NA, Pham D, Luo J, Hsieh CS. Antigen-specific peripheral shaping of the natural regulatory T cell population. J Exp Med. 2008;205:3105–3117. doi: 10.1084/jem.20081359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Darrasse-Jeze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, Masilamani RF, Dustin ML, Rudensky A, Liu K, Nussenzweig MC. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med. 2009;206:1853–1862. doi: 10.1084/jem.20090746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim JK, Klinger M, Benjamin J, Xiao Y, Erle DJ, Littman DR, Killeen N. Impact of the TCR signal on regulatory T cell homeostasis, function, and trafficking. PLoS One. 2009;4:e6580. doi: 10.1371/journal.pone.0006580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Au-Yeung BB, Levin SE, Zhang C, Hsu LY, Cheng DA, Killeen N, Shokat KM, Weiss A. A genetically selective inhibitor demonstrates a function for the kinase Zap70 in regulatory T cells independent of its catalytic activity. Nat Immunol. 2010;11:1085–1092. doi: 10.1038/ni.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, Rowan WC, Sancho S, Walker LS, Vanhaesebroeck B, Okkenhaug K. Cutting edge: the phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 2006;177:6598–6602. doi: 10.4049/jimmunol.177.10.6598. [DOI] [PubMed] [Google Scholar]
- 49.Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. 2008;205:565–574. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, Chi H. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol. 2009;10:769–777. doi: 10.1038/ni.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. 2013;499:485–490. doi: 10.1038/nature12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 54.Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, Zheng XX, Strom TB, Bluestone JA. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol. 2003;171:3348–3352. doi: 10.4049/jimmunol.171.7.3348. [DOI] [PubMed] [Google Scholar]
- 55.Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, Turka LA. An obligate cell-intrinsic function for CD28 in Tregs. J Clin Invest. 2013 doi: 10.1172/JCI65013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bar-On L, Birnberg T, Kim KW, Jung S. Dendritic cell-restricted CD80/86 deficiency results in peripheral regulatory T-cell reduction but is not associated with lymphocyte hyperactivation. Eur J Immunol. 2011;41:291–298. doi: 10.1002/eji.201041169. [DOI] [PubMed] [Google Scholar]
- 57.Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam KP, Coyle AJ, Kroczek RA, Hutloff A. ICOS controls the pool size of effector-memory and regulatory T cells. J Immunol. 2008;180:774–782. doi: 10.4049/jimmunol.180.2.774. [DOI] [PubMed] [Google Scholar]
- 58.Herman AE, Freeman GJ, Mathis D, Benoist C. CD4+CD25+ T Regulatory Cells Dependent on ICOS Promote Regulation of Effector Cells in the Prediabetic Lesion. J Exp Med. 2004;199:1479–1489. doi: 10.1084/jem.20040179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ, Fagarasan S, Liston A, Smith KG, Vinuesa CG. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17:975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, Wang YH, Lim H, Reynolds JM, Zhou XH, Fan HM, Liu ZM, Neelapu SS, Dong C. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. 2011;17:983–988. doi: 10.1038/nm.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol. 2013;14:152–161. doi: 10.1038/ni.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoyer KK, Wolslegel K, Dooms H, Abbas AK. Targeting T cell-specific costimulators and growth factors in a model of autoimmune hemolytic anemia. J Immunol. 2007;179:2844–2850. doi: 10.4049/jimmunol.179.5.2844. [DOI] [PubMed] [Google Scholar]
- 63.Zou T, Satake A, Corbo-Rodgers E, Schmidt AM, Farrar MA, Maltzman JS, Kambayashi T. Cutting Edge: IL-2 Signals Determine the Degree of TCR Signaling Necessary To Support Regulatory T Cell Proliferation In Vivo. J Immunol. 2012;189:28–32. doi: 10.4049/jimmunol.1200507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 65.Mori S, Nakano H, Aritomi K, Wang CR, Gunn MD, Kakiuchi T. Mice lacking expression of the chemokines CCL21-ser and CCL19 (plt mice) demonstrate delayed but enhanced T cell immune responses. J Exp Med. 2001;193:207–218. doi: 10.1084/jem.193.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J Exp Med. 2007;204:735–745. doi: 10.1084/jem.20061405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davalos-Misslitz AC, Rieckenberg J, Willenzon S, Worbs T, Kremmer E, Bernhardt G, Forster R. Generalized multi-organ autoimmunity in CCR7-deficient mice. Eur J Immunol. 2007;37:613–622. doi: 10.1002/eji.200636656. [DOI] [PubMed] [Google Scholar]
- 68.Malhotra D, Fletcher AL, Turley SJ. Stromal and hematopoietic cells in secondary lymphoid organs: partners in immunity. Immunol Rev. 2013;251:160–176. doi: 10.1111/imr.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc Natl Acad Sci U S A. 2000;97:12694–12699. doi: 10.1073/pnas.97.23.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sabatos CA, Doh J, Chakravarti S, Friedman RS, Pandurangi PG, Tooley AJ, Krummel MF. A synaptic basis for paracrine interleukin-2 signaling during homotypic T cell interaction. Immunity. 2008;29:238–248. doi: 10.1016/j.immuni.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. 2013;14:1007–1013. doi: 10.1038/ni.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, Debes GF, Lauber J, Frey O, Przybylski GK, Niesner U, de la RM, Schmidt CA, Brauer R, Buer J, Scheffold A, Hamann A. Developmental Stage, Phenotype, and Migration Distinguish Naive- and Effector/Memory-like CD4+ Regulatory T Cells. J Exp Med. 2004;199:303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dudda JC, Perdue N, Bachtanian E, Campbell DJ. Foxp3+ regulatory T cells maintain immune homeostasis in the skin. J Exp Med. 2008;205:1559–1565. doi: 10.1084/jem.20072594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Freyschmidt EJ, Mathias CB, Diaz N, MacArthur DH, Laouar A, Manjunath N, Hofer MD, Wurbel MA, Campbell JJ, Chatila TA, Oettgen HC. Skin inflammation arising from cutaneous regulatory T cell deficiency leads to impaired viral immune responses. J Immunol. 2010;185:1295–1302. doi: 10.4049/jimmunol.0903144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, Campbell DJ. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med. 2007;204:1335–1347. doi: 10.1084/jem.20070081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, Belz GT, Smyth GK, Busslinger M, Nutt SL, Kallies A. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol. 2011;12:304–311. doi: 10.1038/ni.2006. [DOI] [PubMed] [Google Scholar]
- 77.Kallies A, Hawkins ED, Belz GT, Metcalf D, Hommel M, Corcoran LM, Hodgkin PD, Nutt SL. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat Immunol. 2006;7:466–474. doi: 10.1038/ni1321. [DOI] [PubMed] [Google Scholar]
- 78.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr, Muller W, Rudensky AY. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 79.Killebrew JR, Perdue N, Kwan A, Thornton AM, Shevach EM, Campbell DJ. A Self-Reactive TCR Drives the Development of Foxp3+ Regulatory T Cells That Prevent Autoimmune Disease. J Immunol. 2011 doi: 10.4049/jimmunol.1004009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rosenblum MD, I, Gratz K, Paw JS, Lee K, Marshak-Rothstein A, Abbas AK. Response to self antigen imprints regulatory memory in tissues. Nature. 2011;480:538–542. doi: 10.1038/nature10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gratz IK, Truong HA, Yang SH, Maurano MM, Lee K, Abbas AK, Rosenblum MD. Cutting Edge: Memory Regulatory T Cells Require IL-7 and Not IL-2 for Their Maintenance in Peripheral Tissues. J Immunol. 2013;190:4483–4487. doi: 10.4049/jimmunol.1300212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions (*) Annu Rev Immunol. 2009;27:551–589. doi: 10.1146/annurev.immunol.021908.132723. [DOI] [PubMed] [Google Scholar]
- 83.Fulton RB, Meyerholz DK, Varga SM. Foxp3+ CD4 regulatory T cells limit pulmonary immunopathology by modulating the CD8 T cell response during respiratory syncytial virus infection. J Immunol. 2010;185:2382–2392. doi: 10.4049/jimmunol.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pace L, Tempez A, Arnold-Schrauf C, Lemaitre F, Bousso P, Fetler L, Sparwasser T, Amigorena S. Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science. 2012;338:532–536. doi: 10.1126/science.1227049. [DOI] [PubMed] [Google Scholar]
- 86.Benson A, Murray S, Divakar P, Burnaevskiy N, Pifer R, Forman J, Yarovinsky F. Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. J Immunol. 2012;188:800–810. doi: 10.4049/jimmunol.1100769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ballesteros-Tato A, Leon B, Lund FE, Randall TD. CD4+ T helper cells use CD154-CD40 interactions to counteract T reg cell-mediated suppression of CD8+ T cell responses to influenza. J Exp Med. 2013;210:1591–1601. doi: 10.1084/jem.20130097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hanig J, Lutz MB. Suppression of mature dendritic cell function by regulatory T cells in vivo is abrogated by CD40 licensing. J Immunol. 2008;180:1405–1413. doi: 10.4049/jimmunol.180.3.1405. [DOI] [PubMed] [Google Scholar]
- 89.Serra P, Amrani A, Yamanouchi J, Han B, Thiessen S, Utsugi T, Verdaguer J, Santamaria P. CD40 ligation releases immature dendritic cells from the control of regulatory CD4+CD25+ T cells. Immunity. 2003;19:877–889. doi: 10.1016/s1074-7613(03)00327-3. [DOI] [PubMed] [Google Scholar]
- 90.O’Sullivan BJ, Thomas HE, Pai S, Santamaria P, Iwakura Y, Steptoe RJ, Kay TW, Thomas R. IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J Immunol. 2006;176:7278–7287. doi: 10.4049/jimmunol.176.12.7278. [DOI] [PubMed] [Google Scholar]
- 91.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 92.Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, Liu X, Xiao L, Chen X, Wan B, Chin YE, Zhang JZ. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med. 2013;19:322–328. doi: 10.1038/nm.3085. [DOI] [PubMed] [Google Scholar]
- 93.Bolacchi F, Sinistro A, Ciaprini C, Demin F, Capozzi M, Carducci FC, Drapeau CM, Rocchi G, Bergamini A. Increased hepatitis C virus (HCV)-specific CD4+CD25+ regulatory T lymphocytes and reduced HCV-specific CD4+ T cell response in HCV-infected patients with normal versus abnormal alanine aminotransferase levels. Clin Exp Immunol. 2006;144:188–196. doi: 10.1111/j.1365-2249.2006.03048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cabrera R, Tu Z, Xu Y, Firpi RJ, Rosen HR, Liu C, Nelson DR. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology. 2004;40:1062–1071. doi: 10.1002/hep.20454. [DOI] [PubMed] [Google Scholar]
- 95.Guyot-Revol V, Innes JA, Hackforth S, Hinks T, Lalvani A. Regulatory T cells are expanded in blood and disease sites in patients with tuberculosis. Am J Respir Crit Care Med. 2006;173:803–810. doi: 10.1164/rccm.200508-1294OC. [DOI] [PubMed] [Google Scholar]
- 96.Shafiani S, Tucker-Heard G, Kariyone A, Takatsu K, Urdahl KB. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J Exp Med. 2010;207:1409–1420. doi: 10.1084/jem.20091885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shafiani S, Dinh C, Ertelt JM, Moguche AO, Siddiqui I, Smigiel KS, Sharma P, Campbell DJ, Way SS, Urdahl KB. Pathogen-Specific Treg Cells Expand Early during Mycobacterium tuberculosis Infection but Are Later Eliminated in Response to Interleukin- 12. Immunity. 2013;38:1261–1270. doi: 10.1016/j.immuni.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Scott-Browne JP, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot JD, Rudensky AY, Bevan MJ, Urdahl KB. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J Exp Med. 2007;204:2159–2169. doi: 10.1084/jem.20062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hisaeda H, Maekawa Y, Iwakawa D, Okada H, Himeno K, Kishihara K, Tsukumo S, Yasutomo K. Escape of malaria parasites from host immunity requires CD4+ CD25+ regulatory T cells. Nat Med. 2004;10:29–30. doi: 10.1038/nm975. [DOI] [PubMed] [Google Scholar]
- 100.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 101.Sugimoto K, Ikeda F, Stadanlick J, Nunes FA, Alter HJ, Chang KM. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology. 2003;38:1437–1448. doi: 10.1016/j.hep.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 102.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 103.Hesse M, Piccirillo CA, Belkaid Y, Prufer J, Mentink-Kane M, Leusink M, Cheever AW, Shevach EM, Wynn TA. The pathogenesis of schistosomiasis is controlled by cooperating IL-10-producing innate effector and regulatory T cells. J Immunol. 2004;172:3157–3166. doi: 10.4049/jimmunol.172.5.3157. [DOI] [PubMed] [Google Scholar]
- 104.Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J Immunol. 2004;172:4123–4132. doi: 10.4049/jimmunol.172.7.4123. [DOI] [PubMed] [Google Scholar]
- 105.Brusko T, Atkinson M. Treg in type 1 diabetes. Cell Biochem Biophys. 2007;48:165–175. doi: 10.1007/s12013-007-0018-5. [DOI] [PubMed] [Google Scholar]
- 106.Putheti P, Pettersson A, Soderstrom M, Link H, Huang YM. Circulating CD4+CD25+ T regulatory cells are not altered in multiple sclerosis and unaffected by disease-modulating drugs. J Clin Immunol. 2004;24:155–161. doi: 10.1023/B:JOCI.0000019780.93817.82. [DOI] [PubMed] [Google Scholar]
- 107.Lee JH, Wang LC, Lin YT, Yang YH, Lin DT, Chiang BL. Inverse correlation between CD4+ regulatory T-cell population and autoantibody levels in paediatric patients with systemic lupus erythematosus. Immunology. 2006;117:280–286. doi: 10.1111/j.1365-2567.2005.02306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, Nochy D, Debre P, Piette JC, Gorochov G. Global natural regulatory T cell depletion in active systemic lupus erythematosus. J Immunol. 2005;175:8392–8400. doi: 10.4049/jimmunol.175.12.8392. [DOI] [PubMed] [Google Scholar]
- 109.Lord JD, Valliant-Saunders K, Hahn H, Thirlby RC, Ziegler SF. Paradoxically increased FOXP3+ T cells in IBD do not preferentially express the isoform of FOXP3 lacking exon 2. Dig Dis Sci. 2012;57:2846–2855. doi: 10.1007/s10620-012-2292-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. 2004;50:2775–2785. doi: 10.1002/art.20499. [DOI] [PubMed] [Google Scholar]
- 111.Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2008;105:19396–19401. doi: 10.1073/pnas.0806855105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol. 2008;181:7350–7355. doi: 10.4049/jimmunol.181.10.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kelsen J, Agnholt J, Hoffmann HJ, Romer JL, Hvas CL, Dahlerup JF. FoxP3(+)CD4(+)CD25(+) T cells with regulatory properties can be cultured from colonic mucosa of patients with Crohn’s disease. Clin Exp Immunol. 2005;141:549–557. doi: 10.1111/j.1365-2249.2005.02876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lawson JM, Tremble J, Dayan C, Beyan H, Leslie RD, Peakman M, Tree TI. Increased resistance to CD4+CD25hi regulatory T cell-mediated suppression in patients with type 1 diabetes. Clin Exp Immunol. 2008;154:353–359. doi: 10.1111/j.1365-2249.2008.03810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yan B, Ye S, Chen G, Kuang M, Shen N, Chen S. Dysfunctional CD4+,CD25+ regulatory T cells in untreated active systemic lupus erythematosus secondary to interferon-alpha-producing antigen-presenting cells. Arthritis Rheum. 2008;58:801–812. doi: 10.1002/art.23268. [DOI] [PubMed] [Google Scholar]
- 118.van Amelsfort JM, van Roon JA, Noordegraaf M, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. Proinflammatory mediator-induced reversal of CD4+,CD25+ regulatory T cell-mediated suppression in rheumatoid arthritis. Arthritis Rheum. 2007;56:732–742. doi: 10.1002/art.22414. [DOI] [PubMed] [Google Scholar]
- 119.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 120.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 121.Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L, Coukos G. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 122.Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. 2011;470:548–553. doi: 10.1038/nature09707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009;27:186–192. doi: 10.1200/JCO.2008.18.7229. [DOI] [PubMed] [Google Scholar]
- 124.Carreras J, Lopez-Guillermo A, Fox BC, Colomo L, Martinez A, Roncador G, Montserrat E, Campo E, Banham AH. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood. 2006;108:2957–2964. doi: 10.1182/blood-2006-04-018218. [DOI] [PubMed] [Google Scholar]
- 125.Alvaro T, Lejeune M, Salvado MT, Bosch R, Garcia JF, Jaen J, Banham AH, Roncador G, Montalban C, Piris MA. Outcome in Hodgkin’s lymphoma can be predicted from the presence of accompanying cytotoxic and regulatory T cells. Clin Cancer Res. 2005;11:1467–1473. doi: 10.1158/1078-0432.CCR-04-1869. [DOI] [PubMed] [Google Scholar]
- 126.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 127.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 128.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 129.Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, Eberlein TJ, Hsieh CS, Linehan DC. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182:1746–1755. doi: 10.4049/jimmunol.182.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jarnicki AG, Lysaght J, Todryk S, Mills KH. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol. 2006;177:896–904. doi: 10.4049/jimmunol.177.2.896. [DOI] [PubMed] [Google Scholar]
- 131.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59:3128–3133. [PubMed] [Google Scholar]
- 132.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Robert C, Thomas L, Bondarenko I, O’Day S, MDJW, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH, Jr, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 134.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D, Bonnevier J, Workman CJ, Vignali DA. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013;501:252–256. doi: 10.1038/nature12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Long M, Adler AJ. Cutting edge: Paracrine, but not autocrine, IL-2 signaling is sustained during early antiviral CD4 T cell response. J Immunol. 2006;177:4257–4261. doi: 10.4049/jimmunol.177.7.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.O’Gorman WE, Dooms H, Thorne SH, Kuswanto WF, Simonds EF, Krutzik PO, Nolan GP, Abbas AK. The initial phase of an immune response functions to activate regulatory T cells. J Immunol. 2009;183:332–339. doi: 10.4049/jimmunol.0900691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos SL, O’Brien S, Blank R, Lamb E, Natarajan S, Kastenmayer R, Hunter C, Grigg ME, Belkaid Y. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Clark RA, Kupper TS. IL-15 and dermal fibroblasts induce proliferation of natural regulatory T cells isolated from human skin. Blood. 2007;109:194–202. doi: 10.1182/blood-2006-02-002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Koenen HJ, Fasse E, Joosten I. IL-15 and cognate antigen successfully expand de novo-induced human antigen-specific regulatory CD4+ T cells that require antigen-specific activation for suppression. J Immunol. 2003;171:6431–6441. doi: 10.4049/jimmunol.171.12.6431. [DOI] [PubMed] [Google Scholar]
- 141.Xia J, Liu W, Hu B, Tian Z, Yang Y. IL-15 promotes regulatory T cell function and protects against diabetes development in NK-depleted NOD mice. Clin Immunol. 2010;134:130–139. doi: 10.1016/j.clim.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 142.Ben AM, Belhadj HN, Moes N, Buyse S, Abdeladhim M, Louzir H, Cerf-Bensussan N. IL-15 renders conventional lymphocytes resistant to suppressive functions of regulatory T cells through activation of the phosphatidylinositol 3-kinase pathway. J Immunol. 2009;182:6763–6770. doi: 10.4049/jimmunol.0801792. [DOI] [PubMed] [Google Scholar]
- 143.DePaolo RW, Abadie V, Tang F, Fehlner-Peach H, Hall JA, Wang W, Marietta EV, Kasarda DD, Waldmann TA, Murray JA, Semrad C, Kupfer SS, Belkaid Y, Guandalini S, Jabri B. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature. 2011;471:220–224. doi: 10.1038/nature09849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Benito-Miguel M, Garcia-Carmona Y, Balsa A, Perez de AC, Cobo-Ibanez T, Martin-Mola E, Miranda-Carus ME. A dual action of rheumatoid arthritis synovial fibroblast IL-15 expression on the equilibrium between CD4+CD25+ regulatory T cells and CD4+ J Immunol. 2009;183:8268–8279. doi: 10.4049/jimmunol.0900007. [DOI] [PubMed] [Google Scholar]
- 145.Redpath SA, van der Werf N, Cervera AM, MacDonald AS, Gray D, Maizels RM, Taylor MD. ICOS controls Foxp3(+) regulatory T-cell expansion, maintenance and IL-10 production during helminth infection. Eur J Immunol. 2013;43:705–715. doi: 10.1002/eji.201242794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Strauss L, Bergmann C, Szczepanski MJ, Lang S, Kirkwood JM, Whiteside TL. Expression of ICOS on human melanoma-infiltrating CD4+CD25highFoxp3+ T regulatory cells: implications and impact on tumor-mediated immune suppression. J Immunol. 2008;180:2967–2980. doi: 10.4049/jimmunol.180.5.2967. [DOI] [PubMed] [Google Scholar]
- 147.Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, Sanders D, Lacey C, Wang Y, Vence L, Hwu P, Radvanyi L. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J Clin Invest. 2013 doi: 10.1172/JCI46266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gopisetty A, Bhattacharya P, Haddad C, Bruno JC, Jr, Vasu C, Miele L, Prabhakar BS. OX40L/Jagged1 cosignaling by GM-CSF-induced bone marrow-derived dendritic cells is required for the expansion of functional regulatory T cells. J Immunol. 2013;190:5516–5525. doi: 10.4049/jimmunol.1202298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bresson D, Fousteri G, Manenkova Y, Croft M, Mvon H. Antigen-specific prevention of type 1 diabetes in NOD mice is ameliorated by OX40 agonist treatment. J Autoimmun. 2011;37:342–351. doi: 10.1016/j.jaut.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Griseri T, Asquith M, Thompson C, Powrie F. OX40 is required for regulatory T cell-mediated control of colitis. J Exp Med. 2010;207:699–709. doi: 10.1084/jem.20091618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xiao X, Gong W, Demirci G, Liu W, Spoerl S, Chu X, Bishop DK, Turka LA, Li XC. New insights on OX40 in the control of T cell immunity and immune tolerance in vivo. J Immunol. 2012;188:892–901. doi: 10.4049/jimmunol.1101373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, Sugamura K, Ishii N. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol. 2004;172:3580–3589. doi: 10.4049/jimmunol.172.6.3580. [DOI] [PubMed] [Google Scholar]
- 153.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Li XC. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825–839. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Schneider A, Long SA, Cerosaletti K, Ni CT, Samuels P, Kita M, Buckner JH. In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive T(regs) involves IL-6-mediated signaling. Sci Transl Med. 2013;5:170ra15. doi: 10.1126/scitranslmed.3004970. [DOI] [PubMed] [Google Scholar]
- 156.Li L, Kim J, Boussiotis VA. IL-1beta-mediated signals preferentially drive conversion of regulatory T cells but not conventional T cells into IL-17-producing cells. J Immunol. 2010;185:4148–4153. doi: 10.4049/jimmunol.1001536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ, Haddad e, Lehr HA, Schmitt E, Bopp T, Kallen KJ, Herz U, Schmitt S, Luft C, Hecht O, Hohlfeld JM, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Rose-John S, Renz H, Neurath MF, Galle PR, Finotto S. The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313–325. doi: 10.1172/JCI22433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov. 2013;12:147–168. doi: 10.1038/nrd3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol. 2007;179:154–161. doi: 10.4049/jimmunol.179.1.154. [DOI] [PubMed] [Google Scholar]
- 161.Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J Immunol. 2013;190:1076–1084. doi: 10.4049/jimmunol.1202659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, Rajan TV, Clark RB. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J Immunol. 2011;186:6779–6787. doi: 10.4049/jimmunol.1003868. [DOI] [PubMed] [Google Scholar]
- 163.Wehrens EJ, Mijnheer G, Duurland CL, Klein M, Meerding J, van LJ, de JW, Sawitzki B, Coffer PJ, Vastert B, Prakken BJ, van WF. Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c-akt hyperactivation in effector cells. Blood. 2011;118:3538–3548. doi: 10.1182/blood-2010-12-328187. [DOI] [PubMed] [Google Scholar]
- 164.Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108:253–261. doi: 10.1182/blood-2005-11-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med. 2007;204:33–39. doi: 10.1084/jem.20061531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Golding A, Rosen A, Petri M, Akhter E, Andrade F. Interferon-alpha regulates the dynamic balance between human activated regulatory and effector T cells: implications for antiviral and autoimmune responses. Immunology. 2010;131:107–117. doi: 10.1111/j.1365-2567.2010.03280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mozzillo N, Ascierto P. Reduction of circulating regulatory T cells by intravenous high-dose interferon alfa-2b treatment in melanoma patients. Clin Exp Metastasis. 2012;29:801–805. doi: 10.1007/s10585-012-9504-2. [DOI] [PubMed] [Google Scholar]
- 168.Namdar A, Nikbin B, Ghabaee M, Bayati A, Izad M. Effect of IFN-beta therapy on the frequency and function of CD4(+)CD25(+) regulatory T cells and Foxp3 gene expression in relapsing-remitting multiple sclerosis (RRMS): a preliminary study. J Neuroimmunol. 2010;218:120–124. doi: 10.1016/j.jneuroim.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 169.Pace L, Vitale S, Dettori B, Palombi C, La S, Belardelli VF, Proietti E, Doria G. APC activation by IFN-alpha decreases regulatory T cell and enhances Th cell functions. J Immunol. 2010;184:5969–5979. doi: 10.4049/jimmunol.0900526. [DOI] [PubMed] [Google Scholar]
- 170.Riley CH, Jensen MK, Brimnes MK, Hasselbalch HC, Bjerrum OW, Straten PT, Svane IM. Increase in circulating CD4(+)CD25(+)Foxp3(+) T cells in patients with Philadelphia-negative chronic myeloproliferative neoplasms during treatment with IFN-alpha. Blood. 2011;118:2170–2173. doi: 10.1182/blood-2011-03-340992. [DOI] [PubMed] [Google Scholar]
- 171.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Caretto D, Katzman SD, Villarino AV, Gallo E, Abbas AK. Cutting edge: the Th1 response inhibits the generation of peripheral regulatory T cells. J Immunol. 2010;184:30–34. doi: 10.4049/jimmunol.0903412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cox JH, Kljavin NM, Ramamoorthi N, Diehl L, Batten M, Ghilardi N. IL-27 promotes T cell-dependent colitis through multiple mechanisms. J Exp Med. 2011;208:115–123. doi: 10.1084/jem.20100410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wojno ED, Hunter CA. New directions in the basic and translational biology of interleukin-27. Trends Immunol. 2012;33:91–97. doi: 10.1016/j.it.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, Harris TH, Grainger J, Wojno ED, Wagage S, Roos DS, Scott P, Turka LA, Cherry S, Reiner SL, Cua D, Belkaid Y, Elloso MM, Hunter CA. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity. 2012;37:511–523. doi: 10.1016/j.immuni.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity. 2012;37:501–510. doi: 10.1016/j.immuni.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Koenecke C, Lee CW, Thamm K, Fohse L, Schafferus M, Mittrucker HW, Floess S, Huehn J, Ganser A, Forster R, Prinz I. IFN-gamma production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-host disease. J Immunol. 2012;189:2890–2896. doi: 10.4049/jimmunol.1200413. [DOI] [PubMed] [Google Scholar]
- 179.Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142:914–929. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, Kronenberg M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, Rudensky AY. CD4+ Regulatory T Cells Control TH17 Responses in a Stat3-Dependent Manner. Science. 2009 doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, Jack RS, Wunderlich FT, Bruning JC, Muller W, Rudensky AY. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. 2011;34:566–578. doi: 10.1016/j.immuni.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kubo T, Hatton RD, Oliver J, Liu X, Elson CO, Weaver CT. Regulatory T cell suppression and anergy are differentially regulated by proinflammatory cytokines produced by TLR-activated dendritic cells. J Immunol. 2004;173:7249–7258. doi: 10.4049/jimmunol.173.12.7249. [DOI] [PubMed] [Google Scholar]
- 184.Hall JA, Bouladoux N, Sun CM, Wohlfert EA, Blank RB, Zhu Q, Grigg ME, Berzofsky JA, Belkaid Y. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29:637–649. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, Wang DY, Li Y, Wang HY, Wang RF. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–1384. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 186.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:7048–7053. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116:2022–2032. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 188.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;175:8051–8059. doi: 10.4049/jimmunol.175.12.8051. [DOI] [PubMed] [Google Scholar]
- 190.Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, Diamond MS, Gale M., Jr IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010;6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Anz D, V, Koelzer H, Moder S, Thaler R, Schwerd T, Lahl K, Sparwasser T, Besch R, Poeck H, Hornung V, Hartmann G, Rothenfusser S, Bourquin C, Endres S. Immunostimulatory RNA blocks suppression by regulatory T cells. J Immunol. 2010;184:939–946. doi: 10.4049/jimmunol.0901245. [DOI] [PubMed] [Google Scholar]