

Abstract
Angiogenesis is crucial for tumor development, growth and metastasis. Vascular endothelial growth factor (VEGF) has been implicated in promoting solid tumor growth and metastasis via stimulating tumor-associated angiogenesis, and blocking the activity of VEGF can starve tumors. Avastin, which is a humanized anti-VEGF antibody, has been successfully applied in clinics since 2004. However, the price of Avastin is extremely high for Chinese people. Here, we report a novel human anti-VEGF neutralizing antibody, MIL60, which shows an affinity comparable to that of Avastin (the KD value of MIL60 was 44.5 pM, while that of Avastin was 42.7 pM). MIL60 displays favorable actions in inhibiting VEGF-triggered endothelial cell proliferation (the IC50 value of MIL60 was 31±6.4 ng/ml and that of Avastin was 47±8.1 ng/ml), migration (8 µg/ml or 0.8 µg/ml MIL60 versus the control: P<0.05) and tube formation (2 µg/ml or 0.2 µg/ml MIL60 versus the control: P<0.05) via the VEGFR2 signaling pathway. Moreover, MIL60 was shown to inhibit tumor growth and angiogenesis in vivo in xenograft models of human colon carcinoma and ovarian cancer using immunotherapy and immunohistochemistry analysis (MIL60 versus N.S.: P=0.0007; Avastin versus N.S.: P=0.00046). These data suggest that MIL60 is a potential therapeutic, anti-angiogenic agent. Our work provides a novel anti-VEGF antibody, which can be considered an anti-tumor antibody candidate and a new option for patients with various cancers.
Keywords: angiogenesis, anti-VEGF antibody, cancer
Introduction
Angiogenesis, which is the process of new blood vessel formation, is essential for many physiologically important events and critical in a number of diseases, such as ischemic heart disease, diabetic retinopathy and tumor progression. Neovascularization allows cancer development, tumor growth and metastasis due to the tumor eliciting the formation of capillaries to obtain its own blood supply.1 Starvation of tumors by inhibiting angiogenesis has been discussed for a number of years as a strategy to fight cancer. Currently, dozens of anti-angiogenesis drugs are being tested in clinical trials.2
The vascular endothelial growth factor (VEGF) has been implicated in promoting solid tumor growth and metastasis via stimulating tumor-associated angiogenesis.3,4,5 Blocking the activity of VEGF, such as through competitive binding of its receptor and/or downregulation of its signal transduction, has proven to reduce tumor growth.6,7,8,9 Several VEGF inhibitors, such as antibodies, small molecule tyrosine kinase inhibitors and peptides, have been effective in treating patients with cancer.10,11,12,13,14,15 Among these, the anti-VEGF-A monoclonal antibody bevacizumab (Avastin) has been approved by the FDA for the treatment of metastatic colorectal16 and non-small-cell lung cancers,17 when in combination with chemotherapy. These were the first reports that validated a cancer-treatment approach by which tumor starvation was induced by inhibiting VEGF.
Avastin is widely used in the clinical treatment of various tumors. The success of Avastin and it being extremely expensive for most Chinese people promotes the manufacturing of similar antibodies. In this study, we provided a novel human anti-VEGF neutralizing antibody, MIL60. The in vitro activity of MIL60 in inhibiting VEGF-induced pro-angiogenic effects was investigated using human umbilical vein endothelial cells (HUVECs). In addition, its in vivo anti-tumor efficacy was examined in a human colon carcinoma xenograft mouse model. MIL60 neutralized VEGF released from cancer cells and blocked VEGFR2 phosphorylation and downstream signal transduction; therefore, it inhibited angiogenesis and tumor progression. The effect of MIL60 was similar to that of Avastin. In summary, our work provides an anti-tumor antibody candidate to offer more choices to patients with various cancers in the future.
Materials and methods
Reagents
Bevacizumab (Avastin) was purchased from Roche. Rabbit anti-human VEGFR2, phospho-VEGFR2, Erk, phospho-Erk, P38, phospho-P38, NF-κB P65, phosphor-NF-κB P65 (Ser536) and horseradish peroxidase (HRP)-conjugated chicken anti-rabbit IgG were purchased from Cell Signaling Technology (Danvers, MA, USA). Goat anti-human CD31 antibody was obtained from Abcam Biotechnology (Cambridge, MA, USA). Human recombinant VEGF-A was purchased from R&D Systems (Minneapolis, MN, USA).
Cell lines
Human colon carcinoma HT-29 cells and the human ovarian cancer cell line SKOV3 were obtained from the American Type Culture Collection. HT-29 cells were cultured in RPMI-1640 medium (Gibco) and SKOV3 cells were cultured in DMEM (Gibco) (Gibco, Grand Island, NY, USA). Media was supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Grand Island, NY, USA) and penicillin-streptomycin.
Preparation of HUVECs
HUVECs were obtained from human umbilical veins. After 10-min digestion with 0.1% collagenase I, the veins were washed and the cells were transferred to endothelial cell medium (ECM; ScienCell, San Diego, CA, USA) supplemented with 5% heat-inactivated FBS, penicillin-streptomycin and endothelial cell (EC) growth supplement (ScienCell, San Diego, CA, USA). The cells were assessed for the endothelial cell phenotype by morphology and the expression levels of the von Willebrand factor antigen, vascular endothelial growth factor receptor 2 and CD31. Only cells passaged 2–7 times were used for experiments.
ELISA
ELISA plates were coated with 0.5 µg/ml VEGF fusion protein at 4 °C overnight and then blocked with 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) containing 0.05% Tween-20 for one hour at 37 °C. Diluted MIL60 or Avastin were added as the primary antibody and incubated for 2 h at 37 °C. After washing, HRP-conjugated goat anti-human IgG was incubated for one hour at room temperature. Binding signals were visualized using o-phenylenediamine dihydrochloride substrate and the light absorbance was measured using an ELISA reader at 492 nm. For competition ELISA, MIL60, Avastin or a negative control antibody (IgG) were individually mixed at increasing concentrations with an equal volume of the competitors. The antibody mixtures were added as the primary antibodies and all experiments were performed in triplicate.
Neutralizing activity of MIL60 in SKOV3 cells
The ovarian cancer cell line SKOV3 secretes large amounts of VEGF. Therefore, SKOV3 cells were cultivated in six-well plates. After 12 h, the complete media of the adherent cells were replaced with serum-free medium and 10 µg/ml of control antibody IgG or MIL60 were added. After another 24 h, the supernatants were collected and the concentration of free VEGF was detected using a VEGF detecting kit (eBiosciences, San Diego, CA, USA).
Biacore analysis
The antigen was the standard VEGF protein, Avastin was set as the positive control and the solvent (sample buffer) was set as the negative control. The background was deducted according to the V-baseline to obtain the kinetic curves and calculate the dissociation constant (KD value).
Computer-guided modeling
The three-dimensional structure of the MIL60 Fv was constructed using the computer-guided homology modeling method of InsightII version 2005 software (Molecular Simulations, San Diego, CA, USA). Using the crystal structure of VEGF and considering the potential binding sites of VEGF, the three-dimensional complex structures of VEGF and MIL60 were modeled using the molecular docking method. To evaluate the rationality of the modeling structure, the VEGF and MIL60 modeled structures were optimized using the Discover and CHARMM18 modules in InsightII. The non-bonded cutoff was 10 Å, and the non-bonded parameters and atomic charges were used as defaults. A distance-dependent dielectric constant was used as in vacuo calculations. The model was minimized using the steepest descent (2000 steps) and conjugate gradient (5000 steps) methods, respectively.
Cell proliferation assay
HUVECs were resuspended to a density of 1×105/ml and 100 µl were seeded per 96 wells. After serum-free starvation overnight, the cells were treated with diluted MIL60 or Avastin that was pre-incubated with 12.5 ng/ml VEGF for 30 min at room temperature. After cultivation for 72 h at 37 °C, 10 µl of Cell Counting Kit-8 (CCK8; DOJINDO Laboratories, Kumamoto, Japan) was added to each well, and the plate was incubated for another 4 h. The absorbance was measured using a spectrophotometer at 450 nm to determine the cell viability.
Transwell assay
The chemotactic motility of the HUVECs was identified using a permeable transwell support (8 µm pore size; Costar; Corning, Pittsburgh, PA, USA). The HUVECs were serum starved overnight and resuspended in serum-free ECM to a density of 4×105/ml. Then, 250 µl of cells was seeded in upper chambers. Meanwhile, MIL60 or Avastin was diluted in ECM media with 0.5% FBS and incubated with 50 ng/ml VEGF for an hour. Then, 750 µl of the mixture was added to the lower chamber. After incubation for 10 h at 37 °C, non-migrated cells on the upper membrane were removed with cotton swabs. The migrated cells were fixed with 4% paraformaldehyde and stained with Giemsa solution. Cell images were captured using an OLYMPUS BX5 microscope and an UPlanFL N digital camera (10×0.13 numeric aperture objective). The number of migrated cells was counted from five randomly chosen fields.
Endothelial tube formation assay19,20
After being serum starved overnight, the HUVECs were resuspended in ECM with 0.5% FBS and seeded in a 96-well plate (5000 cells per well). Diluted MIL60 (0.02 µg/ml, 0.2 µg/ml or 2 µg/ml) or Avastin (0.02 µg/ml, 0.2 µg/ml or 2 µg/ml) was pre-mixed with VEGF (12.5 ng/ml) for 30 min and added to the 96-well plate. After incubation for 10 h at 37 °C, the cells were fixed with 4% paraformaldehyde and stained with Giemsa solution. Tubular structures were photographed using a NIKON TE2000-U microscope, and the tube length was measured using ImageJ 1.46r software.
Western blotting
HUVECs were seeded in six-well plates at a density of 8×105 cells/ml. After being serum-starved overnight, they were cultivated in serum-free ECM that was supplemented with 50 ng/ml VEGF and different concentrations of MIL60 or Avastin for 30 min. Thereafter, the supernatant of the cell lysate was subjected to SDS–PAGE analysis using a 12% gel and transferred onto a nitrocellulose membrane. The membrane was blocked with non-fat 5% milk for 1 h at room temperature and then incubated with various antibodies (dilution 1∶1000) overnight at 4 °C. After washing, the membranes were incubated with HRP-conjugated antibodies for an hour at room temperature, and the signals of the membrane were detected and visualized using an enhanced chemiluminescence detection system and autoradiography.
In vivo immunotherapy of MIL60 in a xenograft mouse model
Female, 4-week-old BALB/c nude mice were purchased from Vital River Laboratory (Beijing, China), housed in specific pathogen-free rooms and fed a normal mouse laboratory diet. HT-29 or SKOV3 cells were resuspended in serum-free DMEM to a density of 2×107 cells/ml. The mice were inoculated subcutaneously into the back with 2×106/0.1 ml of cells on day 0. When tumors reached a diameter of 3–5 mm (day 4), the mice were divided randomly into three treatment groups: sterile saline as a negative control, Avastin as a positive control, and MIL60 as an experimental group. The antibodies were first injected i.p.21,22 at a dose of 5 mg/kg, then, the mice were observed and the antibodies were administered twice a week another seven times. Perpendicular dimensions were measured using a Vernier scale caliper twice per week, and the tumor volumes were calculated according to the following equation:
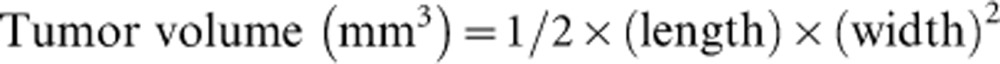 |
All procedures were in strict agreement with the International Guidelines for the Care and Use of Laboratory Animals and approved by the Animal Ethics Committee of the Institute of Basic Medical Sciences.
Immunohistochemistry
One week after the last treatment, the tumors of the mice were removed and fixed with 10% formaldehyde. Paraffin-embedded tissue sections were processed, deparaffinized, rehydrated and quenched of endogenous peroxidase activity. Sections were first stained with anti-CD31 antibody (dilution 1∶100) and then incubated with HRP-conjugated secondary antibody. Finally, the sections were developed with diaminobenzidine and counterstained with hematoxylin. Pictures were captured using an OLYMPUS BX5 microscope and an UPlanFL N digital camera (10×0.13 numeric aperture objective). Any single, brown-stained cell or cluster of EC that was clearly separated from adjacent microvessels, tumor cells and other connective tissue elements was considered a vessel. The number of CD31-position capillaries was counted from five randomly chosen fields.19
Statistical analysis
Data are expressed as the mean±s.d. Pairwise differences between several groups were compared. Significant differences were analyzed by ANOVA, and P values of less than 0.05 were taken as statistically significant.
Results
MIL60 could specifically bind VEGF with high affinity
ELISA analysis was used to screen new antibodies against VEGF and MIL60 from the epitope-specific anti-VEGF antibody phage library that was constructed by our lab was discovered.23 Bevacizumab (Avastin), which is a humanized, anti-human VEGF antibody that was approved for clinical treatment of colorectal cancer, was used as the positive control. As shown in Figure 1a, MIL60 was identified to possess similar binding activity as Avastin. The EC50 value of MIL60 to bind coated VEGF was 44.57 ng/ml, which was similar to that of Avastin (52.17 ng/ml). To further test the specificity of MIL60 in binding VEGF, SKOV3 cells, which secrete VEGF into the supernatant, were incubated with MIL60 or control antibody. The results showed that MIL60 could specifically bind to VEGF, resulting in a reduction in the concentration of free VEGF in the supernatant (Figure 1b, versus control: P<0.05).
Figure 1.

MIL60 binding to VEGF. (a) ELISA analysis of MIL60 binding the coated VEGF antigen. Avastin was set as a positive control. MIL60 showed similar antigen binding activity to that of Avastin. (b) Biofunction of MIL60 in neutralizing free VEGF in the cultural supernatant of SKOV3 cells. *P<0.05, versus IgG control. VEGF, vascular endothelial growth factor.
The binding kinetics of Avastin and MIL60 to VEGF was measured using the Biacore analysis service, which showed that Avastin and MIL60 possessed similar antigen binding affinities. The KD value of MIL60 was 44.5 pM, and the KD value of Avastin was 42.7 pM, which was consistent with results of a previous report (Table 1 & Supplemental Figure 1).24
Table 1. Affinity determination of MIL60 using Biacore analysis.
| Kon(1/Ms) | Koff (1/s) | KD (pM) | |
|---|---|---|---|
| Avastin | 3.15×105 | 1.40×10−5 | 44.5 |
| MIL60 | 3.21×105 | 1.37×10−5 | 42.7 |
The KD value of MIL60 was 44.5 pM, which was nearly the same as that of Avastin (42.7 pM). KD=Kon/Koff.
The epitope of MIL60 was similar to that of Avastin
After sequencing the MIL60 gene, the three-dimensional theoretical Fv structure was modeled using a computer-guided homology modeling method. Under the CVFF and Charmm force fields, the surface properties of VEGF and MIL60 were theoretically analyzed based on their spatial structures. Based on the molecular docking method, the three-dimensional complex structure of MIL60-VEGF was constructed and optimized, as shown in Figure 2a. According to the crystal structures of VEGF and Avastin (PDB code: 2FJH, as shown in Figure 2b), their structural similarities were theoretically analyzed. Based on the distance geometry method and intermolecular hydrogen bond-forming theory, the identified binding domains of MIL60 and Avastin were compared. Residues Lys16, Phe17, Asp19, Tyr21, Arg23, Tyr25, Asn62 and Lys101 in VEGF were the key binding sites of MIL60. Based on the crystal structure of VEGF and Avastin, the binding domain of MIL60 was similar to that of Avastin.
Figure 2.

Theoretical and experimental identification of the epitope of MIL60. (a and b) Theoretical analysis of MIL60 (a) or Avastin (b) in binding VEGF. The left panel shows the structure of VEGF bound to an antibody. In the left panel, the red lines indicate the heavy chains of the variable fragments, the green lines indicate the light chains and the lower pink lines indicated VEGF. The right panel displays the key sites of VEGF that bind the antibody, which are marked with blue balls and sticks. (c and d) Competitive ELISA analysis of the epitope that is recognized by MIL60 and Avastin. (c) Diluted Avastin or negative control antibody (Herceptin) was pre-incubated with HRP-conjugated MIL60, and the mixture was added as the primary antibody. (d) Diluted MIL60 or negative control antibody (Herceptin) was pre-incubated with HRP-conjugated Avastin, and the mixture was added as the primary antibody. It was found that the two antibodies bound the same or similar epitopes because they competed with each other. VEGF, vascular endothelial growth factor; HRP, horseradish peroxidase.
A competition ELISA assay of MIL60 and Avastin to bind human VEGF was then performed. The binding activity of MIL60 to VEGF, as shown in Figure 2c, was affected by Avastin in a dose-dependent manner. When the concentration of Avastin reached 100 µg/ml, the binding activity of MIL60 to VEGF was nearly blocked, while herceptin/trastuzumab, which is an anti-erbB2/HER2 antibody that was set as the negative control, did not compete with Avastin, even at the maximum concentration. Similarly, the binding activity of Avastin to VEGF was also inhibited by MIL60 in a dose-dependent manner (Figure 2d). These results were consistent with the theoretical results.
MIL60 inhibits the VEGF-induced, pro-angiogenic effects of HUVECs
VEGF is an important pro-angiogenic factor that can enhance the proliferation of EC. Here, MIL60 was mixed with VEGF for 30 min and the mixture was added into the supernatant of HUVECs (Supplemental Figure 2) for another 72 h. MIL60 and Avastin displayed dose-dependent inhibitory activity on HUVEC growth (Figure 3a), and MIL60 exhibited similar function as Avastin, with IC50 values of 31±6.4 ng/ml (MIL60) and 47±8.1 µg/ml (Avastin), respectively.
Figure 3.

MIL60 inhibits VEGF-induced cell proliferation, migration and tube formation in vitro in HUVECs. (a) Cell proliferation of HUVECs induced by VEGF was inhibited by MIL60 in a dose-dependent manner. (b and c) MIL60 inhibits the VEGF-induced migration of HUVECs. The MIL60 or Avastin antibodies were diluted to 0.08 µg/ml, 0.8 µg/ml and 8 µg/ml. The migrated cells were counted and are shown in c. (d and e) MIL60 inhibits VEGF-induced tube formation of HUVECs. The MIL60 or Avastin antibodies were diluted to 0.02 µg/ml, 0.2 µg/ml in vitro and 2 µg/ml. The total vessel length per field was counted and is shown in e. Tube length data are presented as the mean±s.d. deviation of three samples for each treatment. *P<0.05, versus control. HUVEC, human umbilical vein endothelial cell; VEGF, vascular endothelial growth factor.
Meanwhile, EC migration is an essential, functional process in angiogenesis. VEGF is known as a chemotactic factor of EC that can activate cytoskeleton remodeling signaling pathways and promote EC migration. Here, a transwell assay was employed to evaluate the inhibition of MIL60 on VEGF-induced chemotaxis of HUVECs. As shown in Figure 3b and c, MIL60 remarkably reduced the number of migrated cells in a dose-dependent manner (8 µg/ml MIL60: 49.4±16.4 cells migrated per field; 0.8 µg/ml MIL60: 87.6±14.3 cells; 0.08 µg/ml MIL60: 142.2±7.3 cells; control: 182.1±18.9 cells. 8 µg/ml or 0.8 µg/ml MIL60 versus the control: P<0.05). These results showed that similar to Avastin, MIL60 could inhibit VEGF-induced chemotaxis and migration of HUVECs.
Neovascularization depends upon angiogenic stimuli to form and organize tubular networks. This capacity to form invasive complex capillary networks can be modeled ex vivo using ECM components as a growth substrate to spontaneously promote the formation of a highly crosslinked network of HUVEC-lined tubes. It was shown that VEGF could promote endothelial tubular morphogenesis and accelerate tube formation; however, the anti-VEGF antibody, MIL60, remarkably decreased the length of the tubes (control: 1734.2±404.2 µm; 2 µg/ml MIL60: 509.8±78 µm; 0.2 µg/ml MIL60: 882.1±145.9 µm; 0.02 µg/ml MIL60: 1420.8±298.4 µm; 2 µg/ml or 0.2 µg/ml MIL60 versus the control: P<0.05), suggesting an effective inhibitory function of MIL60 against VEGF-stimulated tubular structure formation in HUVECs that was similar to that of Avastin (Supplemental Table 1, Figure 3d and e).
MIL60 could inhibit VEGFR2 phosphorylation and downstream signal activation
VEGF binding to its tyrosine kinase receptors (e.g., VEGFR2) causes receptor dimerization and autophosphorylation, which can activate numerous downstream proteins and eventually induce angiogenesis, vascular permeability, endothelial cell proliferation and/or migration. As showed in Figure 4, VEGF could induce VEGFR2 phosphorylation followed by the activation of the p38/IKK/NF-κB and ERK pathways, while MIL60 blocked the VEGF-induced activation of downstream signaling pathways. Antibody-specific blocking of VEGFR2 phosphorylation can be effectively suppressed in a dose-dependent manner. When the antibody concentration was 4 µg/ml, MIL60 and Avastin completely blocked the phosphorylation of VEGFR2. When VEGFR2 phosphorylation was blocked, the corresponding p38/IKK/NF-κB and ERK signaling pathways were specifically blocked, and the phosphorylation of key molecules, such as P38, ERK and P65, were inhibited.
Figure 4.

VEGF induces VEGFR2 phosphorylation and activation of the downstream signal cascade. Pre-incubation of MIL60 and 50 ng/ml VEGF blocked its efficacy for activating key signal molecules such as VEGFR2, ERK, P38 and p65 in a dose-dependent manner. The MIL60 or Avastin antibodies were diluted to 0.04 µg/ml, 0.4 µg/ml and 4 µg/ml. VEGF, vascular endothelial growth factor.
MIL60 inhibits tumor growth and angiogenesis in nude mice
It has been well documented that tumor growth is dependent upon angiogenesis. Avastin inhibited tumor growth in animal experiments and clinical trials. To investigate whether MIL60 had a similar effect in inhibiting tumor growth in vivo, a tumor model was established using the human colon carcinoma cell line HT-29. When tumors reached a diameter of 3–5 mm, the mice were treated with MIL60, Avastin or saline twice per week. MIL60 and Avastin possessed remarkable anti-tumor effects because the growth rates of the tumors were significantly reduced in the MIL60- and Avastin-treated groups compared with that of the saline-treated group (Figure 5a). When the experimental observation was completed, the mice were euthanized and the tumors were stripped. The tumor weight of the antibody-treated group was significantly lower than that of the control group (Figure 5b, MIL60 versus N.S.: P=0.00082; Avastin versus N.S.: P=0.0015), and no significant difference between the Avastin- and MIL60-treated groups was evident (MIL60 versus Avastin: P=0.074).
Figure 5.

MIL60 inhibited carcinoma growth and angiogenesis in vivo. (a) MIL60 had satisfactory anti-tumor activity in human colon carcinoma HT-29 xenografts. The mean and s.d. values of the tumor volumes of each treated group (n=6) at different time points after tumor cell implantation are shown as a polygram and confirmed that MIL60 could significantly inhibit tumor growth. Moreover, the inhibitory effect of MIL60 was similar to that of Avastin. (b) The tumor weight of each group is shown in the column diagram, which also shows the P values between the indicated groups. (c) MIL60 inhibits tumor growth in human ovarian carcinoma SKOV3 xenografts, which was also similar to the effect of Avastin. The mice were killed and the xenograft tumors were separated on day 38.
To further verify the efficacy of anti-VEGF antibodies in treating cancers, we chose another ovarian cancer cell line, SKOV3, to establish a xenograft model for evaluating the biofunction of MIL60 in vivo. As shown in Figure 5c, MIL60 exhibited anti-tumor activity consistent with that of Avastin.
Anti-VEGF antibodies exert anti-tumor mechanisms mainly through inhibiting internal tumor angiogenesis. Immunohistochemical examination of tumor sections was performed to determine the microvessel density. Tumor sections were stained with anti-CD31 antibody (a vascular endothelial marker) and a significant reduction of microvessel density was observed in the MIL60- and Avastin-treated groups (Figure 6a). Furthermore, the number of CD31-positive capillaries was counted from five randomly chosen fields. After antibody treatment, the microvessel density in the tumor obviously decreased (Figure 6b, MIL60 versus N.S.: P=0.0007; Avastin versus N.S.: P=0.00046), which further revealed that MIL60 had a similar function as Avastin in effectively inhibiting tumor angiogenesis in vivo (MIL60 versus Avastin: P=0.24).
Figure 6.

The microvessel densities of tumors were reduced by MIL60. (a) Colon carcinoma (HT-29) sections were stained with an anti-CD31 antibody and the microvessel density was calculated and showed similar inhibitory activity of MIL60 against angiogenesis as Avastin in vivo. Arrow points represent new vessels. (b) The vessel length of each group is shown in a column diagram, which also shows the P values between the indicated groups.
Discussion
Angiogenesis is a complicated and dynamic process that occurs during the growth of all solid tumors. It supplies nutrients and oxygen to and disposes waste products from tumors. Anti-angiogenesis has provided a form of cancer therapy that is worthy of serious exploration. Of the variety of pro-angiogenic factors, VEGF is one of the most important in regulating angiogenesis. The interaction between VEGF-A and VEGFR2 is the best characterized ligand-receptor relationship in the angiogenesis process, which is why VEGF is considered to be one of the most promising targets for cancer therapy. Among all inhibitors, Avastin has been one of the most successful therapeutic agents to target VEGF. Avastin is a humanized variant of mouse anti-human VEGF monoclonal antibody A4.6.125 that was initially identified by its ability to block human VEGF-A-stimulated EC proliferation.26 Furthermore, Avastin was confirmed to be able to inhibit the growth of human tumor xenografts in nude mice.7 In 2004, Avastin was approved by the FDA for the treatment of metastatic colorectal16 and nonsquamous, non-small-cell lung cancers,17 when in combination with chemotherapy.
Because Avastin represents a clinically validated therapeutic agent in cancer and other diseases,10,11,27 many groups have attempted to prepare a similar drug.28 Moreover, Avastin is widely used in the clinical treatment of various tumors, but its price is extremely high for Chinese people. Therefore, we chose to screen for a novel or even ‘me-better' human anti-VEGF antibody.
HUVECs are the most commonly used cell model for VEGF-related studies and several articles have been published in which HUVECs were generally the only cell model to verify the anti-angiogenesis activity of new antagonists.29,30 Therefore, we used HUVECs to biologically identify a novel antibody to VEGF.
Because the major drug modalities of antibodies are due to their high specificity and affinity for binding antigens, antibody-dependent cellular cytotoxicity, downmodulation of antigens and inhibition of downstream signaling activation are major mechanisms for achieving the biofunction of antibodies. In contrast to other monoclonal antibodies that directly bind to extracellular antigens of tumor cells, anti-VEGF antibodies are expected to interfere with angiogenesis primarily by preventing free VEGF from binding to its receptors in the tumor vasculature. This mechanism will cause regression of the remaining tumor vasculature and delay tumor growth and metastasis.31 Therefore, tight binding between the anti-VEGF antibody and VEGF may result in a strong therapeutic effect.
Various affinity-purified, anti-VEGF antibodies, however, did not show clear, increased potency or efficacy in blocking tumor growth, when compared with their lower-affinity counterparts, in most in vivo models, even though in vitro studies demonstrated a correlation between antibody-binding affinity and potency in assays measuring the inhibition of VEGF-stimulated EC proliferation. In contrast, increasing antibody affinity did not affect efficacy or potency in most models, but resulted in increased toxicity, including glomerulosclerosis, hypoalbuminemia and ascite formation. Thus, an optimal value for the binding affinity exists where the anti-VEGF antibody binds VEGF tightly enough to inhibit tumor growth. To achieve minimal toxicity, this drug design parameter must be carefully balanced with the occurrence of adverse events. The affinity limits of the anti-VEGF antibodies were not confirmed, although they were reported in many studies.32
Avastin is one of the most important anticancer drugs and is widely used in clinics. Importantly, its clinical safety and effectiveness have been verified. We hoped to obtain a better antibody than Avastin; however, many previous studies have revealed that the affinity of an anti-VEGF antibody is not proportional to its efficacy because higher affinity could sometimes cause side effects. Therefore, we set Avastin as a positive control and screened MIL60, which has a similar affinity for VEGF.23 This principle ensured the efficacy of the antibody and avoided increased side effects. In other words, MIL60 had similar affinity for VEGF as Avastin, although this study did not provide definitive evidence of the clinical safety of MIL60. The verification of the clinical safety and efficacy of Avastin could infer the possible favorable safety of MIL60.
The antigenic epitope of a novel antibody is important for its efficacy and safety. The epitope that overlapped the receptor-binding sites of VEGF-A appeared to be associated with improved efficacy, and this effect achieved statistical significance in some cases. Many studies have shown that Avastin specifically recognizes VEGF, and the epitope in VEGF included the key sites that bound VEGFR2; therefore, when Avastin binds to VEGF, it inhibits VEGF–VEGFR2 complex formation and the downstream signal cascades.32 Thus, Avastin could block the VEGFR2/VEGF signaling pathway that mediates the subsequent biological effects.
In this study, we discovered a highly specific and potent, neutralizing, anti-human VEGF antibody, MIL60, in a synthetic ‘me-better' phage library. MIL60 could specifically block the VEGF–VEGFR2 interaction (Figure 1). MIL60/Avastin was labeled with HRP for competition binding assays, and it was shown that MIL60 recognized the same epitope as Avastin (Figure 2), which suggested that MIL60 may have similar clinical safety as Avastin. According to in vitro experiments, MIL60 was identified to be effective in inhibiting VEGF-induced EC proliferation, migration and tube formation and blocking the VEGF/VEGFR2 signaling pathway, e.g., VEGFR2 phosphorylation, ERK and P38 activation (Figures 3 and 4). Furthermore, similar to Avastin, MIL60 could inhibit xenograft SKOV3 or HT-29 carcinoma growth and angiogenesis of human colon carcinoma in vivo (Figures 5 and 6). These results suggested that MIL60 could be beneficial as a potential anti-angiogenic agent to efficiently generate a high-quality, anti-tumor drug that will offer a new option for patients in the future.
Acknowledgments
This work was supported by grants from the National Sciences Fund (Nos. 31370938 and 81272528) and the National High Technology Research and Development Program (863 Program, No. 2012AA02A302).
Footnotes
Supplementary Information accompanies the paper on Cellular & Molecular Immunology's website. (http://www.nature.com/cmi).
Supplementary Information
References
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihara E, Kiura K, Tanimoto M. Targeting angiogenesis in cancer therapy. Acta Med Okayama. 2011;65:353–362. doi: 10.18926/AMO/47260. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Molecular and biological properties of vascular endothelial growth factor. J Mol Med (Berl) 1999;77:527–543. doi: 10.1007/s001099900019. [DOI] [PubMed] [Google Scholar]
- Klagsbrun M, D'Amore PA. Vascular endothelial growth factor and its receptors. Cytokine Growth Factor Rev. 1996;7:259–270. doi: 10.1016/s1359-6101(96)00027-5. [DOI] [PubMed] [Google Scholar]
- Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- Prewett M, Huber J, Li Y, Santiago A, O'Connor W, King K, et al. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res. 1999;59:5209–5218. [PubMed] [Google Scholar]
- Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Kowalski J, Sherman D, Eberhard DA, Ferrara N. Complete inhibition of rhabdomyosarcoma xenograft growth and neovascularization requires blockade of both tumor and host vascular endothelial growth factor. Cancer Res. 2000;60:6253–6258. [PubMed] [Google Scholar]
- Konner J, Dupont J. Use of soluble recombinant decoy receptor vascular endothelial growth factor trap (VEGF Trap) to inhibit vascular endothelial growth factor activity. Clin Colorectal Cancer. 2004;4 Suppl 2:S81–S85. doi: 10.3816/ccc.2004.s.013. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- Gasparini G, Longo R, Toi M, Ferrara N. Angiogenic inhibitors: a new therapeutic strategy in oncology. Nat Clin Pract Oncol. 2005;2:562–577. doi: 10.1038/ncponc0342. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- McTigue M, Murray BW, Chen JH, Deng YL, Solowiej J, Kania RS. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc Natl Acad Sci USA. 2012;109:18281–18289. doi: 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LA, Brekken RA. The VEGF family in cancer and antibody-based strategies for their inhibition. MAbs. 2010;2:165–175. doi: 10.4161/mabs.2.2.11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Chung YS, Ogawa Y, Takatsuka S, Kang SM, Ogawa M, et al. Prognostic value of vascular endothelial growth factor expression in gastric carcinoma. Cancer. 1996;77:858–863. doi: 10.1002/(sici)1097-0142(19960301)77:5<858::aid-cncr8>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Weidner N, Folkman J, Pozza F, Bevilacqua P, Allred EN, Moore DH, et al. Tumor angiogenesis: a new significant and independent prognostic indicator in early-stage breast carcinoma. J Natl Cancer Inst. 1992;84:1875–1887. doi: 10.1093/jnci/84.24.1875. [DOI] [PubMed] [Google Scholar]
- Higgins B, Kolinsky K, Linn M, Adames V, Zhang YE, Moisa C, et al. Antitumor activity of capecitabine and bevacizumab combination in a human estrogen receptor-negative breast adenocarcinoma xenograft model. Anticancer Res. 2007;27:2279–2287. [PubMed] [Google Scholar]
- Martiny-Baron G, Korff T, Schaffner F, Esser N, Eggstein S, Marme D, et al. Inhibition of tumor growth and angiogenesis by soluble EphB4. Neoplasia. 2004;6:248–257. doi: 10.1593/neo.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao C, Lv M, Li X, Geng J, Li Y, Zhang J, et al. Affinity maturation of antiHER2 monoclonal antibody MIL5 using an epitope-specific synthetic phage library by computational design. J Biomol Struct Dyn. 2013;31:511–521. doi: 10.1080/07391102.2012.706073. [DOI] [PubMed] [Google Scholar]
- Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15:171–185. doi: 10.1007/s10456-011-9249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presta LG, Chen H, O'Connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–4599. [PubMed] [Google Scholar]
- Kim KJ, Li B, Houck K, Winer J, Ferrara N. The vascular endothelial growth factor proteins: identification of biologically relevant regions by neutralizing monoclonal antibodies. Growth Factors. 1992;7:53–64. doi: 10.3109/08977199209023937. [DOI] [PubMed] [Google Scholar]
- Ng EW, Shima DT, Calias P, Cunningham ET, Jr, Guyer DR, Adamis AP. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat Rev Drug Discov. 2006;5:123–132. doi: 10.1038/nrd1955. [DOI] [PubMed] [Google Scholar]
- Yu Y, Lee P, Ke Y, Zhang Y, Yu Q, Lee J, et al. A humanized anti-VEGF rabbit monoclonal antibody inhibits angiogenesis and blocks tumor growth in xenograft models. PLoS ONE. 2010;5:e9072. doi: 10.1371/journal.pone.0009072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferla R, Bonomi M, Otvos L, Jr, Surmacz E. Glioblastoma-derived leptin induces tube formation and growth of endothelial cells: comparison with VEGF effects. BMC Cancer. 2011;11:303. doi: 10.1186/1471-2407-11-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Fei D, Vanderlaan M, Song A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis. 2004;7:335–345. doi: 10.1007/s10456-004-8272-2. [DOI] [PubMed] [Google Scholar]
- Jain RK. Antiangiogenic therapy for cancer: current and emerging concepts. Oncology (Williston Park) 2005;19:7–16. [PubMed] [Google Scholar]
- Gerber HP, Wu X, Yu L, Wiesmann C, Liang XH, Lee CV, et al. Mice expressing a humanized form of VEGF-A may provide insights into the safety and efficacy of anti-VEGF antibodies. Proc Natl Acad Sci USA. 2007;104:3478–3483. doi: 10.1073/pnas.0611492104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.