

Abstract
Aicardi-Goutières syndrome (AGS) is a rare, genetically determined early-onset progressive encephalopathy. To date, mutations in six genes have been identified as etiologic for AGS. Our Japanese nationwide AGS survey identified six AGS-affected individuals without a molecular diagnosis; we performed whole-exome sequencing on three of these individuals. After removal of the common polymorphisms found in SNP databases, we were able to identify IFIH1 heterozygous missense mutations in all three. In vitro functional analysis revealed that IFIH1 mutations increased type I interferon production, and the transcription of interferon-stimulated genes were elevated. IFIH1 encodes MDA5, and mutant MDA5 lacked ligand-specific responsiveness, similarly to the dominant Ifih1 mutation responsible for the SLE mouse model that results in type I interferon overproduction. This study suggests that the IFIH1 mutations are responsible for the AGS phenotype due to an excessive production of type I interferon.
Main Text
Aicardi-Goutières syndrome (AGS [MIM 225750]) is a rare, genetically determined early-onset progressive encephalopathy.1 Individuals affected with AGS typically suffer from progressive microcephaly associated with severe neurological symptoms, such as hypotonia, dystonia, seizures, spastic quadriplegia, and severe developmental delay.2 On brain imaging, AGS is characterized by basal ganglia calcification, white matter abnormalities, and cerebral atrophy.3,4 Cerebrospinal fluid (CSF) analyses show chronic lymphocytosis and elevated levels of IFN-α and neopterin.3–5 AGS-affected individuals are often misdiagnosed as having intrauterine infections, such as TORCH syndrome, because of the similarities of these disorders, particularly the intracranial calcifications.1 In AGS, etiological mutations have been reported in the following six genes: TREX1 (MIM 606609), which encodes a DNA exonuclease; RNASEH2A (MIM 606034), RNASEH2B (MIM 610326), and RNASEH2C (MIM 610330), which together comprise the RNase H2 endonuclease complex; SAMHD1 (MIM 606754), which encodes a deoxynucleotide triphosphohydrolase; and ADAR1 (MIM 146920), which encodes an adenosine deaminase.6–9 Although more than 90% of AGS-affected individuals harbor etiological mutations in one of these six genes, some AGS-affected individuals presenting with the clinical characteristics of AGS still lack a genetic diagnosis, suggesting the existence of additional AGS-associated genes.1
We recently conducted a nationwide survey of AGS in Japan and reported 14 AGS-affected individuals.10 We have since recruited three other Japanese AGS-affected individuals, and among these 17 individuals, we have identified 11 individuals with etiologic mutations; namely, TREX1 mutations in six, SAMHD1 mutations in three, and RNASEH2A and RNASEH2B mutations in one each. Of the remaining six individuals without a molecular diagnosis, trio-based whole-exome sequencing was performed in three whose parents also agreed to participate in further genome-wide analyses (Figure 1A). Genomic DNA from each individual and the parents was enriched for protein-coding sequences, followed by massively parallel sequencing. The extracted nonsynonymous or splice-site variants were filtered to remove those with minor allele frequencies (MAF) > 0.01 in dbSNP137. To detect de novo variants, any variants observed in family members, listed in Human Genetic Variation Database (HGVD), or with MAF > 0.02 in our in-house exome database were removed. To detect autosomal-recessive (AR), compound heterozygous (CH), or X-linked (XL) variants, those with MAF > 0.05 in our in-house database were removed (Figure S1 available online). All samples were collected with the written informed consents by parents, and the study protocol was approved by the ethical committee of Kyoto University Hospital in accordance with the Declaration of Helsinki.
Figure 1.
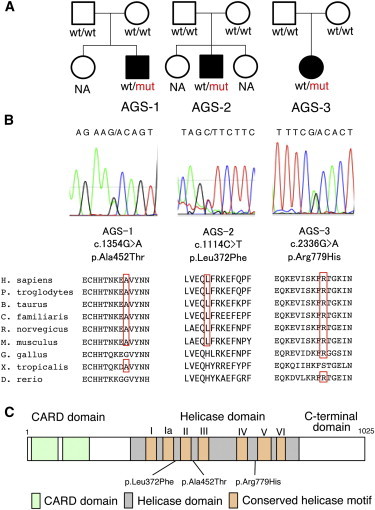
Pedigree Information for the AGS-Affected Individuals and Details of the IFIH1 Mutations Identified
(A) The pedigrees of the three families indicating the AGS probands.
(B) Sanger sequencing chromatograms of the three IFIH1 mutations found in the AGS-affected individuals. The locations of these mutations in the amino acid sequence of the MDA5 protein are shown in alignment with the conserved amino acid sequences from several species. This alignment was obtained via ClustalW2. The amino acids that are conserved with human are circled in red.
(C) The MDA5 protein domain structure with the amino acid substitutions observed in these AGS-affected individuals.
After common polymorphisms were removed, we identified a total of 40, 18, 89, and 22 candidate variants under the de novo, AR, CH, and XL inheritance models, respectively, that were present in at least one of the three individuals (Table S1). Among them, missense mutations were identified in IFIH1 (MIM 606951, RefSeq accession number NM_022168.2), which encodes MDA5 (RefSeq NP_071451.2). These missense mutations are c.1354G>A (p.Ala452Thr) in AGS-1; c.1114C>T (p.Leu372Phe) in AGS-2; and c.2336G>A (p.Arg779His) in AGS-3 (Figure 1B). None of the mutations are found in HGVD, including the 1,208 Japanese samples, or our in-house exome database of 312 Japanese individuals. Multiple-sequence alignment by ClustalW2 revealed that each of the amino acids affected by these mutations are conserved among mammals (Figure 1B). The subsequent amino acid alterations were all suggested to be disease causing in at least one of the four function-prediction programs used (Table 1). None of the other genes identified in the de novo inheritance model, or any of the genes identified in the other three inheritance models, were mutated in all three individuals. The IFIH1 mutations identified were validated by Sanger sequencing. The other coding exons of IFIH1 were also examined by Sanger sequencing, and no other mutations were found.
Table 1.
Functional Predictions of the IFIH1 Variants
| Individuals | Nucleotide Change | Amino Acid Change | SIFT | PolyPhen2 | Mutation Taster | PROVEAN |
|---|---|---|---|---|---|---|
| AGS-1 | c.1354G>A | p.Ala452Thr | tolerated | benign | disease causing | neutral |
| AGS-2 | c.1114C>T | p.Leu372Phe | tolerated | probably damaging | disease causing | neutral |
| AGS-3 | c.2336G>A | p.Arg779His | tolerated | probably damaging | disease causing | deleterious |
The potential functional effects of the IFIH1 variants identified in the AGS-affected individuals were predicted via SIFT, PolyPhen2, Mutation Taster, and PROVEAN.
MDA5 is one of the cytosolic pattern recognition receptors that recognizes double-stranded RNA (dsRNA).11 MDA5 consists of N-terminal tandem CARD domains, a central helicase domain, and a C-terminal domain (Figure 1C). When bound to dsRNA, MDA5 forms a closed, C-shaped ring structure around the dsRNA stem and excludes the tandem CARD as well as creates filamentous oligomer on dsRNA.12 It is hypothesized that the tandem CARD interacts each other and activates MAVS on the mitochondrial outer membrane. Oligomerization of MAVS induces TBK1 activation, IRF3 phosphorylation, and induction of type I interferon transcription, resulting in the activation of a large number of interferon-stimulated genes (ISGs).
The neurological findings of the individuals with these IFIH1 mutations are typical of AGS (Table S2). They were born with appropriate weights for their gestational ages without any signs of intrauterine infection. However, they all demonstrated severe developmental delay in early infancy associated with progressive microcephaly. No arthropathy, hearing loss, or ophthalmological problems were observed. As for extraneural features, all three individuals had at least one of the following autoimmune features: positivity for autoantibodies, hyperimmunoglobulinemia, hypocomplementemia, and thrombocytopenia. Notably, none of the individuals with IFIH1 mutations had chilblain lesions, although all the five individuals with TREX1 mutations and two of the three individuals with SAMHD1 mutations in the Japanese AGS cohort showed chilblain lesions.10 Individuals with SAMHD1 mutations and IFIH1 mutations both show autoimmune features; however, chilblain lesions have been observed only in individuals with SAMHD1 mutations.10
To predict the effects of the identified amino acid substitutions on MDA5, three-dimensional model structures of MDA5 mutants were generated from the crystal structure of human MDA5-dsRNA complex12 (Protein Data Bank [PDB] code 4gl2), using PyMOL (Schroedinger) and MOE (Chemical Computing Group) (Figure S2A). The oligomeric model of MDA5 was generated using the electron microscopy imaging data of MDA5 filament lacking CARD domain13 (Electron Microscopic Data Bank [EMDB] code 5444) (Figure S2B). The three amino acid substitutions in the AGS-affected individuals are all located within the helicase domain (Figures 1C and S2A). Because Ala452 directly contacts the dsRNA ribose O2′ atom, the p.Ala452Thr substitution probably affects the binding affinity to dsRNA due to an atomic repulsion between the side chain of Thr452 and the dsRNA O2′ atom (Figures S2C and S2D). Leu372 is located adjacent to the ATP binding pocket, and the p.Leu372Phe substitution could increase the side chain volume of the binding pocket, affecting its ATP hydrolysis activity (Figures S2E and S2F). In our oligomeric model, Arg779 is located at the interface between the two monomers, which is consistent with the recent report showing that Lys777, close to Arg779, is in close proximity to the adjacent monomer.12 Furthermore, in our model, Arg779 is in close to Asp572 on the surface of the adjacent monomer. We speculate that losing the positive charge due to the p.Arg779His substitution would possibly affect the electrostatic interaction between the MDA5 monomers (Figures S2G and S2H).
To connect the identified IFIH1 mutations with the AGS phenotype, we examined the type I interferon signature in the individuals by performing quantitative RT-PCR (qRT-PCR) of seven ISGs.14 Peripheral blood mononuclear cells (PBMCs) from the three AGS-affected individuals showed upregulation of ISG transcription (Figure 2), confirming the type I interferon signature in the individuals with IFIH1 mutations.
Figure 2.
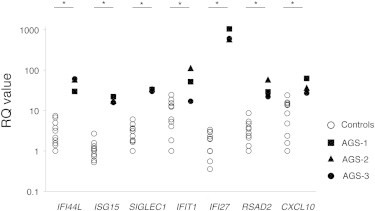
Quantitative RT-PCR of a Panel of Seven ISGs in PBMCs Obtained from the IFIH1-Mutated Individuals and Healthy Control Subject
qRT-PCR was performed as previously described.15 The relative abundance of each transcript was normalized to the expression level of β-actin. Taqman probes used were the same as previous report,14 except for ACTB (MIM 102630). Individual data were shown relative to a single calibrator (control 1). The experiment was performed in triplicate. Statistical significance was determined by Mann-Whitney U test, ∗p < 0.05.
To elucidate the disease-causing capability of the identified IFIH1 mutations, three FLAG-tagged IFIH1 mutant plasmids containing these mutations were constructed via site-directed mutagenesis. These plasmids were transiently expressed on human hepatoma cell line Huh7 and the IFNB1 promoter activity as well as endogenous expression of IFIT1 (MIM 147690) was measured 48 hr after transfection.15 The three mutant plasmids activated the IFNB1 promoter in Huh7 cells more strongly than the wild MDA5 and nearby missense variants reported in dbSNP (Figures 3 and S3). The upregulation of endogenous IFIT1 was also observed in the transfected cells (Figure S4), suggesting that these AGS mutations enhance the intrinsic activation function of MDA5. Recent genome-wide association studies (GWASs) showed association of the IFIH1 with various autoimmune diseases, such as systemic lupus erythematosus (SLE), type I diabetes, psoriasis, and vitiligo.16–19 We examined IFNB1 promoter activity induced by the c.2836G>A (p.Ala946Thr) polymorphism (rs1990760) identified in the GWASs. Although the c.2836G>A polymorphism partially activated the promoter activity, the induced activity was lower than those of the AGS-derived mutants. In addition, the dominantly inherited SLE mouse model in the ENU-treated mouse colony is reported to have the Ifih1 mutation, c.2461G>A (p.Gly821Ser).15 These observations suggest that IFIH1 has strong association with various autoimmune diseases, especially SLE, which also has a type I interferon signature.20 Because alteration of TREX1 has been reported to cause AGS as well as SLE,21 it seems quite plausible for IFIH1 to also be involved in both AGS and SLE. Interestingly, all the individuals identified with IFIH1 mutations had autoantibodies, suggesting the contribution of IFIH1 mutations to autoimmune phenotypes.
Figure 3.
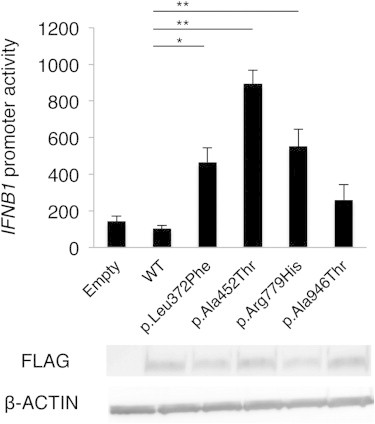
The Effects of the Three MDA5 Variants on IFNB1 Expression
Huh7 cells were transfected with a reporter gene containing IFNB1 promoter (p-55C1B Luc), an empty vector (BOS), and expression vectors for FLAG-tagged human wild-type IFIH1, c.2836G>A polymorphism (p.Ala946Thr) in the GWASs, and the identified IFIH1 mutants. Luciferase activity was measured 48 hr after transfection, and the MDA5 protein accumulation was examined by immunoblotting as previously described.15 FLAG indicates the accumulation of FLAG-tagged MDA5. Each experiment was performed in triplicate and data are mean ± SEM. Shown is a representative of two with consistent results. Statistical significance was determined by Student’s t test. ∗p < 0.05, ∗∗p < 0.01.
To further delineate the functional consequences of the three IFH1 mutations, we measured the ligand-specific Ifnb mRNA induction by stimulating Ifih1null mouse embryonic fibroblasts (MEFs) reconstituted with retrovirus expressing the IFIH1 mutants by an MDA5-specific ligand, encephalomyocarditis virus (EMCV).22 None of the MEF cells expressing the three mutant IFIH1 responded to the EMCV, which suggested that the MDA5 variants lacked the ligand-specific responsiveness. The response of the three AGS mutants against the MDA5-specific EMCV was similar to that of the p.Gly821Ser variant reported in the dominantly inherited SLE mouse model with type I interferon overproduction15 (Figures 4 and S5).
Figure 4.
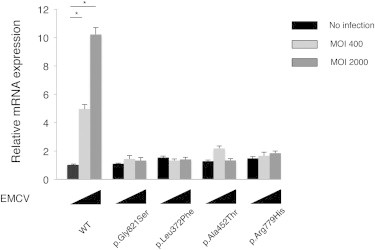
Ifnb mRNA Levels in Ifih1-Deficient MEFs Expressing IFIH1 Mutants
The MEFs were infected with retroviruses encoding mouse wild-type Ifih1, mouse Ifih1 with c.2461G>A (p.Gly821Ser) (RefSeq NM_027835.3) mutation, or the three AGS mutants of human IFIH1. At 48 hr after the retroviral infection, these MEFs were infected with indicated multiplicity of infection (MOI) of EMCV for 6 hr, and Ifnb mRNA levels were measured by qRT-PCR. The relative abundance of each transcript was normalized to the expression level of 18S ribosomal RNA. Data are shown as mean ± SEM of triplicate samples. Shown is a representative of two independent experiments. Statistical significance was determined by Student’s t test, ∗p < 0.001. The expression of the retrovirally transduced FLAG-tagged constructs was confirmed by immunoblotting (Figure S5).
During the revision of this manuscript, Rice et al. identified nine individuals with IFIH1 mutations, including the c.2336G>A mutation we identified, in a spectrum of neuroimmunological features consistently associated with enhanced type I interferon states including AGS.23 Although we agree that the IFIH1 mutations cause constitutive type I interferon activation, Rice et al. show that the mutated MDA5 proteins maintain ligand-induced responsiveness, which was not the case in our study. Because we measured the ligand-specific responsiveness of MDA5 in different experimental conditions, further analysis remains to be performed to reveal the biochemical mechanism of interferon overproduction by the mutated MDA5.
In conclusion, we identified mutations in IFIH1 as a cause of AGS. The individuals with the IFIH1 mutations showed encephalopathy typical of AGS as well as the type I interferon signature with autoimmune phenotypes, but lacked the chilblains. Further analysis remains to elucidate the mechanism of how the IFIH1 mutations identified in AGS cause the type I interferon overproduction.
Acknowledgments
We are very grateful to Y. Takaoka (Kyoto University) and E. Abe (RIKEN Center for Integrative Medical Sciences) for their technical assistance on Sanger sequencing, to E. Hirano (Kyoto University) for her technical assistance on functional analyses of the AGS mutants, to M. Takazawa (Kazusa DNA Research Institute) for his contribution to exome data analysis, and to T. Taylor for his critical reading of the manuscript. This work was supported by the Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology, Japan. This work was supported by Grants-in-aid for Scientific Research from the Japanese Ministry of Health, Labor and Welfare and the Japanese Ministry of Education, Culture, Sports, Science, Technology (MEXT).
Supplemental Data
Web Resources
The URLs for the data presented herein are as follows:
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
ClustalW2, http://www.ebi.ac.uk/Tools/msa/clustalw2/
EMDataBank, http://www.emdatabank.org/index.html
Human Genetic Variation Database (HGVD), http://www.genome.med.kyoto-u.ac.jp/SnpDB/
MutationTaster, http://www.mutationtaster.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
PROVEAN, http://provean.jcvi.org/index.php
RCSB Protein Data Bank, http://www.rcsb.org/pdb/home/home.do
References
- 1.Chahwan C., Chahwan R. Aicardi-Goutieres syndrome: from patients to genes and beyond. Clin. Genet. 2012;81:413–420. doi: 10.1111/j.1399-0004.2011.01825.x. [DOI] [PubMed] [Google Scholar]
- 2.Ramantani G., Kohlhase J., Hertzberg C., Innes A.M., Engel K., Hunger S., Borozdin W., Mah J.K., Ungerath K., Walkenhorst H. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutières syndrome. Arthritis Rheum. 2010;62:1469–1477. doi: 10.1002/art.27367. [DOI] [PubMed] [Google Scholar]
- 3.Orcesi S., La Piana R., Fazzi E. Aicardi-Goutieres syndrome. Br. Med. Bull. 2009;89:183–201. doi: 10.1093/bmb/ldn049. [DOI] [PubMed] [Google Scholar]
- 4.Rice G., Patrick T., Parmar R., Taylor C.F., Aeby A., Aicardi J., Artuch R., Montalto S.A., Bacino C.A., Barroso B. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 2007;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blau N., Bonafé L., Krägeloh-Mann I., Thöny B., Kierat L., Häusler M., Ramaekers V. Cerebrospinal fluid pterins and folates in Aicardi-Goutières syndrome: a new phenotype. Neurology. 2003;61:642–647. doi: 10.1212/01.wnl.0000082726.08631.e7. [DOI] [PubMed] [Google Scholar]
- 6.Crow Y.J., Hayward B.E., Parmar R., Robins P., Leitch A., Ali M., Black D.N., van Bokhoven H., Brunner H.G., Hamel B.C. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 7.Crow Y.J., Leitch A., Hayward B.E., Garner A., Parmar R., Griffith E., Ali M., Semple C., Aicardi J., Babul-Hirji R. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet. 2006;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 8.Rice G.I., Bond J., Asipu A., Brunette R.L., Manfield I.W., Carr I.M., Fuller J.C., Jackson R.M., Lamb T., Briggs T.A. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 2009;41:829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rice G.I., Kasher P.R., Forte G.M., Mannion N.M., Greenwood S.M., Szynkiewicz M., Dickerson J.E., Bhaskar S.S., Zampini M., Briggs T.A. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abe J., Nakamura K., Nishikomori R., Kato M., Mitsuiki N., Izawa K., Awaya T., Kawai T., Yasumi T., Toyoshima I. A nationwide survey of Aicardi-Goutieres syndrome patients identifies a strong association between dominant TREX1 mutations and chilblain lesions: Japanese cohort study. Rheumatology. 2014;53:448–458. doi: 10.1093/rheumatology/ket372. [DOI] [PubMed] [Google Scholar]
- 11.Yoneyama M., Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- 12.Wu B., Peisley A., Richards C., Yao H., Zeng X., Lin C., Chu F., Walz T., Hur S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 13.Berke I.C., Yu X., Modis Y., Egelman E.H. MDA5 assembles into a polar helical filament on dsRNA. Proc. Natl. Acad. Sci. USA. 2012;109:18437–18441. doi: 10.1073/pnas.1212186109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rice G.I., Forte G.M., Szynkiewicz M., Chase D.S., Aeby A., Abdel-Hamid M.S., Ackroyd S., Allcock R., Bailey K.M., Balottin U. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12:1159–1169. doi: 10.1016/S1474-4422(13)70258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funabiki M., Kato H., Miyachi Y., Toki H., Motegi H., Inoue M., Minowa O., Yoshida A., Deguchi K., Sato H. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity. 2014;40:199–212. doi: 10.1016/j.immuni.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 16.Smyth D.J., Cooper J.D., Bailey R., Field S., Burren O., Smink L.J., Guja C., Ionescu-Tirgoviste C., Widmer B., Dunger D.B. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 2006;38:617–619. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- 17.Gateva V., Sandling J.K., Hom G., Taylor K.E., Chung S.A., Sun X., Ortmann W., Kosoy R., Ferreira R.C., Nordmark G. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strange A., Capon F., Spencer C.C., Knight J., Weale M.E., Allen M.H., Barton A., Band G., Bellenguez C., Bergboer J.G., Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin Y., Birlea S.A., Fain P.R., Ferrara T.M., Ben S., Riccardi S.L., Cole J.B., Gowan K., Holland P.J., Bennett D.C. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 2012;44:676–680. doi: 10.1038/ng.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett L., Palucka A.K., Arce E., Cantrell V., Borvak J., Banchereau J., Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee-Kirsch M.A., Gong M., Chowdhury D., Senenko L., Engel K., Lee Y.A., de Silva U., Bailey S.L., Witte T., Vyse T.J. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 22.Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 23.Rice G.I., del Toro Duany Y., Jenkinson E.M., Forte G.M., Anderson B.H., Ariaudo G., Bader-Meunier B., Baildam E.M., Battini R., Beresford M.W. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 2014;46:503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.