

Abstract
Changes in gene regulatory networks are believed to have played an important role in the development of human-specific anatomy and behavior. We identified the human genome regions that show the typical chromatin marks of regulatory regions but cannot be aligned to other mammalian genomes. Most of these regions have become fixed in the human genome. Their regulatory targets are enriched in genes involved in neural processes, CNS development, and diseases such as autism, depression, and schizophrenia. Specific transposable elements contributing to the rewiring of the human regulatory network can be identified by the creation of human-specific regulatory regions. Our results confirm the relevance of regulatory evolution in the emergence of human traits and cognitive abilities and the importance of newly acquired genomic elements for such evolution.
Introduction
Empirical evidence and theoretical arguments suggest that the rewiring of gene regulatory networks plays an important role in the evolution of metazoan anatomy.1 The set of targets of a trans-acting regulatory element can evolve by modifying the cis-regulatory regions (RRs) to which it binds while leaving the trans element unchanged.
Such arguments are supported by a large body of experimental evidence demonstrating, in specific cases, how the evolution of anatomical traits is triggered by the addition or subtraction of targets of a trans-acting regulatory element.2–7 The availability of the complete genome sequence of many organisms has recently allowed the investigation of these issues at a genome-wide scale.
Some of these studies specifically concerned regulatory evolution in the human lineage.8–12 Given a RR in the human genome, these studies variously relied on sequence alignments to identify the orthologous region in other mammals and then proceeded to analyze patterns of divergence and/or variation in the sequence,9,10 the profile of binding affinities for transcription factors (TFs),8,11 or chromatin states.12
On the other hand, genomic regions that have appeared de novo in the human genome, for example, through the insertion of transposable elements (TEs), or that have diverged so extensively since the origin of humans to become unrecognizable by alignment algorithms have not been explored in these studies, even though in principle they could have important regulatory roles. For example, the evolution of CTCF (MIM 604167) binding in mammals was recently shown13 to be largely driven by TEs.
We thus set out to investigate those human genome regions that, on the one hand, show evidence of an active regulatory role and, on the other, are not conserved in other mammals. We called these regions human-specific RRs (HSRRs), and we investigated their variation in human populations, the evolutionary mechanisms at their origin, and the TFs that bind them. Moreover, we analyzed the functional characterization of their putative gene targets and their involvement in genetic diseases.
Material and Methods
Identification of HSRRs
We defined RRs as the human genome regions (obtained from the UCSC Genome Browser, release hg19) assigned by Ernst et al.14 to the following classes: (1) active promoters, (2) weak promoters, (3) poised promoters, (4) strong enhancers, (5) strong enhancers, (6) weak enhancers, (7) weak enhancers, and (8) insulators. We grouped these classes into promoters (classes 1–3), strong enhancers (classes 4 and 5), weak enhancers (classes 6 and 7), and insulators (class 8). The cell lines analyzed by Ernst et al.14 are H1 embryonic stem cells (ESCs), erythrocytic leukemia (K562) cells, B-lymphoblastoid (GM12878) cells, hepatocellular carcinoma (HepG2) cells, human umbilical vein endothelial cells (HUVECs), skeletal muscle myoblasts (HSMMs), normal human lung fibroblasts (NHLFs), normal epidermal keratinocytes (NHEKs), and mammary epithelial cells (HMECs). We generated a meta-cell line, ALL, by merging the RRs of the same type from the cell lines analyzed.
A RR was considered conserved (CRR) if any portion of the region could be aligned to the genome of one or more of the following species (the UCSC Genome Browser version is in parentheses): A. melanoleuca (ailMel1), B. taurus (bosTau4), C. familiaris (canFam2), C. jacchus (calJac3), C. porcellus (cavPor3), E. caballus (equCab2), G. gorilla (gorGor3), M. mulatta (rheMac2), M. musculus (mm9), N. leucogenys (nomLeu1), P. pygmaeus abelii (ponAbe2), P. troglodites (panTro3), and R. rnorvegicus (rn4). All other RRs were considered HSRRs. We used the precomputed net alignments downloaded from UCSC Genome Browser to compare the human genome (hg19) with those reported above. Adjacent RRs belonging to the same RR class, cell line, or human-specificity status were merged.
The same pipeline was applied to data on DNase hypersensitive sites (DHSs): we selected all DHS peaks collected in the ENCODE Project and whose karyotype was flagged as “normal.” For DHS data, we defined a single RR class (“open”). To these we added DHS data from human fetal brain obtained by the NIH Roadmap Epigenomics Mapping Consortium.15 These were downloaded from the Gene Expression Omnibus (samples GSM595913, GSM595920, GSM595922, GSM595923, GSM595926, and GSM595928) as .bam files, on which peaks were detected with MACS16 with default parameters.
Definition of a Neutral Control
A putatively neutral subset of the genome was defined by the removal of (1) regions considered open according to Ernst et al.14 (i.e., classes 1–11) in any ENCODE cell line, (2) DHSs from ENCODE, and (3) sequence gaps derived from the UCSC Genome Browser. The neutral control was composed of regions belonging to this neutral genome; for each RR, we included in the control a region of the same length, completely included in the neutral genome, as close as possible to the RR. The neutral control regions were divided into HSRRs and CRRs and analyzed in the same way as the RRs.
Analysis of Intraspecies Variation of HSRRs
We used variation data inferred by exome and full genome sequencing of 1,092 individuals from the 1000 Genomes Project.17 To maximize the specificity, the 1000 Genomes Project applied a strict procedure to define regions of structural variants (SVs). Given that we were mostly interested in high sensitivity to ensure that the regions we studied could be considered fixed in the human genome, we also considered low-quality, nongenotyped SVs absent in the integrated variant call format. Variation was classified into two classes: SNPs, including indels and small polymorphisms, and SVs, including long deletions or insertions (including those due to mobile elements and tandem duplications). Each RR identified by Ernst et al.14 was classified as “fixed” if it did not overlap a SV or “variant” otherwise. Adjacent RRs belonging to the same RR class, cell line, human-specificity status, or SV status were merged.
To compare SNP density, heterozygosity, and Tajima’s D of fixed HSRRs (FHSRRs) to their conserved counterparts while controlling for potential confounders,18–20 we used a linear model with SNP density, heterozygosity, or Tajima’s D, respectively, as the dependent variable. The independent variables were GC content, CpG content, CpG island overlap, accessibility to sequencing, and human-specificity status. To build this model, we broke each region into 200 bp fragments to avoid overweighing smaller regions. Accessibility was defined as the overlap with “strict” regions from the 1000 Genomes “Ph1 Accsbl” track taken from the UCSC Genome Browser. The sign of the fitted coefficient of the human-specificity status then revealed whether (given all the same confounding factors) SNP density or heterozygosity was higher (positive sign) or lower (negative) than in the CRRs. We evaluated the dispersion of the coefficients with a resampling procedure: for each resampling step, we picked a random subset of CRRs as large as the set of HSRRs, and we fit a linear model to the data set thus obtained. We repeated the procedure 1,000 times and represented the distribution of the 1,000 values of the coefficient as a box plot in Figures 2B–2D.
Figure 2.
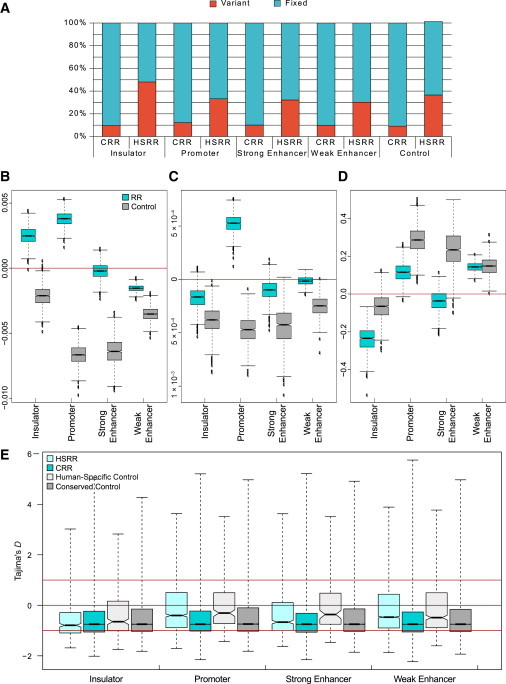
Variation of HSSRs
(A) SVs identified in the 1000 Genomes Project were more prevalent in each class of HSRRs than in their conserved counterparts.
(B–D) Comparison of SNP density (B), heterozygosity, and (C) Tajima’s D between fixed HSRRs and their conserved counterparts. The box plots show the coefficient of the human-specificity status in a linear model including CG content, CpG content, overlap with annotated CpG islands, and DNA accessibility as independent variables. A positive or negative coefficient implies that when all other variables were the same, the independent variable (SNP density or heterozygosity) was higher or lower, respectively, in HSRRs than in CRRs. For each class of RR, we also show the corresponding neutral control. The distribution of the coefficients in the box plot was obtained by resampling. All coefficients are significantly different from 0 (p < 0.05).
(E) Distribution of Tajima’s D. For each class of RR, we show CRRs and HSRRs and the corresponding neutral controls.
Functional Analysis with GREAT
We used the “createRegulatoryDomains” program from the Genomic Regions Enrichment of Annotations Tool (GREAT)21 to associate a regulatory domain with each Ensembl protein-coding gene (with the default “basalPlusExtension” rule). We then associated each RR with a gene if the RR overlapped the corresponding regulatory domain. In this way, we obtained for each cell line and RR class a list of genes associated with FHSRRs; we tested this list for functional enrichment against a universe defined as all genes associated with a fixed RR (HSRR or CRR) of the same type active in the same cells. We assessed functional enrichment by using the GOstats22 and DOSE Bioconductor packages with default parameters for Gene Ontology (GO) and Disease Ontology, respectively. We performed our own enrichment analysis instead of using GREAT to be able to perform a gene-based (rather than region-based) enrichment analysis while specifying a universe. In Figure 3, we used the GOSemSim package23 to remove redundant GO terms, including all terms with a semantic similarity24 higher than 0.7 with a term with a more significant p value. We obtained NPC-specific genes from Table S1 in Xie et al.25 and performed enrichment analysis with the gene-region associations generated by GREAT.
Figure 3.
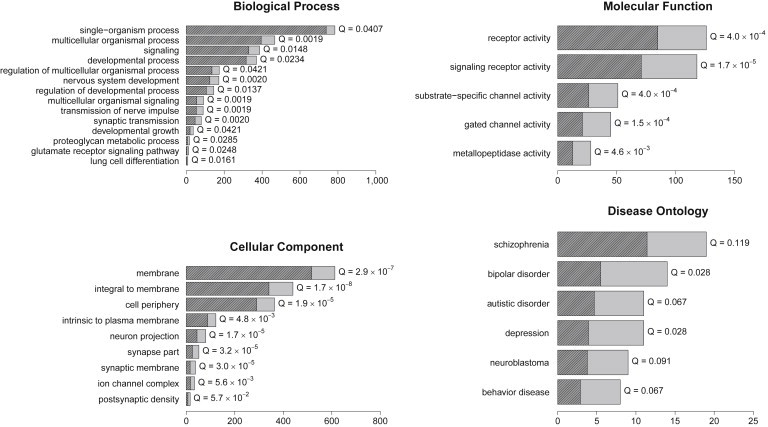
Functional Enrichment of FHSRR Targets
The GO and Disease Ontology terms enriched in targets of human-specific weak enhancers active in ESCs. The length of the bar represents the number of targets, and the shaded part is the number of targets expected by chance. The Q value was evaluated with the Benjamini-Hochberg procedure. We show all terms with Q < 0.05 (GO) or Q < 0.20 (Disease Ontology). Redundant GO terms were removed as described in the Material and Methods.
Overlap with ASD-Related CNVs
We obtained data about copy-number variations (CNVs) in autism spectrum disorders (ASDs) from two different papers: Pinto et al.26 (Table S8: ASD_cases_European) and Sanders et al.27 (Table S8). We converted the UCSC hg18 genomic CNV coordinates used in these papers to UCSC hg19 coordinates. We performed independent Fisher’s exact tests to evaluate the enrichments of (1) CNVs overlapping FHSRRs with respect to all fixed RRs in each individual data set and (2) CNVs overlapping FHSRRs with respect to all fixed RRs in both data sets. For this analysis, we merged RRs of different cell lines into a single list.
Analysis of TF Binding
We used chromatin-immunoprecipitation-sequencing (ChIP-seq) peak data from the ENCODE/HAIB, ENCODE/UChicago, and ENCODE/Sydh tracks downloaded from the UCSC Genome Browser. A TF T was considered bound to RR R if a peak of T overlapped any portion of R. For each TF and cell line, we used a Fisher’s exact test to evaluate the enrichment of TF binding in FHSRRs in comparison to the enrichment in all fixed RRs.
The Role of TEs
We downloaded coordinates for TEs from the UCSC RepeatMasker track (hg19). For each R, we associated the transposon T if R and T overlapped and there was only one overlapping transposon. If there were more overlapping transposons, we chose the one with the longest overlap. We used a Fisher’s exact test to evaluate enrichments of each repeat type (e.g., MIR3), family (e.g., MIR), and class (e.g., SINE) in FHSRRs with respect to enrichments in all fixed RRs overlapping any TE.
Software
All analyses were performed with software available from Bioconductor,28 BEDTools,29 and GREAT21 (see Web Resources).
Results
HSRRs
Our starting point was classifying HSRRs on the basis of chromatin marks in nine cell lines obtained by Ernst et al.14 Independently for each cell line, the authors divided the genome into 15 classes, eight of which were of regulatory significance. We merged some of their classes to obtain four classes of RRs: insulators, promoters, strong enhancers, and weak enhancers.
We defined a HSRR as one that does not appear in genome-wide alignments with any of 13 mammalian genomes (listed in the Material and Methods), including those of six primates and four apes. We used the Net alignments provided by the UCSC Genome Browser. Figure 1 shows the genomic portion occupied by HSRRs that we found in each of the nine investigated cell lines in both absolute terms and as a fraction of the regulatory genome.
Figure 1.
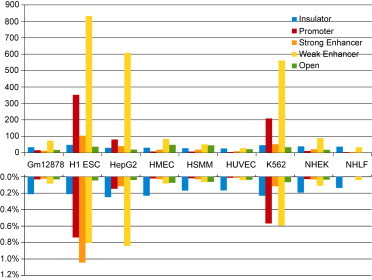
HSRRs
Upper histogram: the size of HSRRs in thousands of base pairs for the various cell types and RR classes. The first four classes were obtained from the data of Ernst et al.,14 whereas the “open” class refers to DHS data.
Lower histogram: the human-specific fraction of each class of regulatory DNA. H1 ESCs, HepG2 cells, and K562 cells showed a higher number of HSRRs in both absolute and relative terms.
Intraspecies Variation of HSRRs
To begin studying the functional relevance of the HSRRs, we investigated their patterns of variation within human populations by using data from the 1000 Genome Project.17 We classified variation in two large classes: (1) SNPs, including SNPs and small indels, and (2) SVs, including long deletions or insertions, insertions due to mobile elements, and tandem duplications.
SVs appeared to be more common in HSRRs than in other RRs, as might be expected given the young evolutionary age of these regions (see Figure 2A). However, for all RR classes and all cell lines, most HSRRs did not overlap any known SV. In the following sections, we will focus on these SV-free HSRRs, which we refer to as FHSSRs, given that these are the ones most likely to have a functional role. The number of FHSSRs found in the nine cell lines is shown in Table 1. Table S1, available online, contains the list of all FHSRRs for each cell line. As a negative set, we defined a putatively neutral control made of regions that are not regulatory in any cell line and are located in the vicinity of a FHSSR.
Table 1.
Numbers of FHSRRs
| Cell Line | Origin |
Number of FHSRRs |
|||
|---|---|---|---|---|---|
| Insulators | Promoters | Strong Enhancers | Weak Enhancers | ||
| Gm12878 | lymphoblastoid cells | 66 | 19 | 11 | 114 |
| H1 ESC | embryonic stem cells | 78 | 535 | 173 | 1,146 |
| HepG2 | liver carcinoma cells | 49 | 135 | 78 | 1,103 |
| HMEC | mammary epithelial cells | 49 | 8 | 23 | 146 |
| HSMM | skeletal muscle myoblasts | 51 | 13 | 17 | 127 |
| HUVEC | human umbilical vein endothelial cells | 47 | 7 | 12 | 73 |
| K562 | leukemia cells | 78 | 413 | 62 | 1,072 |
| NHEK | epidermal keratinocytes | 78 | 14 | 28 | 179 |
| NHLF | lung fibroblasts | 88 | 2 | 7 | 80 |
The nine cell lines studied by Ernst et al.14 are shown with the corresponding numbers of FHSRRs in the four RR classes.
We compared the rate of intraspecies variation in FHSRRs to that in fixed RRs that are not human specific by looking at their SNP density and heterozygosity (Figures 2B and 2C). Using a linear model to control for potential confounding factors18–20 (such as GC content, CpG content, and DNA accessibility), we found that human-specific promoters had higher SNP density and heterozygosity than their conserved counterparts, suggesting that the selective pressure on human-specific promoters is weaker than that on conserved ones. On the other hand, both strong and weak enhancers showed lower SNP density and heterozygosity than did conserved enhancers, instead suggesting stronger negative selection. Finally, human-specific insulators showed higher SNP density but lower heterozygosity than did conserved ones. Note that Ward et al.18 reported higher SNP density and heterozygosity in nonconserved RRs than in CRRs. However, our FHSRRs are a different (and much smaller) set of regions than the nonconserved ones in Ward et al.,18 and their nonconserved regions are actually included in our conserved (i.e., nonhuman specific) regions (at least conceptually, given that the genome-wide alignments we used do not coincide with those used by Ward et al.).
It is difficult to interpret these results in terms of selective pressure alone; indeed, the putatively neutral human-specific controls also showed lower SNP density and heterozygosity than did the conserved ones, possibly because the factors included in our linear model did not completely capture a difference in mutation rate between HSRRs and CRRs. Therefore, we analyzed a quantity that has a direct interpretation in terms of selective pressure, namely Tajima’s D.30 The expected value of D is 0 for the null hypothesis of neutrality; negative values of D indicate purifying selection or population expansion, whereas positive values indicate balancing selection or a decrease in population size.
Overall, the D values were significantly less than 0 for all classes of RRs, particularly HSRRs (Figure 2E; p values from 1.64 × 10−10 for promoters to 4.52 × 10−44 for weak enhancers, Wilcoxon signed-rank test). This implies that HSRRs are under selective pressure. Moreover, for all classes of FHSRRs, the mean D values were more negative than those of the respective controls, even though this reached statistical significance only for insulators (p = 4.56 × 10−4, Mann-Whitney U test) and strong enhancers (p = 2.12 × 10−4). When comparing the D values of HSRRs and CRRs (Figure 2D) while controlling for the same confounding factors considered for SNP density and heterozygosity, we saw that for insulators and strong enhancers, HSRRs had a smaller D, whereas the opposite was true for promoters and weak enhancers.
Regulatory Targets
Overall, the results of the previous section neither exclude nor prove that FHSRRs are functional. We thus investigated their functional relevance by examining their target genes, particularly their functional annotation. We reasoned that overrepresentation of some functional categories in the regulatory targets of FHSRRs would strongly argue for their biological relevance.
We used GREAT21 to associate FHSRRs to putative gene targets. We computed all enrichments by comparing the targets of fixed HSRRs active in a given cell line to all the targets of the same type of RRs active in the same cell line. Weak enhancers active in human ESCs showed the most significant enrichments: the significantly enriched terms are shown in Figure 3, where redundant terms were removed as described in the Material and Methods. Overall, they showed a strong enrichment of genes involved in neural processes and development. Several of these enrichments were also found in the human-specific promoters active in ESCs, in which rather specific terms, such as “serotonin receptor activity,” were also enriched. The functional enrichment of human-specific promoters was especially significant given that this is the class of FHSRRs that appear to be under the weakest selective pressure from the analysis of intraspecies variation (Figure 2). Complete enrichment results are included in Table S2.
Also, the analysis of enrichment in Disease Ontology31 terms gave the most significant results for FHSRRs active in ESCs. Results for weak enhancers are shown in Figure 3, and they confirm the strong neural characterization of the target genes. These terms were also enriched in human-specific promoters active in ESCs, suggesting that many human-specific promoters are functional notwithstanding their relatively high variability, shown in Figure 2. These results prompted us to investigate whether FHSRRs are involved in the CNVs known to be associated with personality diseases, specifically autism.26,27 However, FHSRRs did not show a stronger enrichment in disease-related CNVs than in nonregulatory HSRRs (Figure S1). Other diseases not directly linked to the CNS and enriched in HSRR targets include obesity (enriched in promoters, 13 genes), pancreatitis (promoters, six genes), and abortion (promoters, six genes). Other cell lines gave, in general, a much smaller number of enrichments, often of difficult interpretation. Complete results are shown in Table S3.
FHSRRs are strongly overrepresented in the X chromosome (see Figure S2), possibly because of its enrichment of repeated elements.32 However, the GO and Disease Ontology enrichments we found above were mostly unchanged when we restricted the analysis to autosomes (Table S4 and S5).
These functional enrichments provide strong evidence of the functional relevance of FHSRRs and suggest that they play a role in the very early development of the CNS. We thus hypothesized that the target genes of FHSRRs could be expressed in neural progenitor cells (NPCs). We obtained a list of genes specifically expressed in NPCs from a recent RNA-sequencing experiment,25 and we compared this list to the putative targets of FHSRRs. We found that there was indeed a strong overrepresentation of NPC-specific genes among the targets of human-specific weak enhancers and promoters active in ESCs (weak enhancers: 94 genes, p = 2.5 × 10−7; promoters: 52 genes, p = 1.4 × 10−4; Fisher’s exact test). The NPC-specific genes that are targets of FHSRRs are shown in Table S7. However, human-specific promoters active in K562 cells were also enriched in NPC-specific genes (45 genes, p = 6.5 × 10−5).
Figure 4 shows two examples of FHSRRs near SRGAP1 (MIM 606523) and HTR2C (MIM 312861). SRGAP1, whose expression in the cerebellum is different in humans than in other primates,33 is involved in the early development of the human fetal neocortex.34 HTR2C encodes a serotonin receptor that has been involved in several mental disorders, including schizophrenia, bipolar disorder, and major depression.35 Notably, HTR2C was recently suggested to show human-specific patterns of X-chromosome-inactivation status.36
Figure 4.
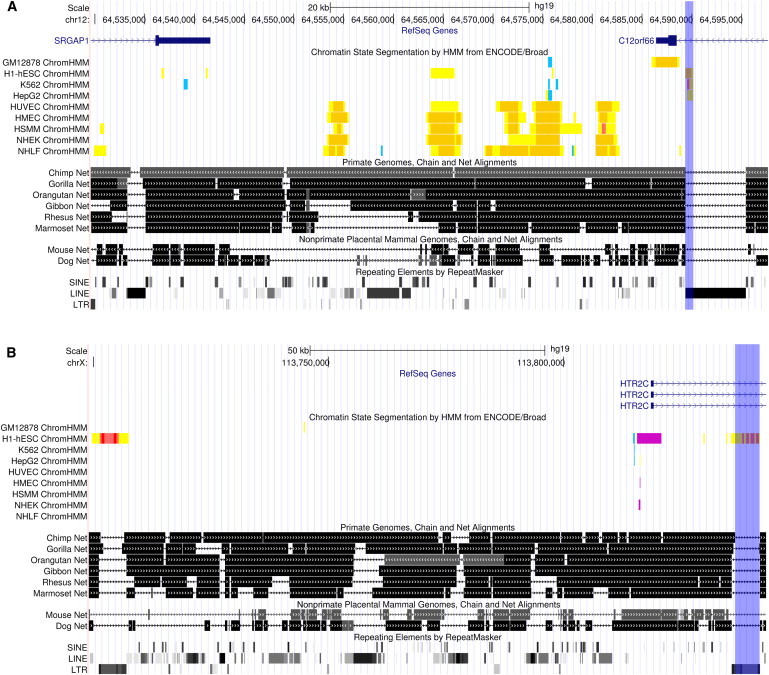
Examples of FHSSRs
Two FHSRRs (indicated by the blue shade) and the genomic landscape around them (which includes putative targets) as depicted by the UCSC Genome Browser. The tracks represent (top to bottom) base position, RefSeq genes, Broad ChromHMM (RRs), Primate Chain/Net alignment, Placental Chain/Net alignment, and RepeatMasker. Color coding for Broad ChromHMM is as follows: yellow, weak enhancer; orange, strong enhancer; red, promoter; light red, weak promoter; purple, poised promoter; and blue, insulator.
(A) SRGAP1, a gene whose expression in humans is shifted from that in other primates.
(B) HTR2C, encoding a serotonin receptor involved in several mental illnesses.
These results suggest that the appearance of new sequence with regulatory potential in the human genome contributed to many of the phenotypic differences that most prominently separate us humans from our closest relatives, particularly those differences concerning the development and physiology of the CNS.
Using DHSs to Define RRs
A major limit of the previous analysis is that it was based on a limited number of cell lines for which chromatin data are available. To widen the scope of our analysis, we turned to DHSs, which are available for a wide variety of cell lines and primary tissues, as an alternative definition of active regulatory sequences. We considered all DHS data available in ENCODE and whose karyotype is flagged as “normal.” The list of DHS data used is provided in Table S7.
Because DHS data are also available for the cell lines used in the previous analysis, we first asked what fraction of the various classes of RRs are represented in DHS data. In general, DHSs tend to cover a smaller fraction of the genome than do chromatin-based RRs. Moreover, as expected, promoters are overrepresented in DHSs, whereas enhancers are underrepresented (Figure S3). This leads to a decrease in statistical power to detect the functional signals discussed above.
However, the functional characterization of FHSRRs expressed in ESCs is confirmed by DHS data: the most significant GO Biological Process term is indeed “neurogenesis” (19 genes). Cells and tissues other than ESCs show relatively few enrichments. A potentially interesting result is the overrepresentation of “transferase activity, transferring hexosyl groups” in hepatocytes (six genes: ALG10 [MIM 603313], ALG10B, B4GALT7 [MIM 603313], FUT3 [MIM 111100], FUT5 [MIM 136835], and FUT6 [MIM 136836]).
Given that FHSRRs seem to be involved specifically in the development of the CNS, we obtained DHS data from six fetal brain samples from the NIH Roadmap Epigenomics Mapping Consortium15,37 and performed the same analysis. The enrichment of “axon extension” was independently found in three different samples, even though it was based (in all three samples) only on three genes (FOXD4 [MIM 601092], FOXD4L1 [MIM 611084], and MAP1B [MIM 157129]). In contrast, DHS data from adult brain did not lead to any GREAT enrichment, again suggesting that HSRRs play their most important role in the development, rather than in the adult physiology, of the CNS.
TF Binding
We took advantage of the large collection of ChIP-seq experiments generated by the ENCODE Project38 to investigate whether specific TFs bind the FHSRRs. When joining all cell lines together, we found significant enrichment (false-discovery rate [FDR] < 0.05) of six TFs. The most significant enrichment was found for NR2F2, which binds 617 out of 2,495 human-specific weak enhancers and 83 out of 359 human-specific strong enhancers. This TF, also known as COUP-TFII, is particularly involved in the migration of neurons during brain development.39,40 The liver-specific TF FOXA1 was also enriched in human-specific weak enhancers (376 bound) and promoters (235 bound out of 815 human-specific promoters). Human-specific insulators were enriched with binding sites for ZBTB33 (a transcriptional repressor, also known as Kaiso, that interacts with CTCF and negatively regulates the insulator activity of the latter41) and the homeobox TF SIX5. Finally, weak enhancers were enriched in TAF1 binding, suggesting significant transcriptional activity, which might be related to the fact that many of these regions originate as retrotransposons (see below). Complete results are available in Table S9.
When analyzing individual cell lines, we considered only the peaks derived from ChIP-seq experiments performed in the same cell line. Complete results are available in Tables S10, S11, S12, S13, and S14. For ESCs, the strongest enrichment was for Pol2 and TAF1, suggesting that the transcriptional activity of FHSRRs is especially notable in these cells.
The Role of TEs
TEs are important sources of genomic evolution.42 We sought to determine which TEs play a role in the appearance of FHSRRs. Specifically, we considered the RRs overlapping each TE class, and we looked at which of such classes are significantly enriched in FHSRRs. Note that an overall enrichment of TEs in FHSRRs is expected because of how FHSRRs are defined; here, we restricted the analysis to RRs overlapping a TE to determine which classes of TEs are associated with FHSRRs.
Overall, considering all cell lines and RR classes, we found 199 significant overlaps at a 5% FDR and 25 different classes of repeated elements. The significant results for ESCs are shown in Table 2, and complete results are available in Table S14. Of particular interest is the appearance of HERVH and LTR7, given that these elements were recently shown25 to play an important role in the regulation of long noncoding RNAs in human ESCs. Our results suggest that this phenomenon might be largely human specific.
Table 2.
TEs Overlapping FHSRRs
| Repeat Class |
Number of Overlapping FHSRRs (FDR) |
|||
|---|---|---|---|---|
| Promoters | Strong Enhancers | Weak Enhancers | Insulators | |
| LINE | 351 (3.93 × 10−60) | 140 (2.47 × 10−41) | 643 (6.29 × 10−66) | 29 (>0.05) |
| LTR | 183 (3.87 × 10−46) | 31 (>0.05) | 479 (2.22 × 10−58) | 39 (1.03 × 10−5) |
| ERV1 | 179 (3.79 × 10−76) | 15 (>0.05) | 381 (7.28 × 10−122) | 11 (>0.05) |
| ERVK | 3 (>0.05) | 15 (1.20 × 10−4) | 95 (4.25 × 10−63) | 25 (3.33 × 10−27) |
| L1 | 351 (5.70 × 10−167) | 140 (3.80 × 10−82) | 643 (1.17 × 10−235) | 29 (2.70 × 10−3) |
| HERVH-int | 164 (8.46 × 10−156) | 12 (3.16 × 10−6) | 343 (<1.0 × 10−300) | 2 (>0.05) |
| HERVK-int | 1 (>0.05) | 0 (>0.05) | 11 (4.20 × 10−11) | 20 (5.11 × 10−41) |
| L1HS | 74 (1.13 × 10−82) | 38 (2.94 × 10−47) | 131 (5.30 × 10−176) | 1 (>0.05) |
| L1PA2 | 243 (2.91 × 10−221) | 92 (4.60 × 10−101) | 402 (<1.0 × 10−300) | 8 (1.70 × 10−11) |
| L1PA3 | 25 (>0.05) | 6 (>0.05) | 74 (6.56 × 10−32) | 6 (7.01 × 10−4) |
| LTR5_Hs | 2 (>0.05) | 15 (5.22 × 10−9) | 77 (1.39 × 10−75) | 0 (>0.05) |
| LTR7 | 13 (3.50 × 10−3) | 1 (>0.05) | 16 (1.31 × 10−2) | 0 (>0.05) |
Featured are TE classes showing significant overlap with FHSRRs active in ESCs. For each TE class and RR class, we report the number of overlapping FHSRRs. In parentheses is the Benjamini-Hochberg FDR from the Fisher’s exact test comparing the FHSRRs overlapping the specific TEs to all TE-overlapping RRs of the same class.
Discussion
By integrating the genomic sequences of a large number of mammals and chromatin-state data on human cell lines, we were able to identify those human genome portions that were acquired after the split from our closest relatives and that perform a regulatory function in our genome. Many of these regions originated from mobile DNA elements, an extremely efficient vehicle for the rewiring of regulatory networks. Most of these regions have been fixed in the human genome, and their functional relevance is suggested by the strong functional characterization of their putative targets.
As originally suggested by King and Wilson,43 the divergence in coding sequence between human and chimpanzee seems too low to account for the extensive differences in cognitive abilities, behavior, and metabolism between the two species. It is therefore natural to postulate that a relevant part of these differences is explained by differences in gene regulation rather than in gene products. HSRRs have most likely played a role in generating such differences, as shown by the enrichment of genes involved in neural development and psychiatric diseases, such as bipolar disorder, schizophrenia, and autism.
Such strong functional characterization of HSRRs is to be contrasted with their rather weak selective pressure at the sequence level: this suggests a model in which regulatory rewiring is more effectively performed by the relocation of whole regulatory sequences to new genomic regions and target genes rather than by a succession of point mutations on existing sequences. This mechanism was recently shown to be largely responsible for the evolution of CTCF binding in mammals.13
Our approach has two main technical limitations. On the one hand, the genomes of nonhuman mammals, particularly primates, are at a much lower stage of completeness than the human genome. Therefore, lack of alignment between a human sequence and the chimp genome might be due to a gap in the sequence of the latter. The fact that we used four nonhuman apes in our comparison should mitigate the consequences of these technical problems, because it is quite unlikely that sequencing gaps happen in the same place in several genomes.
However, the possibility remains that some of the regions that we classify as human specific are in fact shared by humans and chimps. While this manuscript was being prepared, the genome-wide alignments of the human genome to a newer version of the chimp genome (panTro4) were published in the UCSC Genome Browser. We reasoned that if a significant fraction of our FHSRRs were due to the preliminary status of the chimp genome, some should disappear when these newer alignments are used. However, none of the FHSRRs, which were originally derived from version panTro3 of the chimp genome, appear in the alignment with panTro4. This suggests that at least a large majority of our FHSRRs are indeed human specific.
The second limitation concerns the definition of RRs. Data on chromatin modification are available only for cell lines that are not necessarily the most suitable context for the study of human-specific biological processes. For example, ideally, a collection of RRs active in the human brain would be needed for studying the regulation of cognition-related genes and its evolution. As we have shown, DHSs only partially fulfill this function.
Notwithstanding these limitations, we believe that we have shown that the appearance of HSRRs had an important role in shaping our regulatory network and thus the phenotypic features that distinguish humans from other animals.
Acknowledgments
We are grateful to Ugo Ala, Davide Cittaro, Ferdinando Di Cunto, Mattia Forneris, Antonio Lembo, and Elia Stupka for discussions, comments, and suggestions.
Supplemental Data
We defined RR classes by merging the classes defined by Ernst et al.14 as detailed in the Material and Methods. For DHS data, we defined a single RR class (“open”).
Web Resources
The URLs for the data presented herein are as follows:
Bedtools, http://bedtools.readthedocs.org
Bioconductor, http://www.bioconductor.org
Genomic Regions Enrichment of Annotations Tool (GREAT), http://great.stanford.edu
Online Mendelian Inheritance in Man (OMIM), http://omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Carroll S.B. Evolution at two levels: on genes and form. PLoS Biol. 2005;3:e245. doi: 10.1371/journal.pbio.0030245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tishkoff S.A., Reed F.A., Ranciaro A., Voight B.F., Babbitt C.C., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 2007;39:31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller C.T., Beleza S., Pollen A.A., Schluter D., Kittles R.A., Shriver M.D., Kingsley D.M. cis-Regulatory changes in Kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell. 2007;131:1179–1189. doi: 10.1016/j.cell.2007.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wray G.A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- 5.Chan Y.F., Marks M.E., Jones F.C., Villarreal G., Jr., Shapiro M.D., Brady S.D., Southwick A.M., Absher D.M., Grimwood J., Schmutz J. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science. 2010;327:302–305. doi: 10.1126/science.1182213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peter I.S., Davidson E.H. Evolution of gene regulatory networks controlling body plan development. Cell. 2011;144:970–985. doi: 10.1016/j.cell.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rebeiz M., Jikomes N., Kassner V.A., Carroll S.B. Evolutionary origin of a novel gene expression pattern through co-option of the latent activities of existing regulatory sequences. Proc. Natl. Acad. Sci. USA. 2011;108:10036–10043. doi: 10.1073/pnas.1105937108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donaldson I.J., Göttgens B. Evolution of candidate transcriptional regulatory motifs since the human-chimpanzee divergence. Genome Biol. 2006;7:R52. doi: 10.1186/gb-2006-7-6-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haygood R., Fedrigo O., Hanson B., Yokoyama K.-D., Wray G.A. Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nat. Genet. 2007;39:1140–1144. doi: 10.1038/ng2104. [DOI] [PubMed] [Google Scholar]
- 10.Torgerson D.G., Boyko A.R., Hernandez R.D., Indap A., Hu X., White T.J., Sninsky J.J., Cargill M., Adams M.D., Bustamante C.D., Clark A.G. Evolutionary processes acting on candidate cis-regulatory regions in humans inferred from patterns of polymorphism and divergence. PLoS Genet. 2009;5:e1000592. doi: 10.1371/journal.pgen.1000592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molineris I., Grassi E., Ala U., Di Cunto F., Provero P. Evolution of promoter affinity for transcription factors in the human lineage. Mol. Biol. Evol. 2011;28:2173–2183. doi: 10.1093/molbev/msr027. [DOI] [PubMed] [Google Scholar]
- 12.Shibata Y., Sheffield N.C., Fedrigo O., Babbitt C.C., Wortham M., Tewari A.K., London D., Song L., Lee B.-K., Iyer V.R. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet. 2012;8:e1002789. doi: 10.1371/journal.pgen.1002789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt D., Schwalie P.C., Wilson M.D., Ballester B., Gonçalves A., Kutter C., Brown G.D., Marshall A., Flicek P., Odom D.T. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell. 2012;148:335–348. doi: 10.1016/j.cell.2011.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng J., Liu T., Qin B., Zhang Y., Liu X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward L.D., Kellis M. Evidence of abundant purifying selection in humans for recently acquired regulatory functions. Science. 2012;337:1675–1678. doi: 10.1126/science.1225057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green P., Ewing B. Comment on “Evidence of abundant purifying selection in humans for recently acquired regulatory functions”. Science. 2013;340:682. doi: 10.1126/science.1233195. [DOI] [PubMed] [Google Scholar]
- 20.Ward L.D., Kellis M. Response to comment on “Evidence of abundant purifying selection in humans for recently acquired regulatory functions”. Science. 2013;340:682. doi: 10.1126/science.1233366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falcon S., Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- 23.Yu G., Li F., Qin Y., Bo X., Wu Y., Wang S. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010;26:976–978. doi: 10.1093/bioinformatics/btq064. [DOI] [PubMed] [Google Scholar]
- 24.Wang J.Z., Du Z., Payattakool R., Yu P.S., Chen C.-F. A new method to measure the semantic similarity of GO terms. Bioinformatics. 2007;23:1274–1281. doi: 10.1093/bioinformatics/btm087. [DOI] [PubMed] [Google Scholar]
- 25.Xie W., Schultz M.D., Lister R., Hou Z., Rajagopal N., Ray P., Whitaker J.W., Tian S., Hawkins R.D., Leung D. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153:1134–1148. doi: 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schriml L.M., Arze C., Nadendla S., Chang Y.-W.W., Mazaitis M., Felix V., Feng G., Kibbe W.A. Disease Ontology: a backbone for disease semantic integration. Nucleic Acids Res. 2012;40(Database issue):D940–D946. doi: 10.1093/nar/gkr972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey J.A., Carrel L., Chakravarti A., Eichler E.E. Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: the Lyon repeat hypothesis. Proc. Natl. Acad. Sci. USA. 2000;97:6634–6639. doi: 10.1073/pnas.97.12.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brawand D., Soumillon M., Necsulea A., Julien P., Csárdi G., Harrigan P., Weier M., Liechti A., Aximu-Petri A., Kircher M. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- 34.Ip B.K., Bayatti N., Howard N.J., Lindsay S., Clowry G.J. The corticofugal neuron-associated genes ROBO1, SRGAP1, and CTIP2 exhibit an anterior to posterior gradient of expression in early fetal human neocortex development. Cereb. Cortex. 2011;21:1395–1407. doi: 10.1093/cercor/bhq219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwamoto K., Bundo M., Kato T. Serotonin receptor 2C and mental disorders: genetic, expression and RNA editing studies. RNA Biol. 2009;6:248–253. doi: 10.4161/rna.6.3.8370. [DOI] [PubMed] [Google Scholar]
- 36.Hernando-Herraez I., Prado-Martinez J., Garg P., Fernandez-Callejo M., Heyn H., Hvilsom C., Navarro A., Esteller M., Sharp A.J., Marques-Bonet T. Dynamics of DNA methylation in recent human and great ape evolution. PLoS Genet. 2013;9:e1003763. doi: 10.1371/journal.pgen.1003763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M., ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tripodi M., Filosa A., Armentano M., Studer M. The COUP-TF nuclear receptors regulate cell migration in the mammalian basal forebrain. Development. 2004;131:6119–6129. doi: 10.1242/dev.01530. [DOI] [PubMed] [Google Scholar]
- 40.Reinchisi G., Ijichi K., Glidden N., Jakovcevski I., Zecevic N. COUP-TFII expressing interneurons in human fetal forebrain. Cereb. Cortex. 2012;22:2820–2830. doi: 10.1093/cercor/bhr359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Defossez P.-A., Kelly K.F., Filion G.J.P., Pérez-Torrado R., Magdinier F., Menoni H., Nordgaard C.L., Daniel J.M., Gilson E. The human enhancer blocker CTC-binding factor interacts with the transcription factor Kaiso. J. Biol. Chem. 2005;280:43017–43023. doi: 10.1074/jbc.M510802200. [DOI] [PubMed] [Google Scholar]
- 42.Levin H.L., Moran J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011;12:615–627. doi: 10.1038/nrg3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King M.C., Wilson A.C. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
We defined RR classes by merging the classes defined by Ernst et al.14 as detailed in the Material and Methods. For DHS data, we defined a single RR class (“open”).