

Abstract

DNA interstrand cross-links are an important family of DNA damage that block replication and transcription. Recently, it was discovered that oxidized abasic sites react with the opposing strand of DNA to produce interstrand cross-links. Some of the cross-links between 2′-deoxyadenosine and the oxidized abasic sites, 5′-(2-phosphoryl-1,4-dioxobutane) (DOB) and the C4-hydroxylated abasic site (C4-AP), are formed reversibly. Chemical instability hinders biochemical, structural, and physicochemical characterization of these cross-linked duplexes. To overcome these limitations, we developed methods for preparing stabilized analogues of DOB and C4-AP cross-links via solid-phase oligonucleotide synthesis. Oligonucleotides of any sequence are attainable by synthesizing phosphoramidites in which the hydroxyl groups of the cross-linked product were orthogonally protected using photochemically labile and hydrazine labile groups. Selective unmasking of a single hydroxyl group precedes solid-phase synthesis of one arm of the cross-linked DNA. The method is compatible with commercially available phosphoramidites and other oligonucleotide synthesis reagents. Cross-linked duplexes containing as many as 54 nt were synthesized on solid-phase supports. Subsequent enzyme ligation of one cross-link product provided a 60 bp duplex, which is suitable for nucleotide excision repair studies.
Introduction
A number of cytotoxic
pharmacological agents, such as nitrogen
mustards and mitomycin C, produce DNA interstrand cross-links (ICLs).1,2 Other exogenous and endogenous bis-electrophiles also produce ICLs
by alkylating the nitrogenous DNA bases in either the major or minor
groove.3 ICLs are absolute blocks to replication
and transcription, making the agents that produce them potent DNA
damaging agents. Cells are protected from the deleterious effects
of ICLs by nucleotide excision repair (NER).4 How ICLs are recognized and removed by NER is an active area of
research.5−7 The importance of these subjects is exemplified by
recent observations of ICLs that are misrepaired by the bacterial
NER system (UvrABC), which converts them to double-strand breaks.8−10 A double-strand break is the most deleterious form of DNA damage.
Recently, ICLs resulting from the reaction of three abasic lesions
(AP, C4-AP, DOB) with native nucleotides on the opposing strand have
been discovered.11−15 Cross-links 1 (Scheme 1) and 2 (Scheme 2) form from the reaction
of the C4′-oxidized abasic site (C4-AP) and 5′-(2-phosphoryl-1,4-dioxobutane)
(DOB) with the N6-amino group of the dA that is opposite a 3′-adjacent
thymidine.13−15 These ICLs revert to the oxidized abasic lesions
in phosphate buffer and decompose upon reaction with NaBH3CN, which is often used as a stabilizing agent. The chemical properties
of 1 and 2 make characterization of their
repair and other chemical and structural properties impractical and
motivated us to synthesize stable analogues.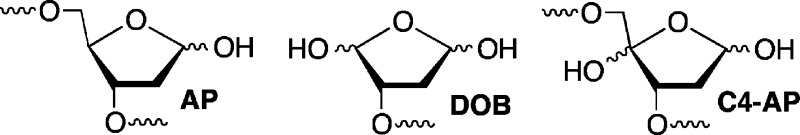
Scheme 1.

Scheme 2.

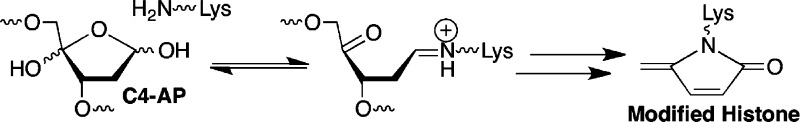
C4-AP and DOB are oxidized abasic lesions that are produced by several antitumor agents, as well as γ-radiolysis following C4′- or C5′-hydrogen atom abstraction, respectively.16 For instance, C4-AP is a major product formed by bleomycin under O2 limiting conditions.17 The enediyne antitumor antibiotics produce C4-AP and DOB.18 These oxidized abasic sites share a common 1,4-dicarbonyl structural feature that is required for forming cross-links 1 and 2 (Schemes 1 and 2) and is responsible for other biochemically interesting reactions. In duplex DNA, C4-AP (following hydrolysis of its 5′-phosphate) and DOB efficiently, irreversibly inactivate DNA polymerase β.19,20 In addition to inactivating this vital enzyme involved in DNA repair, the oxidized abasic sites also inactivate DNA polymerase λ, which is believed to be a back-up of DNA polymerase β.21 C4-AP has also been shown to react with histone proteins in nucleosome core particles.22 Histone proteins catalyze DNA cleavage at C4-AP lesions with half-lives as short as ∼15 min. Furthermore, the lysine residue(s) responsible for strand scission is modified in the process (Scheme 3). Examination of these biochemical reactions was facilitated by the availability of synthetic substrates containing DOB or C4-AP.14,23−25 As mentioned above, ICLs 1 and 2 revert to the respective lesions. The half-life for 1 at 25 °C is ∼10 h and that for 2 under the same conditions is ∼3 h.13,15 Although ICLs 1 and 2 are stable enough to be isolated and characterized by mass spectrometry, they do not survive long enough for more thorough characterization. Consequently, we pursued the synthesis of stable analogues 3 and 4.
Scheme 3.
A number of strategies for DNA–DNA interstrand cross-link synthesis have been reported. Some combine solid-phase oligonucleotide synthesis with postsynthetic chemistry that enables cross-linking by a latent electrophile upon hybridization with a complementary oligonucleotide.26−33 Other methods involve highly reactive intermediates that may produce mixtures of products.34 Total synthesis of cross-linked DNA on solid-phase support is an alternative, more linear approach. Some methods take advantage of sequence symmetry to reduce the number of steps.35,36 Complete control over the sequence of the cross-linked product by solid-phase synthesis can be achieved by using orthogonal protecting groups that enable selective unmasking of appropriate nucleophiles. Recently, Damha employed such an approach for preparing branched RNA molecules.37 One advantage of this approach was that the modified phosphoramidite was compatible with commercially available solid-phase synthesis reagents. We have taken a similar approach for synthesizing stabilized analogues of DOB and C4-AP interstrand cross-links.
Results and Discussion
To stabilize ICLs derived from the reaction of DOB or C4-AP with the N6-amino group of dA, we targeted the lower oxidation state analogues (3, 4, Schemes 1 and 2) that lack the labile hemiaminal functional groups. These analogues could be used to synthesize cross-linked oligonucleotides on solid-phase supports by employing orthogonal protecting groups for the respective hydroxyl groups. This approach requires one orthogonal protecting group for preparing DOB ICLs (3, Scheme 1) and two for C4-AP ICLs (4, Scheme 2). To maximize use of the most common commercially available reagents, protecting groups were considered that are stable to acidic conditions used for detritylation and whose own cleavage will not affect the β-cyanoethyl phosphate protecting group, the amides used to protect nucleobase exocyclic amines, or the ester linkages employed for capping aborted syntheses and tethering the oligonucleotides to the solid phase support.38,39 Three orthogonal functional groups that have been successfully used in oligonucleotide synthesis were initially considered. These were the levulinyl (Lev) group mentioned above,37,40−42 the o-nitrobenzyl photoredox (ONV) reaction,43−45 and Pd(0)-mediated cleavage of allyloxycarbonyl groups.46 After briefly experimenting with allyloxycarbonyl chemistry (data not shown), we chose to investigate the Lev and ONV groups with respect to the synthesis of DOB ICLs (3).
Synthesis of Oligonucleotides Containing an Analogue of a DOB ICL (3)
We approached the synthesis of oligonucleotides containing 3 (Scheme 4) by first preparing the entire (“template”) strand containing the cross-linked dA (6) using commercially available 5′-dimethoxytrityl-protected phosphoramidites and 5a,b (Scheme 5). The secondary alcohol of the DOB analogue containing the orthogonal protecting group (7) was revealed following detritylation and capping of the 5′-hydroxyl group of the template strand, and the solid-phase synthesis was completed using 3′-dimethoxytrityl 5′-β-cyanoethyl (“reverse”) phosphoramidites to obtain 8.
Scheme 4.

Scheme 5.

The requisite phosphoramidites were readily synthesized from an activated form of 2′-deoxyinosine (9) using the methodology established by Lakshman (Scheme 5).47 The hydroxypyrrolidine (11) substitution product (10) was a common precursor to the levulinylated (5a) and o-nitroveratrol carbonate (5b) phosphoramidites. Following desilylation, the nucleoside analogues (12a,b) were carried on to the phosphoramidites via the dimethoxytritylated products (13a,b) using standard procedures.
Oligonucleotides were prepared using 5a and standard commercially available β-cyanoethyl phosphoramidites containing standard amine protecting groups, with the exception of dC. N4-Benzoyl-dC was used instead of the N-acetyl analogue to prevent deprotection during hydrazinolysis. A variety of coupling conditions were experimented with, but ultimately good yields (typically between 50–65%, but as high as 98%) were obtained when the levulinylated phosphoramidite (5a, 0.15 M) was double-coupled for 15 min. The 5′-terminus of the template strand (8, Scheme 4) was manually capped with a solution containing 5% (vol/vol) each of acetic anhydride, N-methylimidazole, and pyridine in THF following detritylation. The resin was typically incubated 10 times with the reagents for 4 min each cycle for oligonucleotides ≤25 nucleotides long. Twenty-five cycles were used for longer oligonucleotides. Complete capping and its stability to the delevulinylation conditions were crucial to prevent unwanted extension during opposing strand synthesis. Delevulinylation was carried out using 0.25 M hydrazine in 3:2 pyridine/AcOH at room temperature for 10 min for oligonucleotides ≤25 nucleotides long and 25 min for longer substrates. Capping efficiency and its resistance to cleavage during hydrazinolysis was examined by determining extension in a model oligonucleotide (dT9) that had been capped as described above. A portion of the resin was subjected to additional automated synthesis with or without prior treatment with the delevulinylation reagents. The resin was subsequently deprotected and cleaved from the solid-phase support using concentrated aqueous ammonia, and an aliquot of the crude material was analyzed by denaturing PAGE following 5′-32P-labeling. No extension was detected via phosphorimaging analysis (data not shown). Following delevulinylation, the second strand (“arm”) of the ICL was synthesized using reverse phosphoramidites. The first phosphoramidite was double-coupled (15 min coupling time), and subsequent phosphoramidites were single-coupled for the same amount of time. The final products (14–20, Figure 1) were obtained following deprotection and cleavage using concentrated aqueous ammonia, purified by denaturing PAGE, and analyzed by either MALDI-TOF MS or ESI-MS. Using these procedures cross-linked DNA containing 3 in a template strand as long as 55 nt and an 8 nt long arm (20) was prepared. Cross-linked molecules containing longer template strands (e.g., 19, 20) were synthesized without full-length arms in the complementary strand. Due to the linear process of solid-phase oligonucleotide synthesis, we anticipated using enzyme ligation to assemble the products containing full-length complementary strands (“arms”). This approach is demonstrated below for a cross-linked duplex containing C4-AP analogue 4.
Figure 1.
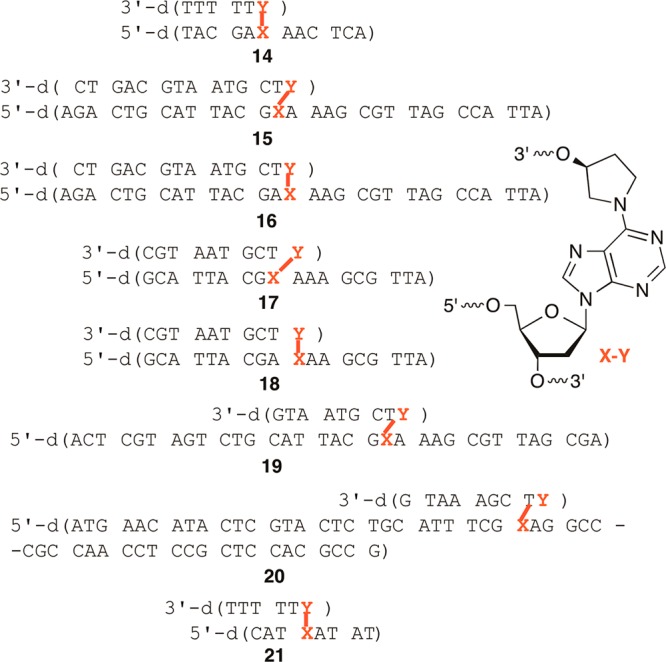
Chemically synthesized oligonucleotides containing 3.
Although the levulinyl group was satisfactory for preparing cross-linked oligonucleotides containing stabilized DOB analogue 3, we also examined the utility of the photolabile 5b in ICL synthesis. This was done in part to establish the viability of the venerable o-nitrobenzyl photoredox reaction for preparing cross-links containing 4, which requires a phosphoramidite whose synthesis is longer than that of 5b. Thinking ahead to the synthesis of 4, we recognized that the necessity of the N4-benzoyl dC protecting group when synthesizing ICLs using the levulinyl group created a conundrum because it was previously found to be incompatible with the o-nitroveratrole photolabile protecting group.48,49 Previous experiments were carried out using a UV light source with maximal output at 350 nm that emitted light close to 300 nm. Rather than search for an alternative orthogonal protecting group, we explored photolysis conditions that would be compatible with the benzoyl protecting group on dC (and likely dA). To direct our search, we measured the absorbance of solutions (52 μM) of an o-nitroveratrole carbonate, as well as benzoyl-dC and benzoyl-dA phosphoramidites between 350 and 400 nm (Figure 2).
Figure 2.

Absorbance of N-benzoylphosphoramidites and o-nitroveratrole (ONV) protected 10 between 350 and 400 nm. All solutions were 54 μM.
Absorption by the benzoyl group was negligible at 375 nm, but the o-nitroveratrole carbonate modestly absorbed light at this wavelength. The utility of the NVOC-protected phosphoramidite (5b) was demonstrated by synthesizing 21. Double-coupling of 5b proceeded in 92% yield, and following photolysis using a long pass filter (Newport no. CGA-375) that had zero transmittance at 370 nm and 50% at 375 nm the complementary strand was extended in 53% overall yield (determined via trityl cation release). With the exception of the first phosphoramidite, which was coupled for 15 min, standard coupling cycles were used to prepare the cross-linked strand. Approximately 4.5 nmol of 21 was isolated following concentrated ammonium hydroxide deprotection and denaturing PAGE purification from ∼1/5th of the resin from a 1 μmol scale commercial column.
Synthesis of Oligonucleotides Containing an Analogue of a C4-AP ICL (4)
Having established the compatibility of the levulinyl and o-nitroveratrole groups with solid-phase synthesis conditions, we sought to implement a strategy for synthesizing ICLs containing 4 that utilizes these orthogonal protecting groups (Scheme 6). We envisioned synthesizing the template strands of the ICLs using standard solid-phase synthesis and the capping conditions described for the preparation of oligonucleotides containing the DOB analogue (Scheme 4). The o-nitroveratrole group would be removed from 22, and the primary alcohol extended using 5′-dimethoxytrityl phosphoramidites. Following capping of 23, 24 would be delevulinylated and the 3′-arm of 25 extended using reverse phosphoramidites. Finally, 26 would be cleaved from the solid-phase support and 4 purified by denaturing PAGE. The synthesis of 4 from 24 was anticipated to follow the general procedure developed for preparing 3 (Scheme 4).
Scheme 6.

The strategy for the synthesis of the necessary phosphoramidite (32, Scheme 7) was based upon the chemistry developed for the DOB analogue. One previously reported procedure for pyrollidine 27 did not work in our hands.50 Instead, 27 was synthesized as described by Merino and reacted with 9 to produce the fully functionalized cross-link analogue (28).51 We were unable to selectively functionalize the primary alcohol in 28 using o-nitroveratryl chloroformate. Hence, 29 was prepared in a straightforward, albeit circuitous, manner and selectively detritylated to provide the primary alcohol necessary for the formation of 30. Following desilylation, 31 was carried on to phosphoramidite 32 via standard methods.
Scheme 7.

The utility of 32 for synthesizing cross-linked oligonucleotides was demonstrated using one short (8 bp, 33, Figure 3) duplex and two longer ones. In each instance, 32 was double-coupled manually. The coupling yields ranged from 80 to 100%. All other phosphoramidites were double-coupled via automated solid-phase synthesis when preparing the longer template strands in 34 and 35, resulting in 98.2% average stepwise yield. Following synthesis of the template strand, a portion of the resin (typically 1/6 to 1/5 of the total amount of resin present in a 1 μmol scale column) was photolyzed as described above for 1.5–2.0 h. The “5′-arm” was then synthesized manually. The 5′-termini were phosphorylated during solid-phase synthesis using a commercially available phosphoramidite (Solid CPR II, Glen Research) to facilitate subsequent T4 DNA ligase mediated ligation. All phosphoramidites were double-coupled except the first, which was coupled four times. Coupling yields were determined by measuring the amounts of dimethoxytrityl cation released and calculated based upon the theoretical yield, which took into account the amount of resin used, its loading, and the overall yield of the template strand. This analysis indicated that the first phosphoramidite coupled in between 63% and 90%. However, subsequent phosphoramidites were coupled in as high as 99.8% average stepwise yield for the synthesis of the 5′-arm in 34 and as low as 97.9% in 35. To complete the syntheses of the C4-AP cross-link analogue containing oligonucleotides, the levulinyl group was removed as described above for the DOB ICL analogue containing oligonucleotides. The syntheses of the 3′-arms were then carried out manually using the same strategy employed for preparing the 5′-arms. Coupling yields for the 3′-arms could not be determined as described for the 5′-arms because the delevulinylation conditions cleaved some of the o-nitroveratrole group that was not released upon photolysis. Consequently, partial extension of this arm resulted in apparent coupling yields >100%. Cross-linked products 33–35 were deprotected with concentrated aqueous ammonium hydroxide at 55 °C overnight and purified by denaturing PAGE.
Figure 3.

Chemically synthesized oligonucleotides containing 4.
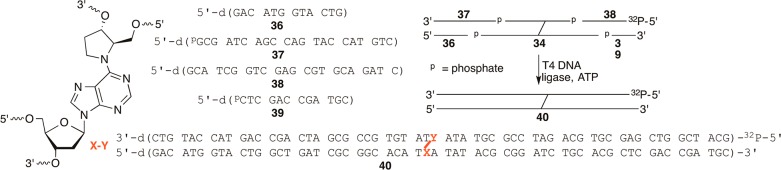
T4 DNA Ligase-Mediated Ligation of 34
To demonstrate the feasibility of transforming cross-linked oligonucleotides containing incomplete arms (e.g., 34, 35) into ones with fully complementary strands, 40 was obtained from 34 and 36–39 using T4 DNA ligase (Scheme 8). Cross-link product 40 is sufficiently long (60 bp) to be used in nucleotide excision repair studies. The one 5′-terminus of 34 that was not already phosphorylated was phosphorylated using T4 polynucleotide kinase and ATP. The 5′-terminus was of 38 also enzymatically phosphorylated except γ-32P-ATP was used. These oligonucleotides were hybridized using 34 as the limiting reagent and ligated (16 °C, 12 h) using T4 DNA ligase and ATP. The product was purified by denaturing PAGE and isolated by the crush and soak method. Isolated yields were determined using the specific activity of 38 and varied between 10% and 20%, based upon the amount of 34 used in the reaction. Following desalting and reannealing, the integrity of 40 was examined using restriction enzymes.52
Scheme 8.
Summary
The methods described enable chemical synthesis of cross-linked DNA. By utilizing orthogonal protecting groups, there are no sequence limitations. By combining the chemical synthesis with DNA ligase, cross-links that are sufficiently long (e.g., 60 bp) for subsequent nucleotide excision repair studies are accessible. In this particular study, stabilized analogues of chemically labile DNA cross-links were synthesized. The true cross-links (1, 2) contain epimerizable centers at the cross-linked nucleotide. The analogues prepared (3 and 27) contain the stereochemistry of native DNA at the cross-linked sites. These were chosen because it was anticipated that these would be the least distorting and most stable stereoisomers. However, the method is amenable to synthesizing other stereoisomers and is generally applicable to the synthesis of cross-linked DNA (and RNA).
Experimental Section
General Methods
Solvents used in reactions were purified and dried (using CaH2 or Na/benzophenone) by distillation before use. Reagents were purchased from commercial sources and were used without further purification. Reactions were carried out under a positive pressure of argon atmosphere and monitored by TLC on silica gel G-25 UV254 (0.25 mm). Spots were detected using UV light and/or by charring with a solution of either ammonium molybdate, ceric ammonium sulfate in water and H2SO4 or p-anisaldehyde in ethanol and H2SO4. Flash chromatography was performed on silica gel 60 (40–60 μm). The ratio between silica gel and crude product ranged from 100:1 to 20:1 (w/w).
Oligonucleotides were synthesized via automated DNA synthesis on an Applied Biosystems model 394 instrument. Commercially available DNA synthesis reagents including phosphorylation reagent were purchased from Glen Research. Phosphoramidites 5a, 5b, and 32 were dried for 12 h in a lyophilizer prior to the experiment. Oligonucleotides containing cross-link analogues were deprotected using concentrated NH4OH at 55 °C for 12 h. Oligonucleotides containing native nucleotides only were deprotected using 1:1 mixture of aqueous methylamine (40%)–concentrated concentrated NH4OH at 55 °C for 1 h.53 Oligonucleotides were purified by 20% denaturing polyacrylamide gel electrophoresis (PAGE), isolated by the crush and soak method, and desalted using C-18-Sep-Pak cartridges (Waters). Oligonucleotides containing modified nucleotides were characterized by MALDI-TOF MS or ESI-MS. Oligonucleotides were 5′-32P-labeled by T4 polynucleotide kinase (New England Biolabs) and γ-32P-ATP (Perkin Elmer) using standard protocols.54 Experiments involving radiolabeled oligonucleotides were analyzed following PAGE using a Storm 840 phosphorimager and Imagequant TL software. RSaI, TaqαI, and CutSmart buffer were from NEB.
Synthesis of 10
The hydrochloride salt of 11 (667 mg, 5.5 mmol) was added to a stirred solution of 9 (1.6 g, 2.7 mmol) in THF (50 mL), followed by addition of Cs2CO3 (4.2 g, 13.5 mmol). The reaction was allowed to stir for 3.5 h at room temperature, at which time the solvent was evaporated in vacuo. The crude material was dissolved in DCM (100 mL) and washed with saturated sodium bicarbonate (70 mL). The organic layer was collected, dried over sodium sulfate, and concentrated. Flash chromatography (70% EtOAc in DCM to 100% EtOAc) yielded 11.2 g of 10 (82%). Rf (5% MeOH in DCM) = 0.3. 1H NMR (CDCl3): δ 8.3 (s, 1H), 8.0 (s, 1H), 6.41 (t, 1H, J = 6.4 Hz), 4.60 (m, 1H), 4.56 (m, 1H), 3.84–4.54 (m, 5H), 3.81 (dd, 1H, J = 3.9, 11 Hz), 3.72 (dd, 1H, J = 3.7, 11 Hz), 3.05 (bd s, 1H), 2.59 (dt, 1H, J = 5.6, 12.2 Hz), 2.38 (m, 1H), 2.13 (m, 2H), 1.89 (bd s, 1H), 0.88 (s, 18H), 0.074 (s, 6H), 0.070 (s, 6H). 13C NMR (CDCl3): δ 153.2, 152.7, 149.8, 137.2, 120.8, 87.8, 84.2, 72.3, 63.1, 45.8, 41.1, 26.1, 25.9, 18.6, 18.2, 8.7, −4.5, −4.6, −5.2, −5.3. IR (neat): 3300 (bd), 2951, 2856, 1591, 1471, 1252, 1105, 1031 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C26H48N5O4Si2 550.3239, found 550.3250.
Synthesis of Levulinyl-Protected 10
Levulinic acid (604 mg, 529 μL, 5.1 mmol), DMAP (731 mg, 5.9 mmol), and EDCI (989 mmol, 5.2 mmol) were added to a stirred solution of 10 (405 mg, 0.7 mmol) in DMF (9 mL). The reaction mixture was stirred for 4 h at room temperature under inert atmosphere. The resulting light orange mixture was diluted with EtOAc (80 mL) and then washed with water (50 mL), saturated sodium bicarbonate (50 mL), and brine solution (50 mL). The organic layer was dried over sodium sulfate, concentrated in vacuo, and subjected to flash chromatography using 30% EtOAc in DCM to obtain the desired product (445 mg, 93%). Rf (30% EtOAc in DCM) = 0.31. 1H NMR (CDCl3): δ 8.32 (s, 1H), 7.97 (s, 1H), 6.42 (t, 1H, J = 6 Hz), 5.45 (m, 1H), 4.56 (s, 1H), 3.97–4.54 (m, 5H), 3.76 (dd, 1H, J = 4.6, 10.8 Hz), 3.72 (dd, 1H, J = 2.5, 10.8 Hz), 2.69 (m, 2H), 2.59 (m, 1H), 2.52 (m, 2H), 2.37 (m, 1H), 2.19 (m, 2H), 2.12 (s, 3H), 0.88 (s, 18H), 0.07 (s, 6H), 0.04 (s, 6H). 13C NMR (CDCl3): δ 206.4, 172.3, 153.1, 152.8, 150.1, 137.5, 120.8, 87.8, 84.1, 72.2, 62.9, 41.1, 37.9, 29.9, 28.2, 26.1, 25.8, 18.5, 18.1, −4.6, −4.7, −5.3, −5.4. IR (neat): 2852, 2926, 2856, 1720, 1591, 1471, 1156, 1093, 834, 777 cm–1. HRMS (ESI/APCI-TOF): m/z calcd for [M + H]+ C31H54N5O6Si2 648.3607, found 648.3616.
Synthesis of NVOC-Protected 10
A mixture of 10 (304 mg, 0.55 mmol), nitroveratryloxycarbonyl chloride (615 g, 2.22 mmol), and (dimethylamino)pyridine (549 mg, 4.43 mmol) was stirred in DCM (6 mL) at room temperature for 2 h, at which time the solution was diluted with 50 mL of DCM. The diluted contents were washed with saturated sodium bicarbonate (2 × 50 mL) followed by washing with brine (50 mL). The combined aqueous layers were washed with DCM (50 mL), and the combined organic layers were dried over sodium sulfate. After the solvent was evaporated in vacuo, the product mixture was dissolved in DCM. It was purified by flash chromatography (10% EtOAc in DCM to 60% EtOAc in DCM) to yield the desired product as a white solid (401 mg, 92%). Rf (50% EtOAc in DCM): 0.6. 1H NMR (CDCl3): δ 8.35 (s, 1H), 7.99 (s, 1H), 7.72 (s, 1H), 7.02 (s, 1H), 6.45 (t, 1H, J = 8 Hz), 5.59 (s, 2H), 5.45 (m, 1H), 4.60 (m, 1H), 4.01- 4.38 (m, 5H), 3.94 (s, 3H), 3.93 (s, 3H), 3.83 (dd, 1H, J = 11.2, 4.8 Hz), 3.75 (dd, 1H, J = 3.2, 10.8 Hz), 2.63 (td, 1H, J = 6, 12.8 Hz), 2.41 (m, 1H), 2.33 (m, 2H), 0.914 (s, 9H), 0.911 (s, 9H), 0.11, (s, 6H), 0.08 (s, 6H). 13C NMR (CDCl3): δ 154.3, 153.8, 153.0, 152.8, 150.1, 148.4, 139.7, 137.6, 126.7, 120.8, 87.9, 84.1, 72.3, 66.6, 63.0, 56.7, 56.6, 41,2, 30.7, 26.1, 25.9, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3. IR (neat): 2953, 2928, 2856, 1749, 1591, 1523, 1470, 1253, 1219, 1064, 834, 776 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C36H57N6O10Si2 789.3669, found 789.3661.
Synthesis of 12a
Triethylamine trihydrofluoride (0.220 mg, 221 μL, 1.37 mmol) was added to a solution of levulinyl-protected 10 (310 mg, 0.47 mmol) in THF (3.0 mL). After being stirred at room temperature for 17 h, TLC (5% MeOH in DCM) showed that a small amount of starting material was still present. An additional equivalent of desilylating reagent was added and the reaction stirred for 6 additional h at which time no starting material remained. The reaction was evaporated to dryness in vacuo, and 140 mg of a white foamlike product (71% yield) was collected after purification by flash chromatography (2% MeOH in DCM to 5% MeOH in DCM). Rf (5% MeOH in DCM) = 0.3. 1H NMR (MeOH-d4): δ 8.23 (s, 1H), 8.18 (s, 1H), 6.42 (t, 1H, J = 6.8 Hz), 5.44 (m, 1H), 4.88 (m, 4H), 4.56 (d, 1H, J = 3.6 Hz), 4.10–4.50 (1H), 4.05 (d, 1H, J = 2.4 Hz), 3.87–4.02 (1H), 3.84 (dd, 1H, J = 11.2, 2.6 Hz), 3.73 (dd, 1H, J = 12.4, 2.9 Hz), 2.70–2.86 (3H), 2.52 (t, 2H, J = 6.4 Hz), 2.37 (m, 1H), 2.24 (m, 2H), 2.06 (s, 3H). 13C NMR (MeOD-d4): δ 209.4, 174.0, 154.2, 153.2, 150.4, 140.6, 121.8, 89.8, 87.2, 73.1, 63.6, 42.6, 38.6, 32.9, 31.1, 29.5, 29.0. IR (neat): 3263, 2929, 1713, 1593, 1472, 1204, 1158. 1095 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C19H26N5O6 420.1878, found 420.1887.
Synthesis of 12b
Triethylamine trihydrofluoride (208 mg, 209 μL, 1.28 mmol) was added to a solution of NVOC protected 10 (202 mg, 0.26 mmol) in THF (2.7 mL) and the mixture was stirred overnight at room temperature. After the solvent was evaporated in vacuo, the residue was dissolved in DCM and purified by flash chromatography (18 × 2.2 cm) (2% methanol in DCM to 4% methanol in DCM) to yield the desired product as a white foam (131 mg, 89%). Rf (4% methanol in DCM) = 0.15. 1H NMR (CDCl3): δ 8.29 (s, 1H), 7.77 (s, 1H), 7.72 (s, 1H), 7.01 (s, 1H), 6.86 (d, 1H, J = 8 Hz), 6.31 (dd, 1H, J = 5.6, 8 Hz), 5.59 (s, 2H), 5.45 (d, 1H, J = 16 Hz), 4.8 (d, 1H, J = 4 Hz), 4.57 (m, 1H), 3.96–4.34 (m, 4H), 3.95 (s, 3H), 3.94 (s, 3H), 3.79 (t, 1H, J = 8 Hz), 3.14 (m, 1H), 2.63 (s, 1H), 2.28 (d, 1H, J = 5.2 Hz), 2.25 (d, 1H, J = 5.5 Hz). 13C NMR (CDCl3): δ 154.3, 153.8, 153.4, 152.1, 149, 148.5, 139.8, 139.1, 126.5, 122.2, 110.2, 108.3, 89.9, 87.9, 77.4, 73.8, 66.6, 63.7, 56.6, 56.5, 54.7, 53.2, 46.5, 45.4, 40.6, 32.3, 30.2, 29.7, 8.9. IR (neat): 3332 (bd), 2934, 2270, 1750, 1596, 1522, 1471, 1387, 1332, 1277, 1220, 1064, 987, 792, 668 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C24H29N6O10 561.1940, found 561.1944.
Synthesis of 13a
A pyridine (2 mL) solution of DMT-Cl (73 mg, 0.19 mmol) and azeotropically dried 12a (75 mg, 0.19 mmol) was stirred overnight, at which time the reaction was quenched with MeOH and evaporated to dryness in vacuo. The dry mass was dissolved in EtOAc (20 mL) and washed with saturated sodium bicarbonate (15 mL) and dried over sodium sulfate. A flash silica column was prepared using 1% Et3N in hexanes that was washed with 5 column volumes of 1% MeOH in EtOAc prior to loading the mixture. A solvent combination of 1% MeOH in EtOAc that was increased to 3% MeOH in EtOAc was used to obtain 93 mg of product (68%). Rf (5% MeOH in DCM) = 0.4. 1H NMR (CDCl3): δ 8.29 (s, 1H), 7.86 (s, 1H), 7.38 (d, 2H, J = 8.6 Hz), 7.14–7.34 (m, 7H), 6.78 (d, 4H, J = 8.6 Hz), 6.45 (t, 1H, J = 6.0 Hz), 5.48 (s, 1H), 4.64 (m, 1H), 4.15–4.55 (m, 2H), 4.12 (m, 1H), 3.72–4.09 (m, 2H), 3.78 (s, 3H), 3.75 (s, 3H), 3.36 (m, 2H), 3.05 (bd s, 1H), 2.68–2.85 (m, 3H), 2.44–2.59 (m, 3H), 2.21 (m, 2H), 2.15 (s, 3H). 13C NMR (CDCl3): δ 206.4, 172.3, 158.5, 153.0, 152.6, 149.8, 144.5, 137.2, 135.7, 130.0, 128.0, 127.9, 126.9, 120.7, 113.1, 86.5, 85.8, 83.9, 72.6, 63.8, 55.2, 54.0, 53.1, 47.0, 45.8, 40.3, 37.8, 30.7, 29.8, 28.0, 24.2. IR (neat): 3300 (bd), 2932, 2836, 1720, 1592, 1508, 1443 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C40H44N5O8 722.3184, found 722.3188.
Synthesis of 13b
A pyridine (3 mL) solution of DMT-Cl (108 mg, 0.26 mmol) and azeotropically dried (pyridine) 12b (131 mg, 0.23 mmol) was stirred overnight, at which time the reaction was diluted with 50 mL of EtOAc. The diluted contents were washed with saturated ammonium chloride (2 × 50 mL), followed by washing with brine (50 mL). The aqueous layers were washed with EtOAc (50 mL), and the combined organic layers were dried over sodium sulfate. After the solvent was evaporated in vacuo, the product mixture was dissolved in DCM containing a few drops of Et3N. A silica column packed with 1% Et3N in DCM was used for separation. The crude product was purified by flash chromatography (60% EtOAc in DCM with a few drops of Et3N to 3% methanol with 90% EtOAc in DCM with a few drops of Et3N) to yield the desired product as a faint yellow foam (125 g, 64%). Rf (3% methanol and 90% EtOAc in DCM) = 0.3. 1H NMR (CDCl3): δ 8.30 (s, 1H), 7.88 (s, 1H), 7.72 (s, 1H), 7.41 (d, 2H, J = 5.2 Hz), 7.20–7.38 (m, 7H), 7.01 (s, 1H), 6.81 (d, 4H, J = 12 Hz), 6.45 (t, 1H, J = 6.4 Hz), 5.58 (s, 2H), 5.45 (m, 1H), 4.64 (m, 1H), 4.03–4.58 (m, 5H), 3.94 (s, 3H), 3.91 (s, 3H), 3.77 (s, 6H), 3.38 (m, 2H), 2.76 (m, 1H), 2.51 (m, 1H), 2.31 (bd s, 3H). 13C NMR (CDCl3): δ 158.6, 154.3, 153.9, 153.1, 152.8, 150.0, 148.5, 144.7, 139.8, 137.5, 135.8, 131.1, 130.1, 128.9, 128.2, 128.0, 127.0, 126.6, 120.9, 113.3, 109.9, 108.3, 86.7, 85.9, 84.1, 72.8, 66.6, 63.9, 56.7, 56.5, 55.3, 46.2, 40.4, 38.6, 32.1, 30.5, 29.8, 26.1, 23.9, 23.1, 14.3, 11.1, 8.9. IR (neat): 3314 (bd), 2926, 2853, 1749, 1593, 1508, 1250, 1219, 1064, 909, 729 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C45H47N6O12 863.3246, found 863.3236.
Synthesis of 5a
DIPEA (85 mg, 0.66 mmol) was added to 13a (79.2 mg, 0.11 mmol) that was azeotropically dried with pyridine followed by 0.2 mL of freshly distilled DCM. The solution was cooled in an ice bath, and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (23 mg, 0.11 mmol) was added. After being stirred for 1 h, the reaction mixture was diluted with EtOAc (20 mL), washed with saturated sodium bicarbonate (15 mL), and then dried over sodium sulfate. After the organic layer was concentrated in vacuo, the compound was purified by flash chromatography (15 × 1.5 cm, packed using 0.5% Et3N in hexanes and then washed with 5 column volumes of EtOAc) using EtOAc as eluent to isolate 5a (79.6 mg, 78%). Rf (100% EtOAc) = 0.5. 1H NMR (CDCl3): δ 8.31 (s, 1H), 7.90 (d, 1H, J = 7.7 Hz), 7.35–7.46 (m, 2H), 7.13–7.32 (m, 7H), 6.72 (m, 4H), 6.44 (t, 1H, J = 5.1 Hz), 5.47 (s, 1H), 4.70 (m, 1H), 4.28 (m, 1H), 3.84–4.57 (m, 3H), 3.76 (s, 6H), 3.64 (m, 1H), 3.57 (m, 2H), 3.37 (m, 2H), 3.29 (m, 2H), 2.79 (m, 1H), 2.73 (d, 1H, J = 6.4 Hz), 2.69 (d, 1H, J = 6.4 Hz), 2.63 (m, 1H), 2.55 (m, 2H), 2.12 (m, 2H), 2.14 (s, 3H), 1.07–1.19 (m, 12H). 31P NMR (CDCl3): 149.06, 148.95. IR (neat): 2960, 1720, 1593, 1509, 1469, 1250, 1178, 1077 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C49H60N7O9P 922.4263, found 922.4265.
Synthesis of 5b
DIPEA (94 mg, 0.73 mmol) was added to 13b (118 mg, 0.13 mmol) that was azeotropically dried with pyridine, followed by 1.5 mL of freshly distilled DCM. The solution was cooled in an ice bath, and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (42.6 mg, 0.11 mmol) was added. After the reaction mixture was stirred for 1 h, the reaction mixture was diluted with EtOAc (30 mL), washed with saturated sodium bicarbonate (20 mL), and then dried over sodium sulfate. After the solvent was evaporated in vacuo, the product mixture was dissolved in 60% ethyl in hexanes. A silica column was packed with 1% Et3N in hexanes and then washed with 3 column volumes volume of 60% EtOAc in hexanes. The above solution was loaded onto the column and separated (60% EtOAc in hexanes to 90% EtOAc in hexanes) to yield the desired product (108 mg, 75%). Rf (90% EtOAc in hexanes) = 0.6. 1H NMR (CDCl3): δ 8.31 (s, 1H), 7.92 (d, 1H, J = 6.2 Hz), 7.74 (s, 1H), 7.39 (m, 2H, J = 11.2 Hz), 7.15–7.33 (m, 7H), 7.01 (s, 1H), 6.72–6.84 (m, 4H), 6.45 (t, 1H, J = 8 Hz), 5.59 (s, 2H), 5.45 (bd s, 1H), 4.72 (m, 1H), 4.00–4.60 (m, 5H), 3.95 (s, 3H), 3.92 (s, 3H), 3.77 (s, 6H), 3.26–3.71 (m, 5H), 2.82 (m, 1H), 2.75 (t, 1H, J = 4 Hz), 2.52–2.67 (m, 2H), 2.44 (t, 1H, J = 4 Hz), 2.34 (bd s, 2H), 1.08–1.57 (m, 12H). 31P NMR (CDCl3): δ 148.9, 148.7. IR (neat): 2925, 2852, 1749, 1592, 1509, 1467, 1249, 1065, 1033, 977, 793 cm–1. HRMS (ESI-TOF): m/z [M + H]+ calcd for C54H64N8O13P 1063.4330, found 1063.4309.
Synthesis of 28
A DMF (130 mL) solution of (2R,3S)-2-(hydroxymethyl)-3-hydroxypyrrolidine hydrochloride51 (27, 2.02 g, 13.1 mmol) and Cs2CO3 (17.1 g, 52.6 mmol) were stirred at room temperatures for 30 min, at which time 9 (9.4 g, 15.7 mmol) was added. The reaction was stirred overnight at room temperature followed by dilution with 400 mL EtOAc. The diluted contents were washed with saturated sodium bicarbonate (300 mL), followed by washing with brine (2 × 200 mL). The combined aqueous layers were washed with EtOAc (200 mL), and the combined organic layers were dried over sodium sulfate. After the solvent was evaporated in vacuo, the product mixture was dissolved in DCM. It was purified by flash chromatography (30–80% EtOAc in DCM to 3% methanol and 7% DCM in EtOAc) to yield the desired product as a white solid (3.54 g, 46%). Rf (90% EtOAc in DCM) = 0.2. 1H NMR (CDCl3): δ 8.27 (s, 1H), 8.04 (s, 1H), 6.42 (t, 1H, J = 8.4 Hz), 4.58 (bd s, 2H), 4.02–4.43 (m, 3H), 3.99 (dd, 1H, J = 4, 7.1 Hz), 3.85 (dd, 1H, J = 4.8, 11.9 Hz), 3.61–3.80 (m, 3H), 2.51–2.74 (m, 2H), 2.41 (m, 1H), 2.01–2.26 (m, 2H), 1.87 (bd s, 1H), 0.89 (s, 18H), 0.08 (s, 6H), 0.06 (s, 6H). 13C NMR (CDCl3): δ 154.0, 152.5, 150.1, 137.8, 120.6, 88.0, 84.4, 84.4, 73.9, 72.1, 70.5, 65.5, 62.9, 41.3, 26.8, 26.2, 25.99, 25.97, 18.6, 18.2, −4.4, −4.5, −5.1, −5.2. IR (neat): 3331 (bd), 2950, 2929, 2857, 1591, 1471, 1254, 1099, 1071, 836, 778 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ C27H50N5O5Si2 calcd for 580.3345, found 580.3335.
Synthesis of DMT-Protected 28
After azeotropically drying with pyridine, 28 (1.01 g, 1.7 mmol) was dissolved in a mixture of DMT-Cl (859.6 mg, 2.09 mmol), and (dimethylamino)pyridine (43 mg, 0.348 mmol) in pyridine (20 mL) at 0 °C. The reaction mixture was allowed to slowly warm to room temperature and then stirred overnight, at which time it was diluted with 150 mL of EtOAc. The diluted contents were washed with saturated ammonium chloride (2 × 200 mL) and then with brine (100 mL). The combined aqueous layers were washed once with EtOAc (100 mL), and the combined organic layers were dried with sodium sulfate. After the solvent was evaporated in vacuo, the product mixture was dissolved in DCM containing a few drops of Et3N. A flash chromatography on a column packed with 1% Et3N in DCM (30% EtOAc in DCM to 70% EtOAc in DCM, along with a few drops of Et3N) yielded the desired product as a faint yellow foam (1.19 g, 72%). Rf (30% EtOAc in DCM) = 0.2. 1H NMR (CDCl3): δ 8.29 (s, 1H), 7.93 (s, 1H), 7.38 (d, 2H, J = 8.2 Hz), 7.10–7.29 (m, 7H), 6.70 (m, 4H), 6.43 (t, 1H, J = 6.8 Hz), 4.59 (m, 2H), 4.05–4.21 (m, 1H), 4.01 (dd, 1H, J = 4, 7.2 Hz), 3.81 (m, 1H), 3.75–3.80 (m, 4H), 3.74 (m, 5H), 3.58 (dd, 1H, J = 3.6, 9.2 Hz), 3.27 (dd, 1H, J = 9.2, 6.8 Hz), 2.59 (td, 1H, J = 7.2, 12.4 Hz), 2.39 (m, 1H), 2.33 (m, 1H), 2.00 (bd s, 1H), 1.75 (bd s, 1H), 0.92 (s, 9H), 0.90 (s, 9H), 0.11 (s, 6H), 0.08 (s, 3H), 0.07 (s, 3H). 13C NMR (CDCl3): δ 158.35, 158.33, 153.2, 152.4, 144.8, 137.1, 136.7, 136.2, 136.0, 130.0, 129.9, 128.2, 127.6, 126.6, 120.6, 112.96, 112.92, 87.7, 86.4, 84.1, 77.2, 74.5, 72.1, 67.0, 62.9, 60.3, 55.1, 46.9, 41.0, 32.3, 25.9, 25.7, 18.4, 18.0, −4.67, −4.79, −5.3, −5.4. IR (neat): 3301 (bd), 3034, 2952, 2856, 1737, 1589, 1508, 1469, 1250, 1093, 1033, 834, 779 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C48H68N5O7Si2 882.4652, found 882.4635.
Synthesis of 29
1-Ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (1.15 g, 6 mmol) and DMAP (655 mg, 5.26 mmol) in DMF (10 mL) were added to DMT-protected 28 (715 mg, 0.75 mmol). After the mixtur was stirred for 10 min at room temperature, levulinic acid (608 mg, 533 μL, 5.25 mmol) was added. The reaction mixture was stirred for 2 h followed by dilution with 70 mL of EtOAc. The solution was washed with saturated sodium bicarbonate (1 × 70 mL) and then with brine (2 × 50 mL). The combined aqueous layers were washed once with EtOAc (50 mL), and the combined organic layers were dried with sodium sulfate. After the solvent was evaporated in vacuo, the product mixture was dissolved in DCM containing a few drops of Et3N. It was purified by flash chromatography (10% EtOAc in DCM with few drops of Et3N to 35% EtOAc in DCM with a few of drops of Et3N) to yield the desired product as a white foam (714 mg, 89%). Rf (30% EtOAc in DCM) = 0.4. 1H NMR (CDCl3) at 39 °C: δ 8.27 (bd s, 1H), 7.91 (bd s, 1H), 7.15–7.26 (m, 9H), 6.66–6.71 (m, 4H), 6.43 (bd s, 1H), 5.51 (bd s, 1H), 4.62 (bd s, 1H), 4.03–5.21 (m, 1H), 4.13 (m, 1H), 4.02 (m, 2H), 3.82 (m, 1H), 3.79 (m, 1H), 3.75 (s, 3H), 3.74 (s, 3H), 3.57 (m, 2H), 2.60–2.78 (m, 3H), 2.47–2.59 (m, 3H), 2.43 (m, 1H), 2.13 (m, 4H), 0.94 (s, 9H), 0.91 (s, 9H), 0.11 (s, 6H), 0.08 (s, 6H). 13C NMR (CDCl3) at 39 °C: δ 206.3, 172.3, 158.67, 158.64, 153.4, 152.7, 150.7, 145.1, 137.4, 136.4, 136.2, 130.29, 130.20, 128.4, 127.8, 126.8, 121.2, 113.2, 113.2, 88.0, 86.9, 84.3, 77.2, 72.5, 64.6, 63.2, 55.3, 41.2, 38.1, 29.9, 28.5, 26.3, 26.0, 18.6, 18.2, −4.4, −4.5, −5.1, −5.2. IR (neat): 2961, 2929, 2856, 1725, 1720, 1588, 1509, 1468, 1251, 1177, 1088, 1033, 835 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C53H74N5O9Si2 980.5020, found 980.5028.
Synthesis of DMT Deprotected 29
Acetic acid, water, and tetrahydrofuran (3:1:1, 50 mL) were added to 29 (715 mg, 0.67 mmol). After the mixture was stirred for 22 min at room temperature, methanol (2 mL) was added, and the solution was stirred for an additional 8 min, at which time 300 mL of EtOAc was added. The solution was washed with water (3 × 100 mL). The combined aqueous layers were washed with fresh EtOAc (200 mL). The combined organic layers were carefully washed with saturated sodium bicarbonate (400 mL) and then dried over sodium sulfate. After the solvent was evaporated in vacuo, the product mixture was dissolved in 30% EtOAc in hexanes and purified by flash chromatography (50% EtOAc in hexanes to 80% EtOAc in hexanes) to yield the desired product as a white foam (403 mg, 89%). Rf (70% EtOAc in hexanes) = 0.3. 1H NMR (CDCl3): δ 8.32 (s, 1H), 8.06 (s, 1H), 6.44 (t, 1H, J = 6.8 Hz), 5.22 (bd s, 1H), 4.75 (bd s, 1H), 4.60 (td, 1H, J = 5.2, 3.2 Hz), 4.23 (m, 2H), 3.99 (dt, 1H, J = 3.2, 7.1 Hz), 3.93 (m, 1H), 3.84 (dd, 1H, J = 4.3, 10.6 Hz), 3.75 (dd, 1H, J = 3.1, 10.6 Hz), 3.71 (dd, 1H, J = 7.1, 11.24 Hz), 2.69 (m, 2H), 2.61 (td, 1H, J = 6.4, 12.4 Hz), 2.52 (m, 2H), 2.42 (m, 1H), 2.14–2.39 (m, 3H), 2.12 (s, 3H), 0.92 (s, 18H), 0.10 (s, 6H), 0.08 (s, 6H). 13C NMR (CDCl3): δ 206.7, 206.2, 172.1, 153.7, 152.3, 150.1, 137.8, 87.8, 84.7, 77.2, 71.9, 67.0, 65.3, 62.7, 41.1, 37.7, 30.85, 30.81, 29.79, 29.73, 29.6, 28.0, 25.9, 25.7, 18.3, 17.9, −4.6, −4.8, −5.4, −5.5. IR (neat): 3363 (bd), 2953, 2928, 2856, 1719, 1589, 1470, 1253, 1157, 1110, 1071, 1029, 836, 778 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C32H56N5O7Si2 678.3726, found 678.3729.
Synthesis of 30
4,5-Dimethoxy-2-nitrobenzyl chloroformate (326 mg, 1.18 mmol), DMAP (277 mg, 2.23 mmol), and DCM (6 mL) were added to azeotropically dried (pyridine) DMT-deprotected 29 (310 mg, 0.447 mmol). After being stirred at room temperature for 2 h, the reaction mixture was diluted with 100 mL of DCM. The diluted solution was washed with sodium bicarbonate (50 mL) and brine (50 mL). The aqueous layers were washed with DCM (50 mL). The combined organic layers were dried over sodium sulfate. After the solvent was evaporated in vacuo, the crude was dissolved in 30% EtOAc in hexanes and purified by flash chromatography (30–70% EtOAc in hexanes) to yield the desired product as a yellowish foam (349 mg, 83%). Rf (50% EtOAc in hexanes) = 0.3. 1H NMR (CDCl3): δ 8.35 (s, 1H), 8.01 (s, 1H), 7.71 (s, 1H), 7.02 (s, 1H), 6.43 (t, 1H, J = 8 Hz), 5.55 (s, 2H), 5.42 (bd s, 1H), 4.59 (m, 1H), 4.55 (m, 2H), 4.17 (m, 1H), 3.98 (m, 2H), 3.89–3.97 (m, 7H), 3.81 (dd, 1H, J = 4.4, 11.2 Hz), 3.74 (dd, 1H, J = 4.3, 11.2 Hz), 2.70 (m, 2H), 2.63 (m, 1H), 2.53 (m, 2H), 2.41 (m, 2H), 2.18 (m, 1H), 2.12 (s, 3H), 0.90 (s, 9H), 0.89 (s, 9H), 0.09 (s, 6H), 0.07 (s, 3H), 0.06 (s, 3H). 13C NMR (CDCl3): δ 206.2, 172.0, 154.4, 153.7, 153.0, 152.5, 150.2, 148.3, 139.6, 137.9, 126.6, 120.7, 109.8, 108.1, 87.8, 84.0,72.0, 66.4, 63.0, 62.8, 56.4, 56.3, 47.3, 40.9, 37.7, 29.9, 29.6, 29.6, 28.0, 25.9, 25.7, 18.3, 17.9, 18.3, 17.9, 15.3, −4.7, −4.8, −5.4, −5.5. IR (neat): 2953, 2928, 2856, 1738, 1587, 1524, 1466, 1278, 1253, 1221, 1065, 837, 780 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C42H65N6O13Si2 917.4143, found 917.4128.
Synthesis of 31
A solution of triethylamine trihydrofluoride (291 mg, 293 μL, 1.8 mmol) and 30 (339 mg, 0.363 mmol) in THF (4 mL) were stirred overnight at room temperature. After evaporating the solvent in vacuo the residue was dissolved in dichloromethane and purified by flash chromatography (2–4% methanol in DCM) to yield the desired product as a white foam (271 mg, 93%). Rf (4% methanol in DCM) = 0.15. 1H NMR (CDCl3): δ 8.31 (s, 1H), 7.78 (s, 1H), 7.70 (s, 1H), 7.01 (s, 1H), 6.62 (d, 1H, J = 12 Hz), 6.30 (dd, 1H, J = 6, 8.8 Hz), 5.54 (s, 2H), 5.42 (m, 1H), 4.65–4.86 (m, 2H), 4.55 (m, 2H), 4.19 (s, 1H), 3.86–4.01 (m, 9H), 3.76 (m, 1H), 3.10 (m, 1H), 2.72 (m, 2H), 2.37–2.59 (m, 3H), 2.17–2.33 (m, 3H), 2.13 (s, 3H). 13C NMR (CDCl3): δ 206.3, 172.1, 154.4, 153.6, 153.4, 151.8, 149.3, 148.3, 139.6, 139.4, 129.4, 126.4, 110.0, 108.2, 89.6, 87.7, 77.6, 73.5, 66.5, 63.5, 63.2, 56.4, 56.3, 53.3, 40.4, 37.7, 29.7, 29.6, 27.9. IR (neat): 3341 (bd), 2927, 1717, 1588, 1523, 1468, 1330, 1277, 1220, 1158, 1096, 1065 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C30H37N6O13 689.2413, found 689.2445.
Synthesis of DMT-Protected 31
DMT-Cl (130 mg, 0.32 mmol), (dimethylamino)pyridine (1 mg, 0.01 mmol), and pyridine (4 mL) were added to azeotropically dried (pyridine) 31 (186 mg, 0.26 mmol) at 0 °C. The reaction mixture was allowed to slowly warm to room temperature and stir overnight, at which time it was diluted with 50 mL of ethyl acetate. The solution was washed with saturated ammonium chloride (2 × 50 mL), followed by washing with brine (50 mL). The combined aqueous layers were washed with ethyl acetate (50 mL), and the combined organic layers were dried with sodium sulfate. After the solvent was evaporated in vacuo, the crude mixture was dissolved in dichloromethane containing a few drops of Et3N. The product was purified by flash chromatography (20–60% ethyl acetate in dichloromethane to 3% methanol in ethyl acetate, with a few drops of Et3N) using a column packed with 1% Et3N in DCM. The desired product was isolated as a faint yellow foam (182 mg, 64%). Rf (5% methanol in DCM) = 0.4. 1H NMR (CDCl3): δ 8.30 (s, 1H), 7.88 (s, 1H), 7.71 (s, 1H), 7.39 (d, 2H, J = 7.8 Hz), 7.19–7.33 (m, 7H), 7.03 (s, 1H), 6.79 (d, 4H, J = 9 Hz), 6.44 (t, 1H, J = 5.2 Hz), 5.55 (s, 2H), 5.43 (bd s, 1H), 4.63 (bd s, 1H), 4.25–4.82 (m, 5H), 4.07–4.19 (m, 2H), 3.95 (s, 3H), 3.94 (s, 3H), 3.77 (m, 7H), 3.40 (dd, 1H, J = 3.6, 10.8 Hz), 3.38 (dd, 1H, J = 5.4, 10.8 Hz), 2.67–2.80 (m, 3H), 2.36–2.53 (m, 3H), 2.23 (m, 1H), 2.12 (s, 3H). 13C NMR (CDCl3): δ 206.2, 172.1, 158.5, 154.4, 153.7, 153.1, 152.5, 150.2, 148.3, 144.5, 139.6, 137.7, 135.7, 130.0, 128.0, 127.8, 126.9, 126.5, 120.8, 113.2, 109.9, 108.2, 86.6, 85.7, 83.9, 80.2, 77.7, 72.7, 66.4, 63.8, 63.1, 60.5, 56.5, 56.4, 55.2, 47.5, 40.1, 37.7, 30.6, 29.7, 28.0, 26.1. IR (neat): 2982–2939 (bd), 1734, 1587, 1509, 1372, 1236, 1176, 1157, 1043, 1064 cm–1. HRMS (ESI/APCI-TOF): m/z [M + H]+ calcd for C51H55N6O15 (M + H+) 991.3720, found 991.3726.
Synthesis of 32
DMT-protected 31 (110 mg, 0.11 mmol) was azeotropically dried with pyridine. 2-Cyanoethyl N,N-diisopropylchlorophosphoramidite (42.2 mg, 44 μL, 0.19 mmol), diisopropylethylamine (120 mg, 161 μL, 0.9 mmol), and dichloromethane (3 mL) were added, and the reaction mixture was stirred for 45 min at room temperature, at which time it was diluted with 60 mL of ethyl acetate. The solution was washed with saturated sodium bicarbonate (25 mL), followed by brine (25 mL), and then dried with sodium sulfate. After the solvent was evaporated in vacuo, the mixture was dissolved in 60% ethyl acetate in hexanes and purified (60–90% ethyl acetate in hexanes) on a silica column packed with 1% Et3N in hexanes and washed with 3 column volumes of 60% ethyl acetate in hexanes. The yield of the desired product was 101.1 mg (78%). Rf (90% EtOAc in hexanes) = 0.6. 1H NMR (CDCl3): δ 8.31 (s, 1H), 7.91 (d, 1H, J = 7.1 Hz), 7.72 (s, 1H), 7.40 (m, 2H), 7.15–7.34 (m, 7H), 7.04 (s, 1H), 6.79 (m, 4H), 6.44 (t, 1H, J = 6.4 Hz), 5.55 (s, 2H), 5.42 (bd s, 1H), 4.57–4.71 (m, 4H), 4.29 (m, 1H), 3.94 (m, 7H), 3.77 (s, 6H), 3.58 (m, 2H), 3.38 (m, 3H), 3.29 (m, 3H), 2.63–2.84 (m, 4H), 2.36–2.63 (m, 4H), 2.21 (bd s, 1H), 2.11 (s, 3H), 1.05–1.20 (m, 12H). 31P NMR (CDCl3): δ 148.7, 148.5. IR (neat): 2965, 2932, 1736, 1586, 1623, 1508, 1465, 1364, 1329, 1278, 1246, 1221, 1178, 1156, 1065, 1032, 975, 823, 793, 727 cm–1. HRMS (ESI-TOF): m/z [M + H]+ calcd for C60H72N8O16P 1191.4804, found 1191.4780.
General Procedure for Synthesis of Template Strand Containing 6 and 22
Native nucleotides were coupled using standard synthesis cycles (25 s coupling, 5 s capping with acetic anhydride, 15 s oxidation with 0.02 M I2 in THF/H2O/pyridine, 95 s detritylation with 3% TCA in DCM) for all oligonucleotides except for the 34 and 35, where double coupling was utilized. Oligonucleotides 15, 16, 19, 20, 34, and 35 were prepared on 1000 Å resin. All other oligonucleotides were synthesized using 500 Å resin. Modified phosphoramidites 5a and 5b were coupled using automated double coupling cycles (15 min coupling time), while 32 was coupled manually, as described below. The synthesizer was programmed to pause when it reached the point where the manual coupling was utilized.
General Procedure for Manual Coupling
Phosphoramidite 32 (13 mg, 11 μmol) was dissolved in 300 μL of activator solution (5-ethylthio-1H-tetrazole in acetonitrile, 0.25 M). The solution was repeatedly passed through a column for 10 min containing the partially synthesized oligonucleotide on CPG resin that was fitted with two 1 mL plastic syringes. The resin was washed with dry acetonitrile (5 mL) and dried under vacuum for 10 min. For manual double coupling, this procedure was repeated using a fresh solution of phosphoramidite and activator prior to carrying out the remaining solid-phase synthesis steps on the automated synthesizer.
General Procedure for Trityl Quantification
The trityl output collected from the DNA synthesis was diluted in a volumetric flask using 0.1 M pTsOH in acetonitrile. The synthetic yield of the corresponding step was calculated based on the absorbance of trityl cation measured at 495 nm (ε495 = 70000).
General Procedure for Photolysis of Resin Containing ONV-Protected 6 and 22
Photolysis of the resin was carried out in 6–8 mg portions in toluene for 2 h in a Pyrex tube using a Schoeffel 1000 W short-arc lamp (model LH151N/2) fitted with an optical filter (Newport no. CGA-375). The resin was stirred using a pipe cleaner (to prevent crushing the resin) during the photolysis. After the photolysis, the resin was washed sequentially with hexanes (3 × 3 mL), DCM (3 × 3 mL), and methanol (10 × 3 mL) within the Pyrex tube. After drying on a rotovap, the resin was transferred to a DNA synthesis column and dried under vacuum before any further synthetic modification. Methanol wash and drying in the previous step was repeated until complete recovery of resin was achieved.
General Procedure for Delevulinylation of Levulinyl-Protected 6 and 24
A solution of 0.25 M hydrazine in pyridine/acetic acid (3:2) was passed back and forth through the resin in a DNA synthesis column as described above for the manual coupling procedure for 10 min (for ≤25 nucleotide long) or 25 min (for longer substrates). For longer treatment times, fresh hydrazine solution was used every 6 min. At the end of the treatment, the resin was washed sequentially with methanol (5 × 10 mL) and acetonitrile (5 × 10 mL) and then dried under vacuum before continuing the oligonucleotide synthesis.
General Procedure for Preparing 5′-32P-40
Oligonucleotide 38 (80 pmol) was 5′-32P-labeled in reaction (80 μL) containing 1 × T4 PNK buffer (70 mM TRIS-HCl pH 7.6, 10 mM MgCl2, 5 mM DTT), 50 μCi γ-32P-ATP, and 30 U of T4 PNK at 37 °C for 1 h. Excess g-32P-ATP was removed by passing the reaction through a 1 mL Sephadex G25 column. The specific activity was determined by counting the radioactivity (using a liquid scintillation counter) and measuring the concentration of 38 (A260). Separately, 34 (40 pmol) was 5′-phosphorylated on the template strand in a 25 μL reaction containing 1 × T4 PNK buffer, ATP (80 pmol), and 30 U of T4 PNK at 37 °C for 1 h. Both phosphorylation reactions were stopped by heating at 65 °C for 30 min. The phosphorylated products were hybridized with 160 pmol each of 36, 37 ,and 39 in 10 mM sodium phosphate (pH 7.2) and 100 mM sodium chloride solution by slowly cooling from 90 to 16 °C. The reaction was incubated overnight at 16 °C in the presence of T4 DNA ligase (50 U) and 1 × ligase buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10 mM DTT, 1 mM ATP). Formamide loading buffer (50 μL, 90% formamide, 10 mM EDTA, pH 8.0) was added, followed by heating at 90 °C for 5 min and cooling on ice prior to purification by 20% denaturing PAGE (37 × 32 × 0.04 cm). The gel was run under limiting power (55 W) for 18 h. The product band was excised from the gel and crushed, and the DNA was eluted overnight in 5 mL of elution buffer (0.2 M NaCl and 1 mM EDTA). The supernatant was desalted using a 100 mg C-18-Sep-Pak cartridge to provide 40. The yield (10–20%) of 40 was determined based on the specific activity of 5′-32P-38.
General Procedure for Restriction Enzyme Treatment
The appropriate restriction enzyme (10 U) and 5′-32P-40 were incubated in a 10 μL reaction containing of 1 X CutSmart buffer (50 mM potassium acetate, 20 mM TRIS acetate, 10 mM magnesium acetate, 100 μg/mL BSA, pH 7.9) at 37 °C (RsaI) or 65 °C (TaqαI) for 1 h, at which time formamide loading buffer (2 μL, 90% formamide, 10 mM EDTA, pH 8.0) was added followed by heating at 90 °C and cooling on ice. The reactions were analyzed by 20% denaturing PAGE.
Acknowledgments
We are grateful for generous support of this research from the National Institute of General Medical Sciences (GM-063028). We thank Dr. Jonathan Sczepanski (The Scripps Research Institute) for helpful discussions and Dr. Michael Delaney (Dharmacon Research) for advice on the manual coupling procedure. We are grateful to Professor Steven Rokita (Johns Hopkins University) for providing access to the Schoeffel 1000 W arc lamp.
Supporting Information Available
NMR spectra of new compounds and mass spectra of modified oligonucleotides. Restriction enzyme cutting sites and autoradiograms of ligation to produce 40 and restriction cleavage. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kuang Y.; Balakrishnan K.; Gandhi V.; Peng X. J. Am. Chem. Soc. 2011, 133, 19278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpato M.; Seargent J.; Loadman P. M.; Phillips R. M. Eur. J. Cancer 2005, 41, 1331. [DOI] [PubMed] [Google Scholar]
- Stone M. P.; Cho Y.-J.; Huang H.; Kim H.-Y.; Kozekov I. D.; Kozekova A.; Wang H.; Minko I. G.; Lloyd R. S.; Harris T. M.; Rizzo C. J. Acc. Chem. Res. 2008, 41, 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truglio J. J.; Croteau D. L.; Van Houten B.; Kisker C. Chem. Rev. 2006, 106, 233. [DOI] [PubMed] [Google Scholar]
- Clauson C.; Scharer O. D.; Niedernhofer L. Cold Spring Harbor Perspect. Biol. 2013, 5, a012732/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakotiprapha D.; Samuels M.; Shen K.; Hu J. H.; Jeruzalmi D. Nat. Struct. Mol. Biol. 2012, 19, 291. [DOI] [PubMed] [Google Scholar]
- Guainazzi A.; Scharer O. D. Cell. Mol. Life Sci. 2010, 67, 3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X.; Ghosh A. K.; Van Houten B.; Greenberg M. M. Biochemistry 2010, 49, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M.-w.; Zheng Y.; Jasti V. P.; Champeil E.; Tomasz M.; Wang Y.; Basu A. K.; Tang M.-s. Nucleic Acids Res. 2010, 38, 6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczepanski J. T.; Jacobs A. C.; Van Houten B.; Greenberg M. M. Biochemistry 2009, 48, 7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price N. E.; Johnson K. M.; Wang J.; Fekry M. I.; Wang Y.; Gates K. S. J. Am. Chem. Soc. 2014, 136, 3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. M.; Price N. E.; Wang J.; Fekry M. I.; Dutta S.; Seiner D. R.; Wang Y.; Gates K. S. J. Am. Chem. Soc. 2013, 135, 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczepanski J. T.; Jacobs A. C.; Majumdar A.; Greenberg M. M. J. Am. Chem. Soc. 2009, 131, 11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczepanski J. T.; Jacobs A. C.; Greenberg M. M. J. Am. Chem. Soc. 2008, 130, 9646. [DOI] [PubMed] [Google Scholar]
- Guan L.; Greenberg M. M. J. Am. Chem. Soc. 2009, 131, 15225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitié M.; Pratviel G. Chem. Rev. 2010, 110, 1018. [DOI] [PubMed] [Google Scholar]
- Rabow L. E.; Stubbe J.; Kozarich J. W. J. Am. Chem. Soc. 1990, 112, 3196. [Google Scholar]
- Xi Z.; Goldberg I. H. In Comprehensive Natural Products Chemistry; Kool E. T., Ed.; Elsevier: Amsterdam, 1999; Vol. 7, p 553. [Google Scholar]
- Guan L.; Greenberg M. M. J. Am. Chem. Soc. 2010, 132, 5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan L.; Bebenek K.; Kunkel T. A.; Greenberg M. M. Biochemistry 2010, 49, 9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens A. J.; Guan L.; Bebenek K.; Kunkel T. A.; Greenberg M. M. Biochemistry 2013, 52, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.; Sczepanski J. T.; Greenberg M. M. J. Am. Chem. Soc. 2013, 135, 5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Gil J. M.; Greenberg M. M. Angew. Chem., Int. Ed. 2003, 42, 5882. [DOI] [PubMed] [Google Scholar]
- Usui K.; Aso M.; Fukuda M.; Suemune H. J. Org. Chem. 2008, 73, 241. [DOI] [PubMed] [Google Scholar]
- Aso M.; Usui K.; Fukuda M.; Kakihara Y.; Goromaru T.; Suemune H. Org. Lett. 2006, 8, 3183. [DOI] [PubMed] [Google Scholar]
- Op de Beeck M.; Madder A. J. Am. Chem. Soc. 2012, 134, 10737. [DOI] [PubMed] [Google Scholar]
- Hentschel S.; Alzeer J.; Angelov T.; Schärer O. D.; Luedtke N. W. Angew. Chem., Int. Ed. 2012, 51, 3466. [DOI] [PubMed] [Google Scholar]
- Angelov T.; Guainazzi A.; Scharer O. D. Org. Lett. 2009, 11, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X.; Hong I. S.; Li H.; Seidman M. M.; Greenberg M. M. J. Am. Chem. Soc. 2008, 130, 10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong I. S.; Greenberg M. M. J. Am. Chem. Soc. 2005, 127, 10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsugi F.; Kawasaki T.; Usui D.; Maeda M.; Sasaki S. J. Am. Chem. Soc. 1999, 121, 6753. [Google Scholar]
- Tsarouhtsis D.; Kuchimanchi S.; DeCorte B. L.; Harris C. M.; Harris T. M. J. Am. Chem. Soc. 1995, 117, 11013. [Google Scholar]
- Ferentz A. E.; Keating T. A.; Verdine G. L. J. Am. Chem. Soc. 1993, 115, 9006. [Google Scholar]
- Qiu Z.; Lu L.; Jian X.; He C. J. Am. Chem. Soc. 2008, 130, 14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll D. M.; Noronha A. M.; Wilds C. J.; Miller P. S. Frontiers Biosci. 2004, 9, 421. [DOI] [PubMed] [Google Scholar]
- Noronha A. M.; Wilds C. J.; Miller P. S. Biochemistry 2002, 41, 8605. [DOI] [PubMed] [Google Scholar]
- Katolik A.; Johnsson R.; Montemayor E.; Lackey J. G.; Hart P. J.; Damha M. J. J. Org. Chem. 2014, 79, 963. [DOI] [PubMed] [Google Scholar]
- Beaucage S. L.; Iyer R. P. Tetrahedron 1992, 48, 2223. [Google Scholar]
- Beaucage S. L.; Reese C. B. Curr. Prot. Nucleic Acid Chem. 2009, 2.16.1. [DOI] [PubMed] [Google Scholar]
- Lackey J. G.; Mitra D.; Somoza M. M.; Cerrina F.; Damha M. J. J. Am. Chem. Soc. 2009, 131, 8496. [DOI] [PubMed] [Google Scholar]
- Ueno Y.; Shibata A.; Matsuda A.; Kitade Y. Bioconjugate Chem. 2003, 14, 684. [DOI] [PubMed] [Google Scholar]
- Iwai S.; Sasaki T.; Ohtsuka E. Tetrahedron 1990, 46, 6673. [Google Scholar]
- McGall G. H.; Barone A. D.; Diggelmann M.; Fodor S. P. A.; Gentalen E.; Ngo N. J. Am. Chem. Soc. 1997, 119, 5081. [Google Scholar]
- Kahl J. D.; Greenberg M. M. J. Am. Chem. Soc. 1999, 121, 597. [Google Scholar]
- McMinn D. L.; Greenberg M. M. J. Am. Chem. Soc. 1998, 120, 3289. [Google Scholar]
- Hayakawa Y.; Wakabayashi S.; Kato H.; Noyori R. J. Am. Chem. Soc. 1990, 112, 1691. [Google Scholar]
- Bae S.; Lakshman M. K. J. Am. Chem. Soc. 2007, 129, 782. [DOI] [PubMed] [Google Scholar]
- Venkatesan H.; Greenberg M. M. J. Org. Chem. 1996, 61, 525. [DOI] [PubMed] [Google Scholar]
- Pirrung M. C.; Bradley J.-C. J. Org. Chem. 1995, 60, 6270. [Google Scholar]
- Mascavage L. M.; Lu Q.; Vey J.; Dalton D. R.; Carroll P. J. J. Org. Chem. 2001, 66, 3621. [DOI] [PubMed] [Google Scholar]
- Merino P.; Delso I.; Tejero T.; Cardona F.; Marradi M.; Faggi E.; Parmeggiani C.; Goti A. Eur. J. Org. Chem. 2008, 2008, 2929. [Google Scholar]
- See the Supporting Information.
- Reddy M. P.; Hanna N. B.; Faroqui F. Tetrahedron Lett. 1994, 35, 4311. [Google Scholar]
- Sambrook J.; Fritsch E. F.; Maniatis T.. Molecular Cloning A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.