

Abstract
Obesity and the metabolic syndrome and their associated morbidities are major public health issues, whose prevalence will continue to increase in the foreseeable future. Aberrant signaling by the receptors for leptin and insulin plays a pivotal role in development of the metabolic syndrome. More complete molecular-level understanding of how both of these key signaling pathways are regulated is essential for full characterization of obesity, the metabolic syndrome, and type II diabetes, and for developing novel treatments for these diseases. Phosphorylation of proteins on tyrosine residues plays a key role in mediating the effects of leptin and insulin on their target cells. Here, we discuss the molecular methods by which protein tyrosine phosphatases, which are key physiological regulators of protein phosphorylation in vivo, affect signaling by the leptin and insulin receptors in their major target tissues.
Keywords: obesity, diabetes, metabolic syndrome, insulin resistance, leptin resistance, tyrosine phosphatase, PTP
INTRODUCTION
The burden of obesity is increasing worldwide and is expected to continue to rise with the spread of Western-type sedentary lifestyle. Obesity is associated with a cluster of risk factors for cardiovascular disease and type 2 diabetes mellitus, which have become known as the metabolic syndrome. In addition to obesity, these risk factors include characteristic dyslipidemia (raised triglycerides and lowered high-density lipoprotein cholesterol), raised fasting glucose and elevated blood pressure[1]. The abnormal glucose and lipid homeostasis that characterizes the syndrome includes increased adipose tissue lipolysis, increased hepatic glucose production and hepatic lipogenesis, decreased clearance of triglyceride-rich lipoproteins, and decreased glucose uptake in muscle. The pathophysiology of the metabolic syndrome is complex and only partially understood, but there is ample evidence to suggest that defects in insulin and leptin signaling in target cells of these hormones lead to obesity and to abnormal glucose and lipid homeostasis that characterize the syndrome[2]-[4]
Individuals who suffer from the metabolic syndrome exhibit resistance to the effects of leptin and insulin, manifested as reduced response by the target cells to the action of these hormones. Resistance affects both the molecular processes that are initiated by the leptin and insulin receptors (LR, IR, respectively), as well as the subsequent physiological responses of these cells. For example, insulin is a potent inhibitor of lipolysis and of release of free fatty acids (FFA) from adipose tissue. In the insulin-resistant state the ability of insulin to inhibit lipolysis is impaired, leading to increased influx of FFA to the liver and development of liver steatosis. Human studies using euglycaemic hyperinsulinaemic clamp techniques demonstrate that in affected individuals, glucose uptake by the muscle is reduced due to attenuated response of the cells to the hormone. If pancreatic beta cells cannot produce more insulin to compensate, this will lead to hyperglycemia[4]. As resistance to insulin and leptin plays a pivotal role in several common diseases including obesity, type II diabetes, cardiovascular diseases, non-alcoholic fatty liver disease, certain types of cancer, and even Alzheimer's disease[5],[6], understanding of the molecular bases of insulin and leptin resistance and developing novel treatments that target them can have a meaningful effect on public health.
Tyrosine phosphorylation of proteins plays a central role in mediating the cellular effects that follow activation of the leptin and insulin receptors. Here, we outline the role of protein tyrosine phosphatases (PTPs), which are central regulators of protein phosphorylation, in regulating IR and LR signaling.
PTPS
Phosphorylation of proteins is among the best-studied mechanisms for regulation of protein structure and function in cells and organisms. Despite being a minority of phosphorylation events in eukaryotic cells, tyrosine phosphorylation plays a critical and well-established role in regulating cellular processes[7],[8]. Phosphorylation of tyrosine residues in proteins is a reversible process. Phosphorylation is carried out by protein tyrosine kinases (PTKs), while dephosphorylation is performed by PTPs; PTKs and PTPs act together to regulate phosphorylation levels of their substrates. The human genome contains 90 PTK genes and over 100 genes that encode PTPs; of these, 85 and 81 genes, respectively, produce products that target protein substrates[9]-[11]. Both enzyme families are therefore much smaller than the number of known and hypothesized phosphorylation sites that they regulate. Consequently, each PTK or PTP usually targets more than a single substrate and carries out distinct physiological roles in different cell types.
The first report of purification and characterization of a tyrosine phosphatase, PTP1B, was published in 1988 by Nicholas Tonks and Edmond Fischer[12],[13]. An outburst of cloning and characterization studies of PTPs soon followed and resulted in our current understanding that PTPs form a large superfamily of enzymes with high activity and specificity in vivo. Of the slightly more than 100 PTP genes known, 38 belong to the “classical” PTP family whose members target phosphotyrosine exclusively. A second major group encodes enzymes that dephosphorylate both tyrosine and serine/threonine residues (the dual-specificity phosphatases, DUSPs). Additional PTPs include the low-molecular weight (LMW) phosphatase and Cdc25 phosphatases[9]-[11],[14]. Classical PTPs can be further classified into receptor-type and non receptor-type families, and either family can be subdivided further according to structural and sequence similarities among its members. Receptor-type PTPs (RPTPs) contain an extracellular domain of varying length and structure, a membrane-spanning domain, and one or two cytosolic PTP domains that either carry out catalysis or regulate it. Although the structures of the extracellular domains of RPTPs resemble those of receptor-type PTKs, few ligands of RPTPs are known. Non receptor-type PTPs typically include a single PTP domain that is flanked by other protein domains, which control the subcellular localization of the enzyme or regulate its activity[9],[11].
Each PTP includes one or two PTP domains of approximately 240 amino acids, which include the HCXXXXXR PTP signature motif. When two such domains are present in a single PTP, the first membrane-proximal domain is active while the second PTP domain often fulfills regulatory roles. PTPs dephosphorylate their substrates in a two-step mechanism that is centered around a conserved critical cysteine residue. Initially, a covalent bond between the sulfur atom of this cysteine and the phosphorous atom of the phosphate group of its phospho-tyrosine substrate is formed. During this step the bond between the phosphate group and the tyrosine residue of the substrate protein is broken, and the dephosphorylated protein is released. The PTP-phosphate bond is then hydrolyzed, releasing the phosphate group and regenerating the active PTP[10],[15].
Recent studies have elucidated the molecular roles of individual PTPs in specific physiological processes such as cellular transformation, differentiation, immune function, bone structure, and others, from which it is clear that PTPs target specific substrate molecules and perform discrete physiological roles. The reader is referred to several recent reviews that discuss the physiological roles of PTPs and their modes of regulation in detail[11],[16]-[23]. Here, we review the roles of PTPs in regulating leptin and insulin signaling, with special emphasis on studies performed in whole-organism model systems.
PTPS AND REGULATION OF THE LEPTIN RECEPTOR AND BODY WEIGHT
Leptin signaling
Leptin is produced by adipocytes and is secreted into the blood; it affects body weight by activating its receptor in specific populations of neurons in the brain, including in the hypothalamus[24],[25]. The leptin receptor is a type I cytokine receptor that lacks catalytic activity, but is constitutively associated with the tyrosine kinase Janus Kinase 2 (JAK2) (Fig. 1). Binding of leptin to its receptor induces a conformational change in the receptor, which activates JAK2; JAK2 undergoes autophosphorylation, and then phosphorylates the LR at Tyr 985, Tyr1077 and Tyr 1138[26]. LR phosphorylation creates binding sites for several SH2 domain-containing proteins to associate with the LR-JAK2 complex and propagate the leptin signal further[26],[27]. A major mediator of LR signaling is Signal Transducer and Activator of Transcription 3 (STAT3), whose SH2 domain binds to the LR at Tyr1138. STAT3 is then phosphorylated by JAK2 and translocates to the nucleus to regulate expression of target genes[24],[25]. In a similar manner, phosphorylation at Tyr 1077 recruits STAT5 and mediates its activation. Phosphorylation of the LR at Tyr985 allows recruitment of the SH2 domain-containing tyrosine phosphatase SHP2, which leads to activation of the ERK-MAP kinase pathway, while JAK2 directly activates the IRS-PI3 kinase pathway[25],[26],[28],[29] (Fig. 2). As expected of a key physiological process, LR signaling is subject to down-regulation. For example, Suppressor of Cytokine Signaling 3 (SOCS3), whose expression is induced by STAT3, inhibits JAK2 and binds to the LR at Tyr985 in a manner that antagonizes activation of the ERK-MAPK pathway[26].
Fig. 1. Hypothalamic lepin receptor signaling.
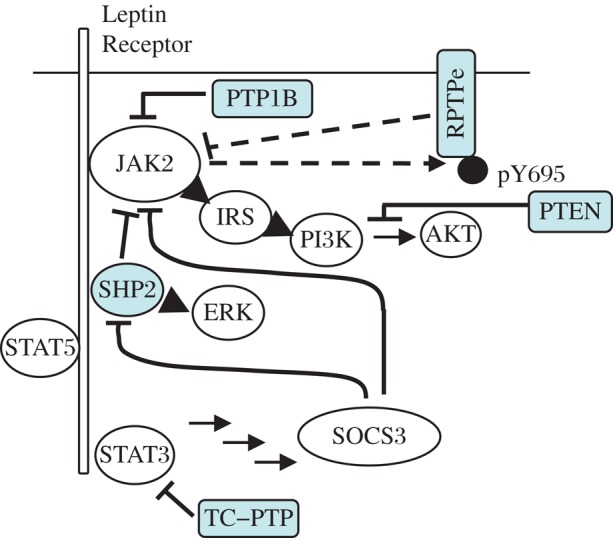
Following LR activation, the associated JAK2 kinase is activated, auto-phosphorylates, and trans-phosphorylates the LR. This creates binding sites for SH2 domain-containing proteins, which activate downstream signaling. JAK2 phosphorylates RPTPe, which deactivates JAK2 (dashed arrows). Also shown are PTP1B that targets JAK2, and TC-PTP that targets STAT3.
Fig. 2. PTPs discussed in this review.
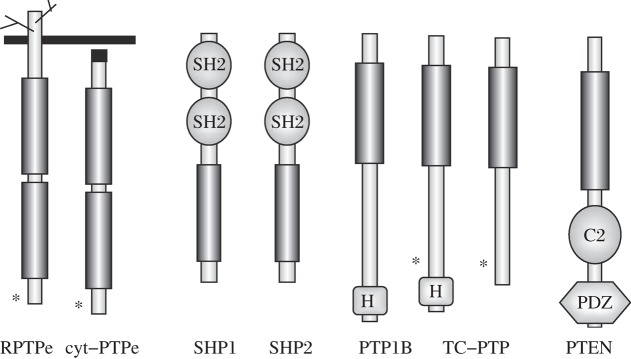
Shown are the receptor (RPTPe) and non-receptor (cyt-PTPe) forms of PTP Epsilon, SHP1, SHP2, PTP1B, the 48kDa and 45 kDa forms of TC-PTP, and PTEN. Vertical rectangles represent PTP domains in all the PTPs. Also shown are the SH2, C2, PDZ, and H (hydrophobic ER-targeting motif) domains of the various PTPs. Asterisks mark critical C-terminal tyrosines (PTPe) or nuclear localization signals (TC-PTP).
The activated hypothalamic LR induces expression of the appetite-inhibiting POMC (pro-opiomelanocortin) and CART (cocaine- and amphetamine-related transcript) in specific neurons, while in others it suppresses the appetite-stimulating neuropeptide Y (NPY) and the agouti-related protein (AgRP). Overall, leptin reduces appetite and increases energy use, thus regulating both main processes that affect body weight. Circulating leptin levels are affected by the mass of adipose tissue in the body, and its concentration in blood indicates the level of fat stores available. Leptin also affects many other systems, such as those that determine bone mass and fertility[30]-[32]; these will not be discussed here. Tyrosine phosphorylation plays central roles in regulating the activities of the LR and its downstream effectors, and indeed several PTPs have been shown to be essential in this respect.
PTP1B
This non-receptor PTP includes an N-terminal catalytic PTP domain that is followed by two proline-rich sequence motifs and a C-terminal hydrophobic tail (Fig. 2). The C-terminal associates with the endoplasmic reticulum (ER) membrane, and localizes the enzyme to the cytosolic side of the ER. Proteolytic cleavage of this tail releases a truncated PTP1B into the cytosol and increases its specific activity[33]-[36]. PTP1B was the first tyrosine phosphatase discovered to regulate body weight and LR signaling in vivo. PTP1B dephosphorylates JAK2 and down-regulates its activity; accordingly, mice that lack PTP1B throughout their bodies are leptin-hypersensitive, exhibit reduced adiposity and are resistant to diet-induced obesity due to increased energy expenditure. As indicated below, these mice also exhibit altered IR signaling[37]-[40]. Further studies of several mouse model systems carrying tissue-specific deletions of PTP1B confirmed the key role of hypothalamic PTP1B in this process. Mice lacking PTP1B in CNS neurons, induced by Cre-mediated recombination in nestin-expressing neurons, exhibit reduced weight and adiposity, increased energy expenditure, decreased food intake, and increased leptin sensitivity despite elevated levels of circulating leptin[41],[42], the latter most likely due to dysregulation of AMP-activated protein kinase (AMPK)[43]. Similar findings were made when PTP1B was targeted by Cre-mediated recombination in neurons specifically expressing the LR or POMC[41],[44]-[46]. By contrast, inactivating PTP1B in skeletal muscle, liver or white adipose tissue did not affect body weight; deleting PTP1B in liver improved lipid metabolism, while mice lacking PTP1B in adipose tissue exhibited larger adipocytes, increased circulating leptin levels and reduced leptin sensitivity[47]-[50]. Interestingly, a high-fat diet and old age increase PTP1B levels in the hypothalamus[51],[52], suggesting that PTP1B activity in that tissue is regulated by control of its protein levels.
TC-PTP
Is a ubiquitous non-receptor PTP that is highly similar in structure and sequence to PTP1B (Fig. 2). Alternative splicing of the TC-PTP mRNA yields a 48 kDa protein that associates with the ER via its C-terminal tail, and a more abundant 45 kDa form that lacks the terminal sequence and is predominantly nuclear. This latter isoform can exit the nucleus following stimulation of cells by any of several factors[36],[53].
Mice lacking TC-PTP throughout their bodies exhibit broad inflammation and die at the age of 3–4 weeks, a fact that necessitated development of tissue-specific deletions of TC-PTP. Indeed, mice lacking TC-PTP in nestin-expressing neurons exhibit leptin hypersensitivity, increased energy expenditure and are resistant to weight gain induced by high-fat food[54]. Interestingly, when fed regular lab chow, neuronal TC-PTP-deficient mice display decreased body weight, reduced levels of circulating leptin, and increased leptin sensitivity, but also increased relative adiposity (when corrected for body weight). In a manner distinct from PTP1B, TC-PTP targets STAT3; accordingly, combined deletion of both PTPs induces stronger protection from diet-induced obesity and a higher level of leptin sensitivity than loss of either PTP alone[54]. Similar to PTP1B, a high-fat diet increases TC-PTP expression in the hypothalamus, suggesting that it is also regulated by control of its expression[54].
PTP Epsilon (PTPe)
The gene for PTPe has two promoters, each of which produces a major isoform of the protein – the receptor-like isoform (RPTPe) or the predominantly cytosolic isoform (cyt-PTPe) (Fig. 2). Both isoforms share most of their sequences, including their PTP domains. They differ only at their N termini, where the transmembranal and extracellular domains of RPTPe are replaced by a sequence of 12 hydrophilic amino acids in cyt-PTPe, thus affecting their cellular localization and physiological roles[55]-[59]. RPTPe is expressed in the hypothalamus and has been implicated in regulating LR signaling and body weight. When fed regular chow, mice lacking both forms of PTPe throughout their bodies exhibit normal body weight and normal energy expenditure. However, when the mice are moved to a high-fat diet, female PTPe-deficient mice exhibit increased energy expenditure levels and are resistant to diet-induced obesity compared to matched controls[60]. RPTPe exerts this activity by inhibiting LR signaling via dephosphorylating JAK2[60]. Although this mode of action appears similar to that of PTP1B, the molecular mechanisms that regulate participation of these two PTPs in LR signaling are distinct. In contrast to PTP1B, which is regulated via its expression levels, RPTPe is regulated by LR-dependent tyrosine phosphorylation. Activation of the LR induces phosphorylation of RPTPe at its C-terminal Y695, most likely by the LR-associated JAK2. Phosphorylation of RPTPe does not appear to change its specific activity[61], but directs it by yet unknown mechanisms to directly dephosphorylate JAK2. RPTPe thus functions also as part of a negative-feedback regulatory mechanism that down-regulates LR signaling following its activation[60]. The gender-specificity of the LR phenotype of mice lacking PTPe requires further research.
SHP2
The non-receptor PTP SHP2 contains two N-terminal SH2 domains followed by a PTP domain and a C-terminal sequence that includes a proline-rich motif and tyrosine phosphorylation sites (Fig. 2). SHP2 can assume an inactive, “closed” conformation when the N-terminal SH2 domain binds its PTP domain in a phosphorylation-independent manner; binding of this SH2 domain to phosphor-tyrosines can release this association and activate the PTP. C-terminal tyrosine phosphorylation of SHP2 can occur following activation of tyrosine kinase-type receptors, which allows recruitment of signaling mediators such as Grb2 to the PTP and support the signaling process. In other cases, SHP2 catalytic activity can suppress signaling, leading to sometimes conflicting reports of its cellular role ([62]; reviewed in[63]).
Mice lacking SHP2 throughout their bodies do not survive[64], making tissue-specific inactivation of SHP2 necessary to examine its role in LR signaling. SHP2 promotes leptin signaling by binding the leptin receptor at pY985 and promoting downstream ERK signaling[28],[29]. However, SHP2 also inhibits JAK2 and STAT3 signaling, thereby down-regulating receptor activity and illustrating the complexity by which regulation of LR signaling is achieved[65]. The overall effect of SHP2 is to upregulate hypothalamic leptin receptor signaling, as shown by the phenotypes of three distinct strains of mice lacking neuronal SHP2 that have been developed. Inactivation of SHP2 using Cre recombinase driven by the promoters for calcium-calmodulin dependent kinase IIα (CamIIα; expressed in the forebrain and hypothalamus,[66]), the pan-neuronal CRE3 promoter[67] or the POMC promoter[44] resulted in increased adiposity, increased serum leptin levels and decreased leptin sensitivity, and decreased energy expenditure. Despite their similarity, the leptin-related phenotypes of these three strains are not identical, possibly due to differences in the tissue specificity and timing of Cre-mediated recombination and to the acknowledged role of SHP2 in neuronal development in general[22]. We note that SHP2 binds the LR at Y985, a site to which SOCS3, which inhibits LR signaling, also binds. Down-regulation of LR signaling by loss of SHP2 may thus reflect not only the balance between the activating and inhibitory roles of SHP2 in LR signaling, but also the opportunity offered to SOCS3 to bind the LR and to inhibit its activity without competition from SHP2. In agreement, expression of dominant-active D61A SHP2 in forebrain neurons of mice increased leptin sensitivity, decreased food uptake, increased energy expenditure, and induced resistance to weight gain caused by high-fat food[68]. Interestingly, this latter effect was detected in females, suggesting that SHP2 links estrogen and leptin signaling[68].
PTEN
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a tumor suppressor in a wide variety of tumor types[69]. PTEN contains an N-terminal PTP domain followed by a C2 domain, which binds the phospholipid cellular membrane, and a C terminal tail that helps regulate phosphatase function[70] (Fig. 2). PTEN is a member of the dual-specificity PTP family, which can target pTyr as well as pSer/pThr, and some of its functions, including in metabolic regulation, have been ascribed to its protein phosphatase activity[71],[72]. However, most of its functions are mediated by its dephosphorylation of the lipid phosphatidylinositol (3,4,5)-trisphosphate (PIP3) at position 3 to generate PI(4,5) bisphosphate (PIP2). As such PTEN counters the activity of PI3 kinase and inhibits signaling pathways, such as the leptin and insulin pathways, that activate this lipid kinase. PTEN affects leptin signaling somewhat distal to the LR and its immediate molecular surroundings, and targets a molecular process that is common to many signaling pathways. It is therefore not surprising that the effects of manipulating PTEN vary considerably depending on the precise cellular compartment that is targeted (reviewed in[73]). For example, targeting PTEN in LR-expressing neurons decreased body weight and adiposity and increased oxygen consumption[74], but overexpressing PTEN in the same subset of neurons did not affect these parameters[75]. Moreover, targeting PTEN in POMC-expressing neurons and in the ventromedial hypothalamic nucleus resulted in the opposite phenotype of increased body weight that in the case of the former model system was sex- and diet-dependent[76],[77]. In these two model systems, the body weight phenotype is most likely the result of dysregulation of neuronal insulin signaling, which affects cell polarization and energy balance, and not a direct effect on LR signaling[22],[76],[77].
Leptin resistance, characterized by a reduced response of the LR to leptin, is a serious physiological phenomenon that is present in most obese humans and precludes use of leptin as a drug to treat most obesity cases. The finding that loss of most of the PTPs discussed above results in a leptin-hypersensitive phenotype suggests that PTPs, either individually or acting in concert, are part of the molecular system that regulates leptin sensitivity in vivo. It is tempting to speculate that inhibition of select PTPs in obese individuals may restore normal leptin sensitivity and allow use of leptin to combat obesity. The complexity that characterizes regulation of physiological processes makes it essential to fully understand the molecular details by which each PTP acts in order to ensure that inhibiting a specific PTP will yield the desired effects.
PTPS AND REGULATION OF INSULIN SIGNALING AND GLUCOSE HOMEOSTASIS
Insulin signaling
Insulin is secreted by the β cells of the pancreas in response to rising concentrations of glucose in the circulation. Insulin binds to the insulin receptor, which is an integral membrane receptor-type PTK (Fig. 3). The IR is composed of two extracellular α-subunits that bind insulin, and two transmembranal β-subunits, whose cytosolic domains possess tyrosine kinase activity. Binding of insulin to the IR α subunits activates the β subunits, leading to their autophosphorylation at tyrosines 1158, 1162, and 1163 in the activation loop and at the juxtamembranal Y972, and induces the IR to phosphorylate target proteins such as insulin receptor substrates (IRS)[78]. Tyrosyl phosphorylated IRS proteins provide docking sites for several SH2 domain-containing proteins, including the p85 regulatory subunit of phosphatidylinositol 3-kinase (PI3K). PI3K, which is recruited to the vicinity of the plasma membrane, phosphorylates PIP2 to generate PIP3, and subsequently activates protein kinase B (PKB/AKT). PI3K/AKT signaling mediates the metabolic actions of insulin, including increasing uptake of glucose into muscle cells and reducing glucose production in the liver[79]. Recruitment of the SH2 domain-containing proteins Grb2 and Shc to the IR activates the Ras-MAPK pathway, which mediates the mitogenic effects of IR signaling[80],[81]. Given the importance of tyrosine phosphorylation in insulin signaling it is not surprising that several PTPs were found to regulate this pathway and may therefore have an important role in insulin resistance, type 2 diabetes and the metabolic syndrome[4],[22],[36].
Fig. 3. Insulin receptor signaling.
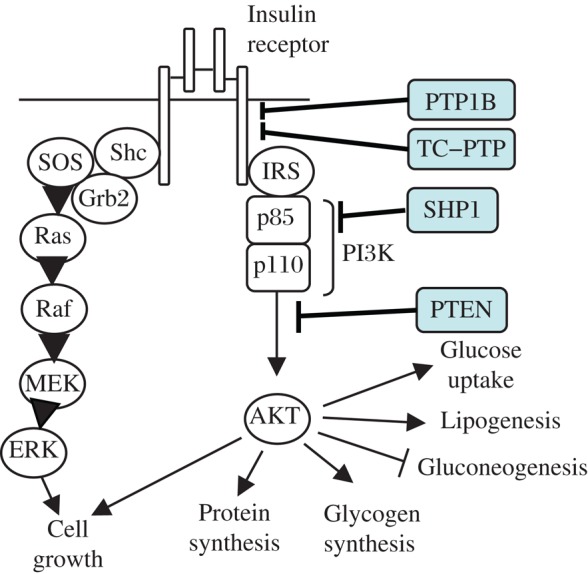
Several PTPs inhibit IR signaling. PTPe and SHP2 also inhibit IR signaling, although the precise molecular mechanisms require added studies.
PTP1B
PTP1B has been a focus of extensive research revealing its central role in obesity, insulin resistance and diabetes[37]-[40],[42],[44],[47],[48],[82]. PTP1B forms a physical complex with the activated IR and dephosphorylates it at Y1162 and Y1163, while expression of exogenous PTP1B in cells can inhibit IR signaling[36]. Starting in 1999, two key studies showed that in addition to reduced adiposity, mice lacking PTP1B throughout their bodies exhibit improved glucose homeostasis with enhanced insulin sensitivity, as well as increased insulin-induced phosphorylation of the IR in liver and muscle tissue compared with wild-type littermates[37],[38]. PTP1B-/- mice also show reduced hepatic secretion of apolipoprotein B (apoB100) lipoproteins, one of the hallmarks of the lipid abnormalities of the metabolic syndrome[83]. Later studies of mice lacking PTP1B in specific tissues enabled the dissection of the role of PTP1B on whole-body glucose homeostasis. Mice lacking PTP1B in their liver exhibit enhanced phosphorylation of the insulin receptor at IR β Y1162/Y1163 and improved glucose homeostasis and lipid profiles, independent of changes in adiposity; similar improvement was obtained when PTP1B was re-expressed in mice lacking PTP1B throughout their bodies[48],[84]. Liver-specific PTP1B knockout mice also exhibited attenuated diet-induced ER stress, an additional mechanism by which high-fat and high calorie Western diets lead to insulin resistance[85]. Muscle-specific PTP1B-deficient mice exhibited significant improvement in whole-body glucose homeostasis on both chow and high-fat diet, despite comparable body weight and adiposity[47]. Interestingly, loss of PTP1B in the brain generated not only leptin hypersensitivity as discussed above, but also insulin hypersensitivity and improved glucose tolerance[42]. Eliminating expression of PTP1B in neurons expressing POMC resulted in substantially improved glucose homeostasis on a high-fat diet irrespective of weight change[44]; similar findings were made with mice lacking PTP1B in LR-expressing neurons[41]. These data agree with other studies demonstrating a brain-liver circuit, by which the central effect of insulin regulates hepatic glucose production[86],[87]. Elimination of PTP1B expression in white adipose tissue reduced IR signaling somewhat, possibly due to interactions with other signaling pathways, but did not alter insulin signaling significantly[49]. Taken together these data imply that PTP1B is a key central negative regulator of insulin signaling in the liver, muscle, and brain and its inhibition in any and all these tissues improves the metabolic profile of insulin resistance[22],[36].
TC-PTP
TC-PTP is expressed ubiquitously, including in insulin-sensitive tissues such the brain, liver, white adipose tissue and skeletal muscle. Like its close structural relative PTP1B, TC-PTP negatively regulates insulin signaling by dephosphorylating IRβ at Y1162/Y1163[88]. In cells, the two PTPs were found to function in a coordinated and temporally distinct manner to regulate IR phosphorylation and signaling[89]. PTP1B can dephosphorylate JAK2, but not JAK1 and JAK3, in contrast to TCPTP that dephosphorylates JAK1/3, but not JAK2[90].
Since mice lacking TC-PTP throughout their bodies die at an early age, the metabolic roles of TC-PTP were examined initially in mice that were heterozygous for the TCPTP null allele[91]. These mice exhibited reduced levels of fasting glucose and of hepatic glucose production, decreased gluconeogenic gene expression and enhanced signaling by STAT3 and PI3K/AKT. Insulin-induced phosphorylation of the IR at Y1162/1163 was also enhanced in hepatocytes isolated from these mice[91]. In contrast to the regulatory role of TC-PTP in the liver, lack of TC-PTP specifically in muscle does not alter insulin signaling and glucose homeostasis in mice[92].
Another tissue that influences systemic insulin sensitivity is bone. Recent data show that insulin signaling in osteoblasts contributes to whole-body glucose homeostasis by increasing the activity of osteocalcin. Interestingly, TC-PTP is similar to another PTP, ESP, that regulates insulin receptor phosphorylation in osteoblasts; osteoblast-specific deletion of TC-PTP increased insulin sensitivity in an osteocalcin-dependent manner[93].
PTPe
Both the receptor-type isoform of PTPe and its cytosolic, soluble form inhibit insulin signaling in cultured cell lines[94]-[97]. Adenovirally-induced expression of PTPe in mouse liver activated PEPCK expression, which catalyzes the rate-controlling step of gluconeogenesis[94]. In addition to their leptin-related body weight phenotypes described above, mice lacking PTPe exhibit improved glucose homeostasis both when fed regular chow or high-fat food, and irrespective of their body weight[60]. Increased AKT phosphorylation was demonstrated in liver and muscle of PTPe-deficient male mice[60]. In another study, both the basal and insulin-induced increase in IR and IRS-1 tyrosine phosphorylation were significantly greater in cultured primary skeletal muscle cells from PTPe knockout mice compared with similar cells from wild type mice[95]. Glucose homeostasis phenotypes were somewhat stronger in male mice, implying that negative regulation of insulin signaling by PTPe may have a sex dependence of unknown basis.
SHP-1
SHP-1 is a hematopoietic PTP, but is also expressed in many tissues including the liver and to a lesser extent the muscle. SHP1 is closely related to SHP2; like SHP2, SHP1 contains two N-terminal SH2 domains, a single PTP catalytic domain, and a C terminal sequence that can undergo phosphorylation (Fig. 2). SHP1 can be inhibited by association of its N-SH2 domain with its PTP domain, an association that is released upon cell stimulation[98]. Viable motheaten mice, which express a naturally-occurring, catalytically defective mutant of SHP-1, exhibited improved glucose tolerance and increased insulin sensitivity in the liver and muscle due to enhanced IR signaling to IRS-PI3K-AKT[99]. Increased insulin sensitivity was also observed with adenoviral expression of a catalytically inert mutant of SHP-1, or after small hairpin RNA–mediated SHP-1 silencing[99]. In another more recent study, liver-specific SHP1 knockout mice develop obesity similar to control wild-type littermates, but they exhibit improved glucose tolerance and insulin sensitivity[100]. SHP-1 therefore inhibits IR signaling, most likely by regulating PI3K signaling. Under basal conditions, the p85 regulatory subunit of PI3K keeps the p110 catalytic subunit stabilized and inactivated. This inhibitory activity of p85 is relieved by binding of its N-terminal SH2 domain to Src family kinases, binding that is reversed by the association of SHP1 to maintain the inhibition of PI3K activity[81],[101].
SHP-2
In vitro experiments regarding the function of SHP2 in IR signaling have yielded conflicting results, suggesting both negative and positive regulatory roles[102]-[104]. However, recent animal studies show that overall, SHP-2 negatively regulates insulin action[105],[106]. Mice with a liver-specific SHP-2 deletion exhibited improved insulin sensitivity, increased glucose tolerance, and increased insulin-dependent suppression of hepatic gluconeogenesis compared with controls[105],[106]. Liver-specific SHP-2-deficiency impeded the development of insulin resistance following exposure to a high-fat diet, and reduced hepatic ER stress, steatosis and inflammation compared with control mice[106]. Mice lacking SHP2 in striated muscle exhibited insulin resistance, glucose intolerance and reduced uptake of glucose following insulin stimulation[107]. Deletion of SHP2 from beta cells of the pancreas resulted in reduced glucose-induced insulin secretion and reduced glucose tolerance[108]. In contrast, deletion of SHP2 from white adipose tissue did not affect IR signaling and glucose homeostasis[109]. Inactivation of a floxed allele of SHP2 in the forebrain and hypothalamus using CamIIα−Cre[66] or the pan-neuronal CRE3-Cre[67] resulted in significant insulin resistance, manifest also as increased levels of glucose and insulin in circulation. Deletion of SHP2 from POMC-expressing neurons yielded similar phenotypes[44]. These mice are obese due to disrupted LR signaling; when mice of similar weight and body fat were compared, no differences were found in circulating glucose levels and hepatic glucose production, suggesting that insulin resistance in these mice might be secondary to their increased weight[44].
PTEN
PTEN affects insulin signaling mainly by dephosphorylating PIP3 into PIP2, leading to reduced activation of its downstream AKT, a key effector kinase for the metabolic effects of insulin. Mice lacking one copy of PTEN exhibited enhanced insulin sensitivity and glucose tolerance[110]. Tissue-specific knockout of PTEN in liver, muscle and adipose tissue increases insulin sensitivity and improves glucose tolerance, but was accompanied by increased fat deposits in the liver[111]-[113]. Interestingly, patients with Cowden syndrome, a rare condition characterized by a high risk for number of different cancers caused by germline mutations in PTEN, exhibited enhanced insulin sensitivity together with increased AKT phosphorylation despite being more obese compared to controls[114].
PTP INHIBITION AND THERAPY
The past several decades have witnessed success in designing small molecule inhibitors of PTKs and their introduction into clinical use as therapies for various diseases. Key factors in making this possible include detailed molecular-level understanding of the roles PTKs play in disease-related processes, design of relatively specific inhibitors of PTKs that are suitable for use in vivo in humans, and the massive commitment of academia and the pharmaceutical industry to this endeavor. The roles of PTPs in regulating key physiological processes and their close functional interactions with PTKs suggested that inhibition of PTPs would also be useful in fighting disease. This notion was strengthened significantly by the reports that PTP1B is a negative regulator of both the IR[37],[38] and LR[39],[40] in vivo. Moreover, data derived mainly from animal models, such as that summarized here, demonstrate that targeting several PTPs leads to increased leptin and insulin sensitivity with amelioration of most metabolic abnormalities of the metabolic syndrome including obesity, hyperglycemia, hyperlipidemia and fatty liver. Consequently, researchers in academia and industry have invested efforts in designing various types of PTP inhibitors (e.g.[82],[115],[116]). While many useful inhibitors for PTPs have been found, it appears that inhibiting PTPs for pharmaceutical use is more complex than believed initially (reviewed in[117]). For example, PTP substrates are typically negatively-charged phospho-tyrosines; a typical competitive inhibitor would also be charged, thereby reducing its ability to penetrate into cells and access PTPs. Specificity of inhibitors to particular PTPs has been another challenge. Consequently, efforts are being invested also to design alternative strategies for PTP inhibition in vivo, such as allosteric inhibitors or inhibition by stabilizing inactive forms of the PTP at hand. For example, PTPs are inhibited transiently by reactive oxygen species that are formed near activated receptors[20],[118]-[121]. Haque and colleagues[82] have designed conformation-sensitive, single-chain variable antibody fragments that can be expressed within cells, and which bind only the oxidized, inactive form of PTP1B. Expression of these fragments in cells inhibited PTP1B activity and increased IR signaling[82]. It is appealing to suggest that small molecules that function similarly and bind oxidized, inactive PTP molecules may sequester these molecules and prevent their re-activation by reduction, thus effectively inhibiting PTP activity. A major challenge in the field is then to transform the significant knowledge of the chemistry and physiological roles of PTPs into clinical therapies.
References
- 1.Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–5. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 2.Achike FI, To NH, Wang H, Kwan CY. Obesity, metabolic syndrome, adipocytes and vascular function: A holistic viewpoint. Clin Exp Pharmacol Physiol. 2011;38:1–10. doi: 10.1111/j.1440-1681.2010.05460.x. [DOI] [PubMed] [Google Scholar]
- 3.Gade W, Schmit J, Collins M, Gade J. Beyond obesity: the diagnosis and pathophysiology of metabolic syndrome. Clin Lab Sci. 2010;23:51–61. [PubMed] [Google Scholar]
- 4.Kashyap SR, Defronzo RA. The insulin resistance syndrome: physiological considerations. Diab Vasc Dis Res. 2007;4:13–9. doi: 10.3132/dvdr.2007.001. [DOI] [PubMed] [Google Scholar]
- 5.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–95. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer's disease. J Alzheimers Dis. 2005;7:45–61. doi: 10.3233/jad-2005-7106. [DOI] [PubMed] [Google Scholar]
- 7.Hunter T. Signaling-2000 and beyond. Cell. 2000;100:113–27. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 8.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 10.Tonks NK. Protein tyrosine phosphatases-from housekeeping enzymes to master regulators of signal transduction. Febs J. 2012;280:346–78. doi: 10.1111/febs.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–46. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 12.Tonks NK, Diltz CD, Fischer EH. Characterization of the major protein-tyrosine-phosphatases of human placenta. J Biol Chem. 1988;263:6731–7. [PubMed] [Google Scholar]
- 13.Tonks NK, Diltz CD, Fischer EH. Purification of the major protein-tyrosine-phosphatases of human placenta. J Biol Chem. 1988;263:6722–30. [PubMed] [Google Scholar]
- 14.Andersen JN, Jansen PG, Echwald SM, Mortensen OH, Fukada T, Del Vecchio R, et al. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J. 2004;18:8–30. doi: 10.1096/fj.02-1212rev. [DOI] [PubMed] [Google Scholar]
- 15.Barford D, Das AK, Egloff MP. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu Rev Biophys Biomol Struct. 1998;27:133–64. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 16.Navis AC, van den Eijnden M, Schepens JT, Hooft van Huijsduijnen R, Wesseling P, Hendriks WJ. Protein tyrosine phosphatases in glioma biology. Acta Neuropathol. 2010;119:157–75. doi: 10.1007/s00401-009-0614-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vang T, Miletic AV, Arimura Y, Tautz L, Rickert RC, Mustelin T. Protein tyrosine phosphatases in autoimmunity. Annu Rev Immunol. 2008;26:29–55. doi: 10.1146/annurev.immunol.26.021607.090418. [DOI] [PubMed] [Google Scholar]
- 18.Hendriks WJ, Elson A, Harroch S, Stoker AW. Protein tyrosine phosphatases: functional inferences from mouse models and human diseases. Febs J. 2008;275:816–30. doi: 10.1111/j.1742-4658.2008.06249.x. [DOI] [PubMed] [Google Scholar]
- 19.Pulido R, Hooft van Huijsduijnen R. Protein tyrosine phosphatases: dual-specificity phosphatases in health and disease. Febs J. 2008;275:848–66. doi: 10.1111/j.1742-4658.2008.06250.x. [DOI] [PubMed] [Google Scholar]
- 20.den Hertog J, Ostman A, Bohmer FD. Protein tyrosine phosphatases: regulatory mechanisms. Febs J. 2008;275:831–47. doi: 10.1111/j.1742-4658.2008.06247.x. [DOI] [PubMed] [Google Scholar]
- 21.St-Pierre J, Tremblay ML. Modulation of leptin resistance by protein tyrosine phosphatases. Cell Metab. 2012;15:292–7. doi: 10.1016/j.cmet.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Tsou RC, Bence KK. Central regulation of metabolism by protein tyrosine phosphatases. Front Neurosci. 2013;6:192. doi: 10.3389/fnins.2012.00192. doi:10.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hendriks WJ, Elson A, Harroch S, Pulido R, Stoker A, den Hertog J. Protein tyrosine phosphatases in health and disease. Febs J. 2013;280:708–30. doi: 10.1111/febs.12000. [DOI] [PubMed] [Google Scholar]
- 24.Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–37. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- 25.Zhang F, Chen Y, Heiman M, Dimarchi R. Leptin: structure, function and biology. Vitam Horm. 2005;71:345–72. doi: 10.1016/S0083-6729(05)71012-8. [DOI] [PubMed] [Google Scholar]
- 26.Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–51. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wauman J, Tavernier J. Leptin receptor signaling: pathways to leptin resistance. Front Biosci. 2011;17:2771–93. doi: 10.2741/3885. [DOI] [PubMed] [Google Scholar]
- 28.Banks AS, Davis SM, Bates SH, Myers MG., Jr Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. 2000;275:14563–72. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- 29.Bjorbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, et al. Divergent roles of SHP-2 in ERK activation by leptin receptors. J Biol Chem. 2001;276:4747–55. doi: 10.1074/jbc.M007439200. [DOI] [PubMed] [Google Scholar]
- 30.Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100:197–207. doi: 10.1016/s0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- 31.Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, Liu X, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434:514–20. doi: 10.1038/nature03398. [DOI] [PubMed] [Google Scholar]
- 32.Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111:305–17. doi: 10.1016/s0092-8674(02)01049-8. [DOI] [PubMed] [Google Scholar]
- 33.Frangioni JV, Oda A, Smith M, Salzman EW, Neel BG. Calpain-catalyzed cleavage and subcellular relocation of protein phosphotyrosine phosphatase 1B (PTP-1B) in human platelets. EMBO J. 1993;12:4843–56. doi: 10.1002/j.1460-2075.1993.tb06174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell. 1992;68:545–60. doi: 10.1016/0092-8674(92)90190-n. [DOI] [PubMed] [Google Scholar]
- 35.Bourdeau A, Dube N, Tremblay ML. Cytoplasmic protein tyrosine phosphatases, regulation and function: the roles of PTP1B and TC-PTP. Curr Opin Cell Biol. 2005;17:203–9. doi: 10.1016/j.ceb.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Tiganis T. PTP1B and TCPTP-nonredundant phosphatases in insulin signaling and glucose homeostasis. Febs J. 2013;280:445–58. doi: 10.1111/j.1742-4658.2012.08563.x. [DOI] [PubMed] [Google Scholar]
- 37.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–8. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 38.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–89. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, et al. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell. 2002;2:497–503. doi: 10.1016/s1534-5807(02)00149-1. [DOI] [PubMed] [Google Scholar]
- 40.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, et al. PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–95. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 41.Tsou RC, Zimmer DJ, De Jonghe BC, Bence KK. Deficiency of PTP1B in Leptin Receptor-Expressing Neurons Leads to Decreased Body Weight and Adiposity in Mice. Endocrinology. 2012;153:4227–37. doi: 10.1210/en.2012-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–24. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 43.Xue B, Pulinilkunnil T, Murano I, Bence KK, He H, Minokoshi Y, et al. Neuronal protein tyrosine phosphatase 1B deficiency results in inhibition of hypothalamic AMPK and isoform-specific activation of AMPK in peripheral tissues. Mol Cell Biol. 2009;29:4563–73. doi: 10.1128/MCB.01914-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banno R, Zimmer D, De Jonghe BC, Atienza M, Rak K, Yang W, et al. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest. 2010;120:720–34. doi: 10.1172/JCI39620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Jonghe BC, Hayes MR, Banno R, Skibicka KP, Zimmer DJ, Bowen KA, et al. Deficiency of PTP1B in POMC neurons leads to alterations in energy balance and homeostatic response to cold exposure. Am J Physiol Endocrinol Metab. 2011;300:E1002–11. doi: 10.1152/ajpendo.00639.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Jonghe BC, Hayes MR, Zimmer DJ, Kanoski SE, Grill HJ, Bence K. K. Food intake reductions and increases in energetic responses by hindbrain leptin and melanotan II are enhanced in mice with POMC-specific PTP1B deficiency. Am J Physiol Endocrinol Metab. 2012;303:E644–51. doi: 10.1152/ajpendo.00009.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delibegovic M, Bence KK, Mody N, Hong EG, Ko HJ, Kim JK, et al. Improved glucose homeostasis in mice with muscle-specific deletion of protein-tyrosine phosphatase 1B. Mol Cell Biol. 2007;27:7727–34. doi: 10.1128/MCB.00959-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delibegovic M, Zimmer D, Kauffman C, Rak K, Hong EG, Cho YR, et al. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes. 2009;58:590–9. doi: 10.2337/db08-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Owen C, Czopek A, Agouni A, Grant L, Judson R, Lees EK, et al. Adipocyte-specific protein tyrosine phosphatase 1B deletion increases lipogenesis, adipocyte cell size and is a minor regulator of glucose homeostasis. PLoS One. 2012;7:e32700. doi: 10.1371/journal.pone.0032700. doi: 10.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Owen C, Lees EK, Grant L, Zimmer DJ, Mody N, Bence KK, et al. Inducible liver-specific knockdown of protein tyrosine phosphatase 1B improves glucose and lipid homeostasis in adult mice. Diabetologia. 2013;56:2286–96. doi: 10.1007/s00125-013-2992-z. [DOI] [PubMed] [Google Scholar]
- 51.Morrison CD, White CL, Wang Z, Lee SY, Lawrence DS, Cefalu WT, et al. Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology. 2007;148:433–40. doi: 10.1210/en.2006-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, Morrison CD. HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am J Physiol Endocrinol Metab. 2009;296:E291–9. doi: 10.1152/ajpendo.90513.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stuible M, Doody KM, Tremblay ML. PTP1B and TC-PTP: regulators of transformation and tumorigenesis. Cancer Metastasis Rev. 2008;27:215–30. doi: 10.1007/s10555-008-9115-1. [DOI] [PubMed] [Google Scholar]
- 54.Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 2011;14:684–99. doi: 10.1016/j.cmet.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakamura K, Mizuno Y, Kikuchi K. Molecular cloning of a novel cytoplasmic protein tyrosine phosphatase PTP epsilon. Biochem Biophys Res Commun. 1996;218:726–32. doi: 10.1006/bbrc.1996.0129. [DOI] [PubMed] [Google Scholar]
- 56.Tanuma N, Nakamura K, Kikuchi K. Distinct promoters control transmembrane and cytosolic protein tyrosine phosphatase epsilon expression during macrophage differentiation. Eur J Biochem. 1999;259:46–54. doi: 10.1046/j.1432-1327.1999.00004.x. [DOI] [PubMed] [Google Scholar]
- 57.Elson A, Leder P. Identification of a cytoplasmic, phorbol ester-inducible isoform of protein tyrosine phosphatase epsilon. Proc Natl Acad Sci USA. 1995;92:12235–9. doi: 10.1073/pnas.92.26.12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gil-Henn H, Volohonsky G, Elson A. Regulation of protein-tyrosine phosphatases alpha and epsilon by calpain-mediated proteolytic cleavage. J Biol Chem. 2001;276:31772–9. doi: 10.1074/jbc.M103395200. [DOI] [PubMed] [Google Scholar]
- 59.Gil-Henn H, Volohonsky G, Toledano-Katchalski H, Gandre S, Elson A. Generation of novel cytoplasmic forms of protein tyrosine phosphatase epsilon by proteolytic processing and translational control. Oncogene. 2000;19:4375–84. doi: 10.1038/sj.onc.1203790. [DOI] [PubMed] [Google Scholar]
- 60.Rousso-Noori L, Knobler H, Levy-Apter E, Kuperman Y, Neufeld-Cohen A, Keshet Y, et al. Protein tyrosine phosphatase epsilon affects body weight by downregulating leptin signaling in a phosphorylation-dependent manner. Cell Metab. 2011;13:562–72. doi: 10.1016/j.cmet.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 61.Berman-Golan D, Elson A. Neu-mediated phosphorylation of protein tyrosine phosphatase epsilon is critical for activation of Src in mammary tumor cells. Oncogene. 2007;26:7028–37. doi: 10.1038/sj.onc.1210505. [DOI] [PubMed] [Google Scholar]
- 62.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–99. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 63.Grossmann KS, Rosario M, Birchmeier C, Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res. 2010;106:53–89. doi: 10.1016/S0065-230X(10)06002-1. [DOI] [PubMed] [Google Scholar]
- 64.Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, et al. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. Embo J. 1997;16:2352–64. doi: 10.1093/emboj/16.9.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feng GS. Shp2 as a therapeutic target for leptin resistance and obesity. Expert Opin Ther Targets. 2006;10:135–42. doi: 10.1517/14728222.10.1.135. [DOI] [PubMed] [Google Scholar]
- 66.Zhang EE, Chapeau E, Hagihara K, Feng G. S. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci USA. 2004;101:16064–9. doi: 10.1073/pnas.0405041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krajewska M, Banares S, Zhang EE, Huang X, Scadeng M, Jhala US, et al. Development of diabesity in mice with neuronal deletion of Shp2 tyrosine phosphatase. Am J Pathol. 2008;172:1312–24. doi: 10.2353/ajpath.2008.070594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He Z, Zhang SS, Meng Q, Li S, Zhu H, Raquil MA, et al. Shp2 controls female body weight and energy balance by integrating leptin and estrogen signals. Mol Cell Biol. 2012;32:1867–78. doi: 10.1128/MCB.06712-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 70.Shi Y, Paluch BE, Wang X, Jiang X.PTEN at a glance. J Cell Sci 2012125(Pt 20), 4687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gupta A, Dey CS. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Mol Biol Cell. 2012;23:3882–98. doi: 10.1091/mbc.E12-05-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chetram MA, Hinton CV. PTEN regulation of ERK1/2 signaling in cancer. J Recept Signal Transduct Res. 2012;32:190–5. doi: 10.3109/10799893.2012.695798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ortega-Molina A, Serrano M. PTEN in cancer, metabolism, and aging. Trends Endocrinol Metab. 2012;24:184–9. doi: 10.1016/j.tem.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Plum L, Rother E, Munzberg H, Wunderlich FT, Morgan DA, Hampel B, et al. Enhanced leptin-stimulated Pi3k activation in the CNS promotes white adipose tissue transdifferentiation. Cell Metab. 2007;6:431–45. doi: 10.1016/j.cmet.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 75.Warne JP, Alemi F, Reed AS, Varonin JM, Chan H, Piper ML, et al. Impairment of central leptin-mediated PI3K signaling manifested as hepatic steatosis independent of hyperphagia and obesity. Cell Metab. 2011;14:791–803. doi: 10.1016/j.cmet.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 76.Klockener T, Hess S, Belgardt BF, Paeger L, Verhagen LA, Husch A, et al. High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nat Neurosci. 2011;14:911–8. doi: 10.1038/nn.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Plum L, Ma X, Hampel B, Balthasar N, Coppari R, Munzberg H, et al. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Invest. 2006;116:1886–901. doi: 10.1172/JCI27123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kasuga M, Karlsson FA, Kahn CR. Insulin stimulates the phosphorylation of the 95,000-dalton subunit of its own receptor. Science. 1982;215:185–7. doi: 10.1126/science.7031900. [DOI] [PubMed] [Google Scholar]
- 79.Le Roith D, Zick Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care. 2001;24:588–97. doi: 10.2337/diacare.24.3.588. [DOI] [PubMed] [Google Scholar]
- 80.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 81.Xu E, Schwab M, Marette A. Role of protein tyrosine phosphatases in the modulation of insulin signaling and their implication in the pathogenesis of obesity-linked insulin resistance. Rev Endocr Metab Disord 2013 [Epub] [DOI] [PubMed] [Google Scholar]
- 82.Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK. Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell. 2011;147:185–98. doi: 10.1016/j.cell.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qiu W, Avramoglu RK, Dube N, Chong TM, Naples M, Au C, et al. Hepatic PTP-1B expression regulates the assembly and secretion of apolipoprotein B-containing lipoproteins: evidence from protein tyrosine phosphatase-1B overexpression, knockout, and RNAi studies. Diabetes. 2004;53:3057–66. doi: 10.2337/diabetes.53.12.3057. [DOI] [PubMed] [Google Scholar]
- 84.Haj FG, Zabolotny JM, Kim YB, Kahn BB, Neel BG. Liver-specific protein-tyrosine phosphatase 1B (PTP1B) re-expression alters glucose homeostasis of PTP1B-/-mice. J Biol Chem. 2005;280:15038–46. doi: 10.1074/jbc.M413240200. [DOI] [PubMed] [Google Scholar]
- 85.Agouni A, Mody N, Owen C, Czopek A, Zimmer D, Bentires-Alj M, et al. Liver-specific deletion of protein tyrosine phosphatase (PTP) 1B improves obesity- and pharmacologically induced endoplasmic reticulum stress. Biochem J. 2011;438:369–78. doi: 10.1042/BJ20110373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ono H, Pocai A, Wang Y, Sakoda H, Asano T, Backer JM, et al. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J Clin Invest. 2008;118:2959–68. doi: 10.1172/JCI34277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thareja S, Aggarwal S, Bhardwaj TR, Kumar M. Protein tyrosine phosphatase 1B inhibitors: a molecular level legitimate approach for the management of diabetes mellitus. Med Res Rev. 2012;32:459–517. doi: 10.1002/med.20219. [DOI] [PubMed] [Google Scholar]
- 88.Galic S, Klingler-Hoffmann M, Fodero-Tavoletti MT, Puryer MA, Meng TC, Tonks NK, et al. Regulation of insulin receptor signaling by the protein tyrosine phosphatase TCPTP. Mol Cell Biol. 2003;23:2096–108. doi: 10.1128/MCB.23.6.2096-2108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Galic S, Hauser C, Kahn BB, Haj FG, Neel BG, Tonks NK, et al. Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Mol Cell Biol. 2005;25:819–29. doi: 10.1128/MCB.25.2.819-829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr Biol. 2002;12:446–53. doi: 10.1016/s0960-9822(02)00697-8. [DOI] [PubMed] [Google Scholar]
- 91.Fukushima A, Loh K, Galic S, Fam B, Shields B, Wiede F, et al. T-cell protein tyrosine phosphatase attenuates STAT3 and insulin signaling in the liver to regulate gluconeogenesis. Diabetes. 2010;59:1906–14. doi: 10.2337/db09-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Loh K, Merry TL, Galic S, Wu BJ, Watt MJ, Zhang S, ey al. T cell protein tyrosine phosphatase (TCPTP) deficiency in muscle does not alter insulin signalling and glucose homeostasis in mice. Diabetologia. 2011;55:468–78. doi: 10.1007/s00125-011-2386-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zee T, Settembre C, Levine RL, Karsenty G. T-cell protein tyrosine phosphatase regulates bone resorption and whole-body insulin sensitivity through its expression in osteoblasts. Mol Cell Biol. 2012;32:1080–8. doi: 10.1128/MCB.06279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakagawa Y, Aoki N, Aoyama K, Shimizu H, Shimano H, Yamada N, et al. Receptor-type protein tyrosine phosphatase epsilon (PTP epsilon M) is a negative regulator of insulin signaling in primary hepatocytes and liver. Zoolog Sci. 2005;22:169–75. doi: 10.2108/zsj.22.169. [DOI] [PubMed] [Google Scholar]
- 95.Aga-Mizrachi S, Brutman-Barazani T, Jacob AI, Bak A, Elson A, Sampson SR. Cytosolic protein tyrosine phosphatase-epsilon is a negative regulator of insulin signaling in skeletal muscle. Endocrinology. 2008;149:605–14. doi: 10.1210/en.2007-0908. [DOI] [PubMed] [Google Scholar]
- 96.Andersen JN, Elson A, Lammers R, Romer J, Clausen JT, Moller KB, et al. Comparative study of protein tyrosine phosphatase-epsilon isoforms: membrane localization confers specificity in cellular signalling. Biochem J 2001354(Pt 3), 581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moller NP, Moller KB, Lammers R, Kharitonenkov A, Hoppe E, Wiberg FC, et al. Selective down-regulation of the insulin receptor signal by protein- tyrosine phosphatases alpha and epsilon. J Biol Chem. 1995;270:23126–31. doi: 10.1074/jbc.270.39.23126. [DOI] [PubMed] [Google Scholar]
- 98.Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev. 2009;228:342–59. doi: 10.1111/j.1600-065X.2008.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dubois MJ, Bergeron S, Kim HJ, Dombrowski L, Perreault M, Fournes B, et al. The SHP-1 protein tyrosine phosphatase negatively modulates glucose homeostasis. Nat Med. 2006;12:549–56. doi: 10.1038/nm1397. [DOI] [PubMed] [Google Scholar]
- 100.Xu E, Charbonneau A, Rolland Y, Bellmann K, Pao L, Siminovitch KA, et al. Hepatocyte-specific Ptpn6 deletion protects from obesity-linked hepatic insulin resistance. Diabetes. 2012;61:1949–58. doi: 10.2337/db11-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cuevas BD, Lu Y, Mao M, Zhang J, LaPushin R, Siminovitch K, et al. Tyrosine phosphorylation of p85 relieves its inhibitory activity on phosphatidylinositol 3-kinase. J Biol Chem. 2001;276:27455–61. doi: 10.1074/jbc.M100556200. [DOI] [PubMed] [Google Scholar]
- 102.Myers MG, Jr, Mendez R, Shi P, Pierce JH, Rhoads R, White MF. The COOH-terminal tyrosine phosphorylation sites on IRS-1 bind SHP-2 and negatively regulate insulin signaling. J Biol Chem. 1998;273:26908–14. doi: 10.1074/jbc.273.41.26908. [DOI] [PubMed] [Google Scholar]
- 103.Ouwens DM, van der Zon GC, Maassen JA. Modulation of insulin-stimulated glycogen synthesis by Src Homology Phosphatase 2. Mol Cell Endocrinol. 2001;175:131–40. doi: 10.1016/s0303-7207(01)00389-6. [DOI] [PubMed] [Google Scholar]
- 104.Noguchi T, Matozaki T, Horita K, Fujioka Y, Kasuga M. Role of SH-PTP2, a protein-tyrosine phosphatase with Src homology 2 domains, in insulin-stimulated Ras activation. Mol Cell Biol. 1994;14:6674–82. doi: 10.1128/mcb.14.10.6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Matsuo K, Delibegovic M, Matsuo I, Nagata N, Liu S, Bettaieb A, et al. Altered glucose homeostasis in mice with liver-specific deletion of Src homology phosphatase 2. J Biol Chem. 2010;285:39750–8. doi: 10.1074/jbc.M110.153734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nagata N, Matsuo K, Bettaieb A, Bakke J, Matsuo I, Graham J, et al. Hepatic Src homology phosphatase 2 regulates energy balance in mice. Endocrinology. 2012;153:3158–69. doi: 10.1210/en.2012-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Princen F, Bard E, Sheikh F, Zhang SS, Wang J, Zago WM, et al. Deletion of Shp2 tyrosine phosphatase in muscle leads to dilated cardiomyopathy, insulin resistance, and premature death. Mol Cell Biol. 2009;29:378–88. doi: 10.1128/MCB.01661-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang SS, Hao E, Yu J, Liu W, Wang J, Levine F. et al. Coordinated regulation by Shp2 tyrosine phosphatase of signaling events controlling insulin biosynthesis in pancreatic beta-cells. Proc Natl Acad Sci USA. 2009;106:7531–6. doi: 10.1073/pnas.0811715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bettaieb A, Matsuo K, Matsuo I, Nagata N, Chahed S, Liu S. et al. Adipose-specific deletion of Src homology phosphatase 2 does not significantly alter systemic glucose homeostasis. Metabolism. 2011;60:1193–201. doi: 10.1016/j.metabol.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wong JT, Kim PT, Peacock JW, Yau TY, Mui AL, Chung SW, et al. Pten (phosphatase and tensin homologue gene) haploinsufficiency promotes insulin hypersensitivity. Diabetologia. 2007;50:395–403. doi: 10.1007/s00125-006-0531-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wijesekara N, Konrad D, Eweida M, Jefferies C, Liadis N, Giacca A, et al. Muscle-specific Pten deletion protects against insulin resistance and diabetes. Mol Cell Biol. 2005;25:1135–45. doi: 10.1128/MCB.25.3.1135-1145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci USA. 2004;101:2082–7. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kurlawalla-Martinez C, Stiles B, Wang Y, Devaskar SU, Kahn BB, Wu H. Insulin hypersensitivity and resistance to streptozotocin-induced diabetes in mice lacking PTEN in adipose tissue. Mol Cell Biol. 2005;25:2498–510. doi: 10.1128/MCB.25.6.2498-2510.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pal A, Barber TM, Van de Bunt M, Rudge SA, Zhang Q, Lachlan KL, et al. PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N Engl J Med. 2012;367:1002–11. doi: 10.1056/NEJMoa1113966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Barr AJ. Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development. Future Med Chem. 2010;2:1563–76. doi: 10.4155/fmc.10.241. [DOI] [PubMed] [Google Scholar]
- 116.Scott LM, Lawrence HR, Sebti SM, Lawrence NJ, Wu J. Targeting protein tyrosine phosphatases for anticancer drug discovery. Curr Pharm Des. 2010;16:1843–62. doi: 10.2174/138161210791209027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.He R, Zeng LF, He Y, Zhang S, Zhang ZY. Small molecule tools for functional interrogation of protein tyrosine phosphatases. Febs J. 2013;280:731–50. doi: 10.1111/j.1742-4658.2012.08718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–72. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lou YW, Chen YY, Hsu SF, Chen RK, Lee CL, Khoo KH, et al. Redox regulation of the protein tyrosine phosphatase PTP1B in cancer cells. Febs J. 2008;275:69–88. doi: 10.1111/j.1742-4658.2007.06173.x. [DOI] [PubMed] [Google Scholar]
- 120.Ostman A, Frijhoff J, Sandin A, Bohmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150:345–56. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 121.Tanner JJ, Parsons ZD, Cummings AH, Zhou H, Gates KS. Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxid Redox Signal. 2011;15:77–97. doi: 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]