

Abstract
Extraocular muscles (EOMs) are a unique group of skeletal muscles with unusual physiological properties such as being able to undergo rapid twitch contractions over extended periods and escape damage in the presence of excess intracellular calcium (Ca2+) in Duchenne’s muscular dystrophy (DMD). Enhanced Ca2+ buffering has been proposed as a contributory mechanism to explain these properties; however, the mechanisms are not well understood. We investigated mechanisms modulating Ca2+ levels in EOM and tibialis anterior (TA) limb muscles. Using Fura-2 based ratiometric Ca2+ imaging of primary myotubes we found that EOM myotubes reduced elevated Ca2+ ~2-fold faster than TA myotubes, demonstrating more efficient Ca2+ buffering. Quantitative PCR (qPCR) and western blotting revealed higher expression of key components of the Ca2+ regulation system in EOM, such as the cardiac/slow isoforms sarcoplasmic Ca2+-ATPase 2 (Serca2) and calsequestrin 2 (Casq2). Interestingly EOM expressed monomeric rather than multimeric forms of phospholamban (Pln), which was phosphorylated at threonine 17 (Thr17) but not at the serine 16 (Ser16) residue. EOM Pln remained monomeric and unphosphorylated at Ser16 despite protein kinase A (PKA) treatment, suggesting differential signalling and modulation cascades involving Pln-mediated Ca2+ regulation in EOM. Increased expression of Ca2+/SR mRNA, proteins, differential post-translational modification of Pln and superior Ca2+ buffering is consistent with the improved ability of EOM to handle elevated intracellular Ca2+ levels. These characteristics provide mechanistic insight for the potential role of superior Ca2+ buffering in the unusual physiology of EOM and their sparing in DMD.
Keywords: extraocular muscle, EOM, calcium, Serca2, phospholamban
Introduction
Extraocular muscles (EOMs) are among the fastest muscles in mammals. They form an integral part of the oculomotor system performing highly diverse eye movements including vergence, pursuit and saccadic eye movements as well as optokinetic and vestibulo-ocular reflexes. Their ability to sustain individual twitch contractions at an extremely fast frequency without tetanic fusion is unparalleled in mammalian muscle (Asmussen and Gaunitz, 1981, Bach-y-Rita and Ito, 1966, Close and Luff, 1974). Furthermore, in contrast to other skeletal muscles, they are able to do this over an extended period without fatigue (Frueh, et al., 1994). Due to the spatial and temporal demands on EOMs, they exhibit unique group-specific characteristics in many of their attributes including mechanical properties, regenerative capacities and fiber composition (Bron, et al., 1997, Demer, 2006, McLoon and Wirtschafter, 2002, Pacheco-Pinedo, et al., 2009, Porter, et al., 1995). In contrast to skeletal limb muscle, where mainly one slow fiber-type and three fast fiber-types are distinguished (Brooke and Kaiser, 1970), in EOM at least six fiber-types are present, which do not fit the fiber-type classification of limb muscle (Porter et al., 1995). The fiber-type patterns differ in the two layers that are distinguished in EOM; the global layer in proximity to the eyeball and the orbital layer adjacent to the bony orbit (Bron et al., 1997, Demer, 2006, Kato, 1938, Porter et al., 1995).
An intriguing EOM group-specific property is their differential involvement in neuromuscular diseases. EOMs are early and prominent targets in myasthenia gravis and mitochondrial myopathies, while being clinically and histologically spared in DMD (Kaminski, et al., 1992, Khurana, et al., 1995, Porter et al., 1995, Wirtschafter, et al., 2004). We and others have shown that EOMs differ from limb muscles in terms of their gene and protein expression at the level of their transcriptome and proteome in both rodents and humans (Fischer, et al., 2005, Fischer, et al., 2002, Fraterman, et al., 2007a, Fraterman, et al., 2007b, Porter, et al., 2001, Porter, et al., 2003). More recently, we have shown that EOMs differ from limb muscles in terms of their miRNA expression at the level of the miRNAome as well (Zeiger and Khurana, 2010). Indeed, based on the myriad of unique characteristics, EOMs have been categorized as having a unique muscle ‘allotype’ (Hoh and Hughes, 1988).
Compared to limb muscles, EOM exhibit a number of features predicting differences in their ability to buffer Ca2+ that would in part, provide mechanistic explanation of their unique patho-physiological properties. For example, EOMs have greater numbers of mitochondria, which are capable of quickly taking up a considerable amount of Ca2+ (Andrade, et al., 2005, Demer, 2002, Felder, et al., 2005, Fischer et al., 2002, Mayr, 1971). Expression patterns of SERCA isoforms in EOMs are more complex and differ widely from those typical of limb muscles (Jacoby and Ko, 1993, Kjellgren, et al., 2003). Several expression profiles implied that mRNA for proteins involved in Ca2+ homeostasis, such as S100 isoforms and Pln are up-regulated in EOM as compared to TA (Fischer et al., 2005, Fischer et al., 2002, Khanna, et al., 2003, Porter et al., 2001). EOMs from DMD patients and animal models, such as mdx mice and dystrophic dogs lack dystrophic calcification seen in dystrophin-deficient limb muscles implying that they are more resistant to elevated Ca2+ levels (Khurana et al., 1995). Based on these findings and their unusual twitch properties it has been hypothesized that EOM are better able to buffer excess intracellular Ca2+ levels and therefore are able to maintain Ca2+ homeostasis within a wider dynamic range of Ca2+ concentrations compared to TA (Andrade, et al., 2000, Khurana et al., 1995). However, lacunae exist in our current knowledge of Ca2+ handling properties of EOM and limb muscles.
In order to test the hypothesis that EOMs have superior Ca2+-buffering properties, we used Fura-2 based Ca2+ imaging to study the dynamics of Ca2+ homeostasis in cultured EOM and TA myotubes in vitro, as well as determined mRNA and protein levels of Ca2+ pumps, channels and buffers, using qPCR and western blotting.
Material and Methods
Animals
EOM and TA muscles from adult Sprague Dawley rats were used. For generating primary myoblasts 8–12 day old pups were used. All animal experiments were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, using protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania School of Medicine.
RNA isolation and SYBR Green based qPCR
RNA isolation was performed using Trizol reagent (Ambion, Austin, TX) in combination with RNAeasy Mini Kit (Qiagen, Valencia, CA). Primers were designed using PrimerExpress 2.0 (Applied Biosystems, Foster City, CA) across exon boundaries (Table S1). qPCR was run on a 7900HT ABI Prism real-time PCR instrument (Applied Biosystems). Gapdh served as reference gene. Fold-change calculations and statistical analysis was performed by randomized statistical testing using REST qPCR analysis software (Pfaffl, et al., 2002) with p < 0.05 considered statistically significant.
Western blot analysis
Western blot analysis was performed using the NuPage System or BioRad precast gels as described by the manufacturer (Invitrogen Carlsbad, CA or Biorad, Hercules, CA). Crude whole muscle homogenates were prepared using TNEC lysis buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 1% Igepal, 2 mM EDTA) containing a complete protease inhibitor cocktail and PhosStop phosphatase inhibitors (Roche, Basel, Switzerland). The protein concentrations were determined using the DC Assay (BioRad). Equal amounts (10–50 μg) of samples were resolved on 4–12% Bis-Tris gels or in the case of the two Casq isoforms on a 7.5% Tris/HCl gels as described before (Paolini et al 2007), transferred onto PVDF membranes (Millipore, Billerica, MA) and probed with the following mouse or rabbit antibodies: monoclonal anti-SERCA1, monoclonal anti-SERCA2, polyclonal anti-Calsequestrin (recognises Casq1 and Casq2), monoclonal anti-Phospholamban, polyclonal anti-Calmodulin (all Affinity Bioreagents, Golden, CO), polyclonal anti-Phospholamban Phospho-Ser16, polyclonal anti-Phospholamban Phospho-Thr17 (Badrilla, Leeds, UK), polyclonal anti-FXYD1 (Phospholemman) (Abcam,) or monoclonal anti-CamkIIB (Abnova, Walnut, CA). Secondary goat-anti-mouse or goat-anti-rabbit antibodies were conjugated with horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA). Protein bands were detected with a LAS-3000 Fuji imaging system (Fujifilm, Tokyo, Japan). Equal loading was confirmed after the transfer by Ponceau S staining (Sigma, St. Louis, MO). Bands were quantified densitometrically using ImageJ software (http://rsb.info.nih.gov/ij/National Institutes of Health). Statistical analysis was done using a non-parametric Mann-Whitney test, with p < 0.05 considered significant.
Immunocytochemistry
Myoblasts or myotubes were stained according to standard procedures. Briefly, after fixation in 4% paraformaldehyde and permeabilization (0.05% Triton X-100) cells were blocked in 10% goat serum. Incubation with antibodies for desmin (Sigma), myogenin (Imgenex, San Diego, CA) or alpha-actinin (Sigma) was followed by incubation with goat-anti-mouse-Alexa-Fluor-546 (Molecular Probes, Carlsbad, CA) and Hoechst 33342 (Sigma). Pictures of stained cells were taken using a fluorescence microscope Olympus BX51 equipped with a MagnaFire CCD camera (Olympus, Center Valley, PA).
Myoblast culture
Primary myoblasts were generated based on the methods of Rando et al (Rando and Blau, 1994). Briefly, freshly dissected EOM and TA muscles from 8–12 day old rat pubs were minced and digested with collagenase IV and dispase (Worthington, Lakewood, NJ). Cell suspensions were strained (BD Biosciences, San Jose, CA) and pre-plated on uncoated dishes. Cells that remained in suspension were plated on collagen I-coated 60 mm cell culture dishes and were allowed to proliferate in Ham’s F10 medium (Gibco, Carlsbad, CA) supplemented with 20% FBS (Hyclone, South Logan, UT) and 1% penicillin/streptomycin (Gibco) until reaching ~ 70% confluence. The cells were split and grown on 12 mm collagen I (Sigma) coated glass cover slips until fusion into multinucleated myotubes was initialized with DMEM/Ham’s F10 (1:1) medium containing 5% horse serum (Gibco). Pre-plating on uncoated dishes was used each time before plating to reduce the amount of fibroblasts. For immunocytochemistry cells were plated on 8-well chamber slides (Nunc, Rochester, NY).
Measurement of Intracellular Ca2+
Fusion of myoblasts was induced 48 hours prior to measurements. Cells were loaded with 10 μM Fura-2 AM and 2% pluronic F-127 (Molecular Probes) for 30 min at room temperature and maintained in Fura-2-free isotonic solution for 30 min before data acquisition at room temperature began. Coverslips were mounted on an inverted microscope (Nikon, Melville, NY) and visualized with a 20X objective. Myotubes of similar size were selected for measurements. The field was alternately excited at 340 and 380 nm with a scanning monochromator, and the fluorescence emitted > 520 nm from the region of interest surrounding an individual myotube was imaged and analyzed (all Photon Technologies International, Inc., Lawrenceville, NJ). Cells were perfused with extracellular solution at the start of Ca2+-imaging experiments containing (in mM) 105 NaCl, 4.5 KCl, 2.8 Na HEPES, 7.2 HEPES acid, 1.3 CaCl2, 0.5 MgCl2, 5 glucose, and 75 mannitol (pH 7.4). For assessment of intracellular Ca2+ changes, cells were permeabilized with 5 μM ionomycin while perfused with extracellular solution containing 300 nM Ca2+ or perfused with extracellular solution containing 60 mM KCl and 50 mM NaCl without ionomycin. Calibration was performed separately on each cell after the experiment using standard techniques (Zhang, et al., 2005); cells were perfused with 5 μM ionomycin in Ca2+-free solution with 5 mM EGTA followed by ionomycin in high-Ca2+ solution (both solutions were at pH 8.0). The 340/380-ratio was converted to Ca2+ concentration using the method of Grynkiewicz (Grynkiewicz, et al., 1985). The peak Ca2+ values were determined by subtracting the baseline from the maximum levels. The “decay 0.5” value denotes the time needed for the Ca2+ to decay from peak to half peak size. Data were statistically analysed using Student’s t-test with p < 0.05 considered statistically siginificant. To correlate cell size with the corresponding Ca2+ peak (Suppl. Fig. S1), the cell size was calculated using an approximation of two cones abutting at the base to represent the spindle shaped cell.
Results
EOM myotubes express myogenic markers
EOM and TA primary cultured cells were morphologically similar (Fig. 1A, B). Myoblasts were smaller, spindle-like and more compact in size than fibroblasts and could be differentiated into multinucleated myotubes as described before (Porter et al 2006, Rando and Blau 1994). EOM myoblasts proliferated somewhat faster and tended to form slightly more and longer myotubes than TA myoblast (observation not quantified). Spontaneous twitching in the myotubes demonstrated that the essential components of the muscle cells contractile apparatus was preserved in culture, which was confirmed by positive alpha-actinin staining (Fig. 1B). Further characterization of the cell cultures showed that undifferentiated myoblasts stained positive with an antibody against the early muscle marker desmin (Fig. 1B), whereas differentiated myotubes stained positive for the differentiation marker myogenin. Co-staining with the nucleic stain Hoechst 33342 (Fig. 1B) showed existence of multinucleated myotubes. This demonstrated that the cultured cells retained myogenic characteristics and were able to form multinucleated myotubes.
Fig. 1. Cultured primary EOM and TA myoblasts and myotubes.
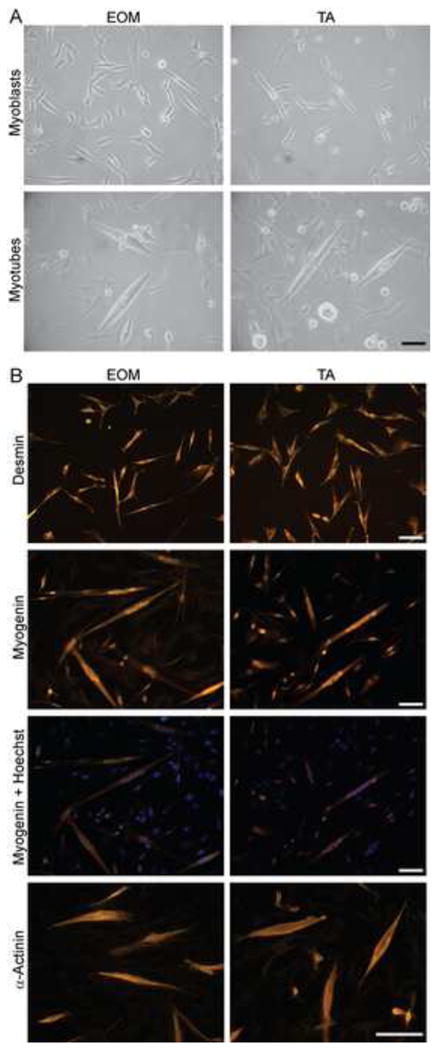
(A) Light microscopic images of myoblasts and differentiated myotubes derived from EOM and TA. (B) EOM and TA myoblasts and myotubes stained for myogenic markers. Top panel: Myoblasts stained for desmin. Middle panels: differentiated myotubes stained for myogenin; the overlay with Hoechst 33342 staining shows localization of myogenin to nuclei. Bottom panel: differentiated myotubes stained for alpha-actinin. Scale bar = 100 μm.
More efficient Ca2+ buffering in EOM myotubes
To test the hypothesis that EOM are better able to buffer high intracellular Ca2+ levels compared to limb muscles, we used Fura-2 based ratiometric imaging to measure Ca2+ inside individual cultured primary EOM and TA myotubes (single cell imaging). Moderate and similar baseline levels of 92.5 ± 9.1 nM Ca2+ in EOM and 90.4 ± 5.0 nM in TA confirmed that the signal originated predominantly from the cytoplasm and implied that the mechanisms for control of resting Ca2+ levels were similar in both cell types.
After reaching stable baseline levels, the cells were perfused with an extracellular solution containing the Ca2+ ionophore ionomycin and 300 nM Ca2+. The lowered Ca2+ concentration (300 nM instead of 1.3 mM) of the perfusion solution was chosen to prevent myotube contraction. Exposure to the ionomycin solution led to a rapid rise in intracellular Ca2+ levels (Fig. 2A) surpassing 300 nM Ca2+ suggesting rapid depletion of Ca2+ from the SR. This rise in Ca2+ was considerably higher in EOM myotubes than in TA, with peak levels of 2.37 ± 1.8 μM in EOM compared with 1.14 ± 0.23 μM in TA (p < 0.01) (Fig. 2B). The decay time was quantified by defining decay 0.5 as the time needed for the Ca2+ levels to decrease by 50 % after reaching the peak. This decay was significantly faster in EOM myotubes, with Ca2+ concentrations taking only 56.6 ± 6.1 s to fall by half. This contrasts with TA cells, where the reduction took nearly twice as long (105.5 ± 19.4 s, p < 0.05) (Fig. 2C, D). Peak-size did not correlate with the size of the myotube, demonstrating that intrinsic cellular properties depending on the EOM vs TA allotype rather than size per se mediated the differences (Suppl. Fig. S1). These findings confirm that excessive Ca2+ is buffered more effectively in EOM than in TA myotubes.
Fig. 2. Intracellular Ca2+ measurements in myotubes showed enhanced buffering in EOM.

Representative Ca2+ traces are shown for perfusion experiments of Fura-2 loaded myotubes with an extracellular solution containing 300 nM Ca2+ and 5 μM ionomycin (A, C). EOM myotubes showed a 2.0-fold larger maximum peak size (A, B) and a 1.9-fold faster half-maximum decay time (C, D) indicating better Ca2+ handling properties compared to TA. Representative positions of the parameters measured are illustrated in A & C. Data are depicted as bar graphs showing significant differences in the evaluated parameters (B, D) (Mean ± SEM, * P < 0.05, ** P < 0.01).
To independently validate these results we evaluated whether similar responses were obtained by inducing membrane depolarization with high extracellular potassium (60 mM K+). High K+ stimulation leads to excitation-contraction (E-C) coupling and triggers immediate Ca2+ release from the SR (Melzer, et al., 1995). EOM and TA myotubes were stimulated for 30 s with high K+. In accordance with the previous observations the Ca2+ peaks recorded from EOM were significantly higher than the peaks recorded from TA myotubes (1.5 ± 0.3 vs. 0.77 ± 0.1 nM, p < 0.05, Fig. 3A, B), likely reflecting a larger amount of Ca2+ released from the SR in EOM. However, the peaks were only a little more than half as high as those triggered by ionomycin, possibly reflecting the differences in physiological vs pharmacological approaches used experimentally, such as the shorter stimulation time with high K+ and the differences in receptor and channel involvement rather than membrane perforation obtained using ionophores. The noted decay 0.5-rate in EOM was much faster than the decay rate with ionomycin but not significantly faster than in TA (14.9 ± 1.8 vs. 13.1 ± 1.6 s, Fig. 3D). Nonetheless, EOM myotubes re-sequestered a larger amount of Ca2+ from the cytosol than TA myotubes within the same time. These results support the notion of a larger Ca2+ release and more efficient buffering in EOMs.
Fig. 3. Efficient buffering of high K+ induced increase of intracellular Ca2+ in EOM myotubes.

Representative Ca2+ traces are shown for perfusion experiments of Fura-2 loaded myotubes stimulated with high K+ (60 mM) solution (A, C). EOM myotubes showed a 1.9-fold large maximum peak size than TA myotubes (A, B) but a similar average half-maximum decay time (C, D). Parameters were measured in the same way as described in Fig. 2. (Mean ± SEM, * P < 0.05).
Increased mRNA expression levels of Ca2+ handling proteins in EOM
Having shown more efficient buffering of high Ca2+ levels in EOM compared to TA myotubes, we determined mRNA levels of genes known to be involved in Ca2+ homeostasis in cultured myotubes by qPCR. Candidate genes from five functionally distinct groups of Ca2+/SR proteins were investigated. As shown in Table 1, the first group contained genes of the sarcoplasmic and plasma membrane Ca2+ pumps and the Na+/Ca2+-exchanger (Ncx). The second group contained genes that function as regulators of the Serca pumps. The third group contained the major Ca2+ binding and buffering proteins of the SR. In the fourth group genes coded for cytosolic and sarcomeric Ca2+ binding proteins, predicted to buffer elevated intracellular Ca2+. The final group of genes examined included Ca2+ channels involved in E-C coupling. mRNA levels of 7 (Pln, Sarcolipin (Sln), Regucalcin (Rgn), Casq2, Parvalbumin (Parv), S100a1 and dihydropyridine-sensitive L-type Ca2+ channel alpha-1 subunit (Cacna1s) of the 18 genes quantified (cf. below) were expressed at significantly higher levels in EOM myotubes than in TA (Table 1 “Myotubes”).
Table 1. Relative mRNA levels of genes encoding Ca2+ handling proteins in EOM myotubes and EOM tissue compared with TA.
Listed are relative mRNA levels of genes encoding Ca2+ handling proteins determined by SYBR Green qPCR in myotubes and muscle tissue. Mean fold-changes ± SEM of three independent samples each were calculated and statistically analysed using REST 2005 and P < 0.05 was considered statistically significant.
|
|
||||||
|---|---|---|---|---|---|---|
| Myotubes | Tissue | |||||
|
| ||||||
| Protein category | Protein Function | Gene | Fold-change EOM vs TA | statistical significance | Fold-change EOM vs TA | statistical significance |
|
| ||||||
| Pumps | SR Ca2+ ATPase, fast-twitch | Serca1 | 1.0 ± 0.65 | n.s. | 1.1 ± 0.20 | n.s. |
| SR Ca2+ ATPase, slow-twitch | Serca2 | 1.2 ± 0.19 | n.s. | 13.2 ± 6.75 | < 0.05 | |
| Ca2+ ATPase 1, plasma membrane | Pmca1/Atp2b1 | 1.1 ± 0.20 | n.s. | 33.3 ± 7.77 | < 0.05 | |
| Ca2+ ATPase 4, plasma membrane | Pmca4/Atp2b4 | 1.1 ± 0.38 | n.s. | 30.0 ± 15.40 | < 0.05 | |
| Na+/Ca2+ exchanger, plasma membrane | Ncx/Scl8a1 | 1.4 ± 0.34 | n.s. | 15.8 ± 6.68 | < 0.05 | |
|
| ||||||
| Regulatory proteins | SR, regulates SERCA2 | Phospholamban | 2.7 ± 0.42 | < 0.05 | 74.7 ± 10.72 | < 0.05 |
| SR, regulates SERCA1 | Sarcolipin | 1.3 ± 0.16 | < 0.05 | 20.6 ± 11.12 | < 0.05 | |
| cytosol, regulates SERCAs | Regucalcin | 1.5 ± 0.33 | < 0.05 | 12.7 ± 6.3 | < 0.05 | |
|
| ||||||
| SR calcium binding proteins | SR, calcium binding protein, skeletal | Calsequestrin 1 | 0.9 ± 0.26 | n.s. | 1.5 ± 0.39 | n.s. |
| SR, calcium binding protein, cardiac | Calsequestrin 2 | 1.5 ± 0.27 | < 0.05 | 29.9 ± 18.30 | < 0.05 | |
| SR, calcium binding protein | Sarcalumenin | 1.4 ± 0.31 | n.s. | 2.6 ± 0.55 | n.s. | |
|
| ||||||
| Cytosolic calcium binding proteins | cytosol, calcium binding protein | Parvalbumin | 2.0 ± 0.35 | < 0.05 | 2.3 ± 0.54 | n.s. |
| cytosol, calcium binding protein | S100a1 | 2.3 ± 0.39 | < 0.05 | 5.4 ± 0.68 | < 0.05 | |
| calcium binding protein | Calmodulin | 0.9 ± 0.10 | n.s. | 10.1 ± 3.64 | < 0.05 | |
| calcium binding protein, slow-twitch/cardiac | Troponin C 1 | 1.0 ± 0.14 | n.s. | 5.1 ± 0.59 | < 0.05 | |
| calcium binding protein, fast-twitch | Troponin C 2 | 1.4 ± 0.29 | n.s. | 2.1 ± 0.49 | n.s. | |
|
| ||||||
| Calcium channels | calcium channel subunit | Cacna1s | 1.5 ± 0.22 | < 0.05 | 0.9 ± 0.12 | n.s. |
| calcium release channel, skeletal | Ryanodine Receptor 1 | 1.3 ± 0.25 | n.s. | 1.0 ± 0.16 | n.s. | |
We also tested the gene expression of Ca2+/SR proteins in fully differentiated adult tissue and we found that 11 out of 18 quantified genes showed significantly higher expression in EOM compared with TA (Table 1 “Tissue”). The highest fold-changes were detected for the Ca2+ pumps (Serca2, plasma membrane Ca2+-ATPase 1 (Pmca1), Pmca4, Ncx), the regulatory proteins (Pln, Sln and Rgn), with fold-changes of up to 75-fold difference (Pln) and the Ca2+ binding proteins Casq2 and Calmodulin (Calm) (29.9 and 10.1 fold, respectively). Only Serca1, Casq1, Cacna1s and Ryr1 were not differentially expressed in EOM vs. TA muscle tissue.
Increased protein levels of Ca2+ handling proteins in EOM
The relative expression levels of selected proteins involved in Ca2+/SR homeostasis were examined using western blots to determine whether message levels correlated with protein levels in muscle tissue (Fig. 4). Levels of the sarcoplasmic Ca2+ storage protein Casq1 (skeletal muscle isoform) were lower in EOM but Casq2 (slow/cardiac isoform) levels were much greater in EOM than in TA (Fig. 4A). Total Casq content was slightly higher in EOM but did not reach statistical significance (Fig. 4A). Protein levels of the sarcoplasmic Ca2+-ATPase Serca1 were less in EOM, while those for Serca2 were considerably higher in EOM (Fig. 4B). However, when considering relative band intensities Serca1 appears to be the dominant isoform.
Fig. 4. Increased protein levels of SR Ca2+ handling proteins in EOM.

Western blot analysis showing relative abundance of indicated proteins in EOM and TA. (A) Casq1 and Casq2 isoforms were resolved on a 7.5% gel. Heart tissue indicates the molecular weight of Casq2. In EOM, Casq2 was more abundant than Casq1 compared with TA. Total Casq showed a tendency toward higher levels in EOM. (B) Serca1 levels were less in EOM whereas Serca2 levels were significantly higher compared with TA. Serca1 was the predominant isoform in both tissues. Bands were quantified using densitometry and data were statistically analysed using a non-parametric test. * P < 0.05 was considered statistically significant. (Mean ± SD; AU = arbitrary units).
Pln has frequently been found to be highly expressed at the mRNA level in EOM. At the protein level, under non-reducing conditions, only monomeric Pln was detected in EOM, while in heart both, monomeric and pentameric Pln were present. No Pln protein was detected in TA (Fig. 5A). Pln, depending on its phosphorylation status, regulates Serca2, which is activated when Pln is phosphorylated on either or both of its phosporylation sites: Ser16 is phosphorylated by PKA and Thr17 is phosphorylated by Ca2+/Calmodulin dependent kinase II (CamkII). Using phosphorylation site specific antibodies, only trace amounts of PKA dependent phosphorylation at Ser16 in Pln from EOM (Fig. 5B) were detected. In vitro PKA treatment only slightly increased these levels in EOM, whereas in heart samples PKA treatment substantially increased endogenous levels of Ser16 phosphorylation (Fig. 5B). On the other hand EOM contained considerable amounts of endogenous CamkII dependent phosphorylation of monomeric Pln at Thr17 (Fig. 5C). Neither of the phospho-Pln specific antibodies detected a Pln pentamer in EOM. In both cases heart samples served as positive control, where endogenous phosphorylation at Ser16 and Thr17 was present in monomeric and pentameric Pln. The 14kDa band detected by the P-Ser16 antibody (Fig. 5B, arrow) was identified as phospholemman as described for this antibody (Suppl. Fig. S2) (Drago and Colyer, 1994).
Fig. 5. Increased protein levels of phospho-Pln, CamkII and Calm in EOM.

Western blot analysis showing relative abundance of indicated proteins in EOM and TA. (A) Monomeric Pln is found in EOM but not in TA under non-reducing conditions. In heart, monomeric (~5kDa) and pentameric (~25kDa) Pln is present. (B) In EOM Pln is phosphorylated in trace amounts at Ser16, which cannot be further induced by PKA treatment. In heart, serving as positive control, phosphorylation at Ser16 was increased after PKA treatment. The band at about 14kD (arrow) detected by the Pln-Phospho-Ser16 antibody could be identified as phospholemman (Suppl. Fig. S2). (C) Phosphorylation of Pln at Thr17 in EOM is endogenously high. Reprobing of blots with the Anti-Pln antibody (lower panels in B and C) showed relative abundance of all Pln versus the phosphorylated forms. (D) CamkII and Calm were more abundant in EOM supporting preferential phosphorylation of Pln at Thr17. Bands were quantified using densitometry and data were statistically analysed using a non-parametric t-test. * P < 0.05 was considered statistically significant. (Mean ± SD; AU = arbitrary units). Red.= reduced.
Furthermore, in support of increased CamkII activity significantly higher protein levels of Calm and CamkIIB were detected in EOM (Fig. 5D). This would support predominant Thr17 phosphorylation in EOM.
These results demonstrate differential expression of components underlying the unique signalling cascades modulating Ca2+ homeostasis of EOM, suggesting that regulation of Serca activity in EOM by Pln relies mainly on CamkII dependent phosphorylation.
Discussion
A combination of physiological, molecular and biochemical methods were used to demonstrate clear differences in the mechanisms used by EOM and TA to handle cytoplasmic Ca2+ and in the ability of these cell types to buffer excess Ca2+. Intracellular measurement of cytoplasmic Ca2+ levels demonstrated more effective buffering of excessive Ca2+ in EOM (Fig. 2 and 3). Molecular and biochemical methods revealed increased expression of mRNAs and proteins encoding numerous Ca2+ handling proteins, in particular Casq2, Serca2 and Pln (Table 1 & Fig. 4, 5A). In vitro kinase studies suggested differential signalling and modulation cascades involving Pln-mediated Ca2+ regulation in EOM (Fig. 5). Combined, these data suggest EOM may be better able to prevent prolonged elevations of cytoplasmic Ca2+ levels: the findings are summarized in the model displayed in Fig. 6.
Fig. 6. Model for superior Ca2+ homeostasis in EOM.

Schematic (A) shows an EOM muscle cell with a larger SR, numerous pumps and transporters in both the sarcolemma and the sarcoplasmic membrane and many Ca2+ binding proteins in the cytosol (e.g. S100a, Parv) and SR (Casq1 and Casq2), as well as numerous mitochondria. CamkII phosphorylates Pln at Thr17, which regulates Serca2. Schematic (B) represents a TA muscle cell containing a smaller SR, fewer pumps and transporters and less Ca2+ binding proteins in the cytosol and SR compared with EOM. Excess Ca2+ ions entering the cell can be bound and/or removed more effectively in EOM than TA, leading to a faster return to resting Ca2+ levels. We propose that the extensive Ca2+ handling capacity of EOM reflects their functional properties and may help to protect this muscle group from Ca2+ mediated damage in DMD. (Symbols: Serca: sarcoplasmic reticulum Ca2+-ATPase, Casq: Calsequestrin, Pln: Phospholamban, Pmca: plasmamembrane Ca2+-ATPase, Parv: Parvalbumin, CamkII: Ca2+/Calmodulin dependent kinase II, Calm: Calmodulin, Mito: Mitochondria, Ncx: Na+/Ca2+-exchanger, Thr17-P: phosphorylated threonine 17 residue).
EOM are superfast muscles with very fast contraction cycles depending on rapid Ca2+ turnover, including both the quick release of Ca2+ from intracellular stores for contraction and the prompt sequestering of Ca2+ to terminate the contraction and allow muscle relaxation (Asmussen and Gaunitz, 1981, Bach-y-Rita and Ito, 1966, Close and Luff, 1974).
Measurements of cytoplasmic Ca2+ levels in EOM and TA myotubes showed larger Ca2+ peaks in EOM cells stimulated with ionomycin or high K+ suggesting that the SR contains and can release more Ca2+ into the cytoplasm as compared to the TA. Interestingly, EOM reduced the higher cytoplasmic levels of Ca2+ more quickly than TA; indeed EOM were nearly twice as effective as TA in terms of the time required to reduce the increased Ca2+ by 50% after ionomycin stimulation. The Ca2+ cycle observed in the cultured EOM myotubes may have been facilitated by the increased levels of Casq2 in the SR and Pln stimulating Serca mediated Ca2+ uptake. However, the only moderately elevated mRNA levels of Ca2+ handling proteins in the cultured myotubes, compared to muscle tissue (Table 1 “Myotubes”) may be related to the fact that the cultured cells only partially recapitulated the physiological characteristics of fully differentiated muscle (Barjot, et al., 1995, Rubinstein and Holtzer, 1979). The similar morphology of EOM and TA myotubes (as opposed to the dissimilar morphology of EOM and TA muscle) further suggests that allotype-specific expression in cultured cells is partially rather than fully maintained. Indeed, this has been demonstrated in cell lines made from EOM and limb muscles and studied at the level of the transcriptome (Porter, et al., 2006). This suggests that our cell culture results likely underestimated the ability of EOM to buffer Ca2+, and that even more dramatic differences may exist in vivo.
The larger Ca2+ release from the EOM myotubes is in accord with an abundant SR in EOM containing potentially more releasable Ca2+ than TA limb muscles (Mayr, 1971). This is also consistent with the elevated levels of Casq isoforms that we observed in EOM. Casq2, normally found in slow-twitch fibers and cardiac muscle, shares about 65% homology with Casq1, the skeletal muscle isoform and has a slightly lower Ca2+ binding capacity (Park, et al., 2004). Interestingly, Casq2 was the predominant isoform in EOM, although the majority of EOM fibers are fast muscle fibers of which most have an abundant SR (Asmussen and Gaunitz, 1981, Mayr, 1971, Porter et al., 1995). This suggests functional differences in terms of Ca2+ storage and turnover in EOM. Moreover, Casq2 has been suggested as target in autoimmune thyroid disease with eye involvement (Wescombe, et al., 2009). In fast EOM fibers Casq2 may also be present and may be co-expressed with the fast fiber isoform Casq1 in at least some EOM fiber-types.
The fast removal of elevated intracellular Ca2+ in muscle fibers is mainly carried out by the Serca pumps (Rossi and Dirksen, 2006). In rat EOM Serca1 was the predominant isoform in EOM and TA, whereas Serca2 showed higher levels in EOM than in TA. In human and rabbit EOMs Serca1 and Serca2 are widely co-expressed (Jacoby and Ko, 1993, Kjellgren et al., 2003). This suggests that Serca1 and Serca2 might be co-expressed in rat EOM as well and that Serca2 is not restricted to the approximately 20% of slow-tonic fibers in EOM, as in the case of slow limb muscle fibers. More detailed studies are needed to investigate the fiber-type specific expression of Ca2+ binding proteins.
The expression and co-expression of cardiac/slow and skeletal muscle isoforms in the same fiber would confer unique SR function and Ca2+ handling to EOM fibers. Furthermore, the SR of fast vs. slow fibers of limb muscles differ regarding their Ca2+ load and release kinetics (Fryer and Stephenson, 1996) and the co-existence of both fast and slow characteristics in EOM fibers, in addition to the varying abundance of SR, add to the complexity of Ca2+ homeostasis in EOM regarding fiber-types (Mayr, 1971, Spencer and Porter, 2005).
Both Serca1 and Serca2 activity can be regulated by the homologous proteins Pln and Sln depending on their phosphorylation status (Slack, et al., 1997, Traaseth, et al., 2008). While Pln was not expressed in TA, the high levels of Pln (mRNA and protein) could play a major role in regulating Serca activity in EOM (Morita, et al., 2008). Serca activity is inhibited by Pln but this inhibition is relieved upon phosphorylation of Pln (Traaseth et al., 2008). In EOM Pln was predominantly phosphorylated at Thr17 and at trace levels at Ser16. Furthermore, phosphorylation at Ser16 could not be increased in vitro suggesting that the Pln-Ser16 site in EOM is not subject to significant functional regulation. This is consistent with results in rabbit soleus muscle where endogenous phosphorylation at Thr17 predominates over phosphorylation at Ser16 (Damiani, et al., 2000). Phosphorylation at Thr17 is mediated by the SR bound CamkII, which is activated by increased Ca2+ levels and Calm (Simmerman, et al., 1986). We demonstrated previously and in this study that CamkII is expressed at increased levels in rat EOM (Fraterman et al., 2007b). This is in accordance with the increased levels of Calm and the predominance of endogenous phosphorylation at Thr17 we found in EOM. Furthermore, it has been proposed that CamkII activity and the resulting phosphorylation of Pln at Thr17 have a predominant role in skeletal muscle endurance exercise, which is consistent with the constant activity of EOMs (Damiani et al., 2000, Rose, et al., 2007, Rose, et al., 2006). Moreover, we found only monomeric Pln in rat EOM, which is believed to be the active form (MacLennan and Kranias, 2003). This suggests that Pln is mainly present in its active form stimulating Serca activity. These results suggest that in contrast to limb muscle, Serca2 and possibly Serca1 take advantage of an extra level of regulation in EOM, which may be modulated dependent on the activity of the fibers.
In addition to the Ca2+ turnover mediated by the SR, the increased levels of cytoplasmic buffering proteins such as Parv and Troponin C observed in EOM may also help to quickly reduce the cytoplasmic concentration of free Ca2+ (Heizmann, et al., 1982, Rome, 2006). Moreover, the greater amount of mitochondria in EOM (Felder et al., 2005, Fischer et al., 2002, Mayr, 1971) have been implicated in playing a substantial role in taking up free Ca2+ (Andrade et al., 2005). Overall, the greater release and more rapid buffering of excess cytoplasmic Ca2+ that we observed provides direct support for the hypothesis that differences in Ca2+ handling dynamics contribute to the fast contraction cycles in EOM.
Our results confirm our hypothesis and demonstrate that rat EOM are endowed with an extraordinary capacity to regulate their Ca2+ homeostasis as illustrated in our model (Fig. 6), which is consistent with the functional demands of very fast and constantly active muscles. In addition, our findings revealed distinct mechanisms involved in the Ca2+ homeostasis of EOMs. These features could help explain the lack of Ca2+-mediated damage in DMD and mdx EOM (Khurana et al., 1995). In the future, it will be of interest to map out the expression of Ca2+ buffering proteins with respect to the different EOM fiber-types as well as differences in Ca2+ buffering properties of these fibers from normal and mdx mice.
Supplementary Material
Primers used for SYBR Green qPCR.
Ca2+ peak and cell size did not correlate when plotted for EOM and TA myotubes.
To identify the 14kDa band detected by the Pln-phospho-Ser16 antibody (A), the blot was stripped and reprobed with anti-phospholemman (B).
Acknowledgments
This work was supported by grants EY013862 to TSK and EY015537 to CHM from the NIH and by a grant from the Pennsylvania Department of Health to TSK. EY013862 support was made possible via American Recovery and Reinvestment Act of 2009 (ARRA) funds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrade FH, McMullen CA, Rumbaut RE. Mitochondria are fast Ca2+ sinks in rat extraocular muscles: a novel regulatory influence on contractile function and metabolism. Invest Ophthalmol Vis Sci. 2005;46:4541–4547. doi: 10.1167/iovs.05-0809. [DOI] [PubMed] [Google Scholar]
- Andrade FH, Porter JD, Kaminski HJ. Eye muscle sparing by the muscular dystrophies: lessons to be learned? Microsc Res Tech. 2000;48:192–203. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<192::AID-JEMT7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Asmussen G, Gaunitz U. Mechanical properties of the isolated inferior oblique muscle of the rabbit. Pflugers Arch. 1981;392:183–190. doi: 10.1007/BF00581270. [DOI] [PubMed] [Google Scholar]
- Bach-y-Rita P, Ito F. In vivo studies on fast and slow muscle fibers in cat extraocular muscles. J Gen Physiol. 1966;49:1177–1198. doi: 10.1085/jgp.0491177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barjot C, Cotten ML, Goblet C, Whalen RG, Bacou F. Expression of myosin heavy chain and of myogenic regulatory factor genes in fast or slow rabbit muscle satellite cell cultures. J Muscle Res Cell Motil. 1995;16:619–628. doi: 10.1007/BF00130243. [DOI] [PubMed] [Google Scholar]
- Bron AJ, Tripathi RC, Tripathi BJ. Wolff’s Anatomy of the eye and the orbit. London: Chapman & Hall Medical; 1997. [Google Scholar]
- Brooke MH, Kaiser KK. Muscle fiber types: how many and what kind? Arch Neurol. 1970;23:369–379. doi: 10.1001/archneur.1970.00480280083010. [DOI] [PubMed] [Google Scholar]
- Close RI, Luff AR. Dynamic properties of inferior rectus muscle of the rat. J Physiol. 1974;236:259–270. doi: 10.1113/jphysiol.1974.sp010434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani E, Sacchetto R, Margreth A. Variation of phospholamban in slow-twitch muscle sarcoplasmic reticulum between mammalian species and a link to the substrate specificity of endogenous Ca(2+)-calmodulin-dependent protein kinase. Biochim Biophys Acta. 2000;1464:231–241. doi: 10.1016/s0005-2736(00)00153-x. [DOI] [PubMed] [Google Scholar]
- Demer JL. The orbital pulley system: a revolution in concepts of orbital anatomy. Ann N Y Acad Sci. 2002;956:17–32. doi: 10.1111/j.1749-6632.2002.tb02805.x. [DOI] [PubMed] [Google Scholar]
- Demer JL. Current concepts of mechanical and neural factors in ocular motility. Curr Opin Neurol. 2006;19:4–13. doi: 10.1097/01.wco.0000198100.87670.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago GA, Colyer J. Discrimination between two sites of phosphorylation on adjacent amino acids by phosphorylation site-specific antibodies to phospholamban. J Biol Chem. 1994;269:25073–25077. [PubMed] [Google Scholar]
- Felder E, Bogdanovich S, Rubinstein NA, Khurana TS. Structural details of rat extraocular muscles and three-dimensional reconstruction of the rat inferior rectus muscle and muscle-pulley interface. Vision Res. 2005;45:1945–1955. doi: 10.1016/j.visres.2005.01.031. [DOI] [PubMed] [Google Scholar]
- Fischer MD, Budak MT, Bakay M, Gorospe JR, Kjellgren D, Pedrosa-Domellof F, Hoffman EP, Khurana TS. Definition of the unique human extraocular muscle allotype by expression profiling. Physiol Genomics. 2005;22:283–291. doi: 10.1152/physiolgenomics.00158.2004. [DOI] [PubMed] [Google Scholar]
- Fischer MD, Gorospe JR, Felder E, Bogdanovich S, Pedrosa-Domellof F, Ahima RS, Rubinstein NA, Hoffman EP, Khurana TS. Expression profiling reveals metabolic and structural components of extraocular muscles. Physiol Genomics. 2002;9:71–84. doi: 10.1152/physiolgenomics.00115.2001. [DOI] [PubMed] [Google Scholar]
- Fraterman S, Zeiger U, Khurana TS, Rubinstein NA, Wilm M. Combination of peptide OFFGEL fractionation and label-free quantitation facilitated proteomics profiling of extraocular muscle. Proteomics. 2007a;7:3404–3416. doi: 10.1002/pmic.200700382. [DOI] [PubMed] [Google Scholar]
- Fraterman S, Zeiger U, Khurana TS, Wilm M, Rubinstein NA. Quantitative proteomics profiling of sarcomere associated proteins in limb and extraocular muscle allotypes. Mol Cell Proteomics. 2007b;6:728–737. doi: 10.1074/mcp.M600345-MCP200. [DOI] [PubMed] [Google Scholar]
- Frueh BR, Hayes A, Lynch GS, Williams DA. Contractile properties and temperature sensitivity of the extraocular muscles, the levator and superior rectus, of the rabbit. J Physiol. 1994;475:327–336. doi: 10.1113/jphysiol.1994.sp020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer MW, Stephenson DG. Total and sarcoplasmic reticulum calcium contents of skinned fibres from rat skeletal muscle. J Physiol. 1996;493:357–370. doi: 10.1113/jphysiol.1996.sp021388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Heizmann CW, Berchtold MW, Rowlerson AM. Correlation of parvalbumin concentration with relaxation speed in mammalian muscles. Proc Natl Acad Sci U S A. 1982;79:7243–7247. doi: 10.1073/pnas.79.23.7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoh JF, Hughes S. Myogenic and neurogenic regulation of myosin gene expression in cat jaw-closing muscles regenerating in fast and slow limb muscle beds. J Muscle Res Cell Motil. 1988;9:59–72. doi: 10.1007/BF01682148. [DOI] [PubMed] [Google Scholar]
- Jacoby J, Ko K. Sarcoplasmic reticulum fast CA(2+)-pump and myosin heavy chain expression in extraocular muscles. Invest Ophthalmol Vis Sci. 1993;34:2848–2858. [PubMed] [Google Scholar]
- Kaminski HJ, al-Hakim M, Leigh RJ, Katirji MB, Ruff RL. Extraocular muscles are spared in advanced Duchenne dystrophy. Ann Neurol. 1992;32:586–588. doi: 10.1002/ana.410320418. [DOI] [PubMed] [Google Scholar]
- Kato T. Uber die histologischen Untersuchungen der Augenmuskeln von Menschen und Saugetieren. Okajimas Folia Anat Jpn. 1938;16:14. [Google Scholar]
- Khanna S, Merriam AP, Gong B, Leahy P, Porter JD. Comprehensive expression profiling by muscle tissue class and identification of the molecular niche of extraocular muscle. Faseb J. 2003;17:1370–1372. doi: 10.1096/fj.02-1108fje. [DOI] [PubMed] [Google Scholar]
- Khurana TS, Prendergast RA, Alameddine HS, Tome FM, Fardeau M, Arahata K, Sugita H, Kunkel LM. Absence of extraocular muscle pathology in Duchenne’s muscular dystrophy: role for calcium homeostasis in extraocular muscle sparing. J Exp Med. 1995;182:467–475. doi: 10.1084/jem.182.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjellgren D, Ryan M, Ohlendieck K, Thornell LE, Pedrosa-Domellof F. Sarco(endo)plasmic reticulum Ca2+ ATPases (SERCA1 and -2) in human extraocular muscles. Invest Ophthalmol Vis Sci. 2003;44:5057–5062. doi: 10.1167/iovs.03-0218. [DOI] [PubMed] [Google Scholar]
- MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- Mayr R. Structure and distribution of fiber types in the external eye muscles of the rat. Tissue Cell. 1971;3:29. doi: 10.1016/s0040-8166(71)80045-9. [DOI] [PubMed] [Google Scholar]
- McLoon LK, Wirtschafter JD. Continuous myonuclear addition to single extraocular myofibers in uninjured adult rabbits. Muscle Nerve. 2002;25:348–358. doi: 10.1002/mus.10056. [DOI] [PubMed] [Google Scholar]
- Melzer W, Herrmann-Frank A, Luttgau HC. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim Biophys Acta. 1995;1241:59–116. doi: 10.1016/0304-4157(94)00014-5. [DOI] [PubMed] [Google Scholar]
- Morita T, Hussain D, Asahi M, Tsuda T, Kurzydlowski K, Toyoshima C, MacLennan DH. Interaction sites among phospholamban, sarcolipin, and the sarco(endo)plasmic reticulum Ca2+-ATPase. Biochemical and Biophysical Research Communications. 2008;369:188–194. doi: 10.1016/j.bbrc.2007.11.098. [DOI] [PubMed] [Google Scholar]
- Pacheco-Pinedo EC, Budak MT, Zeiger U, Jorgensen LH, Bogdanovich S, Schroder HD, Rubinstein NA, Khurana TS. Transcriptional and functional differences in stem cell populations isolated from extraocular and limb muscles. Physiol Genomics. 2009;37:35–42. doi: 10.1152/physiolgenomics.00051.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Park IY, Kim E, Youn B, Fields K, Dunker AK, Kang C. Comparing skeletal and cardiac calsequestrin structures and their calcium binding: a proposed mechanism for coupled calcium binding and protein polymerization. J Biol Chem. 2004;279:18026–18033. doi: 10.1074/jbc.M311553200. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JD, Baker RS, Ragusa RJ, Brueckner JK. Extraocular muscles: basic and clinical aspects of structure and function. Surv Ophthalmol. 1995;39:451–484. doi: 10.1016/s0039-6257(05)80055-4. [DOI] [PubMed] [Google Scholar]
- Porter JD, Israel S, Gong B, Merriam AP, Feuerman J, Khanna S, Kaminski HJ. Distinctive morphological and gene/protein expression signatures during myogenesis in novel cell lines from extraocular and hindlimb muscle. Physiol Genomics. 2006;24:264–275. doi: 10.1152/physiolgenomics.00234.2004. [DOI] [PubMed] [Google Scholar]
- Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, Leahy P, Li J, Andrade FH. Extraocular muscle is defined by a fundamentally distinct gene expression profile. Proc Natl Acad Sci U S A. 2001;98:12062–12067. doi: 10.1073/pnas.211257298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JD, Merriam AP, Leahy P, Gong B, Khanna S. Dissection of temporal gene expression signatures of affected and spared muscle groups in dystrophin-deficient (mdx) mice. Hum Mol Genet. 2003;12:1813–1821. doi: 10.1093/hmg/ddg197. [DOI] [PubMed] [Google Scholar]
- Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rome LC. Design and function of superfast muscles: new insights into the physiology of skeletal muscle. Annu Rev Physiol. 2006;68:193–221. doi: 10.1146/annurev.physiol.68.040104.105418. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Froesig C, Kiens B, Wojtaszewski FP, Jr, Richter EA. Effect of endurance exercise training on Ca2+ calmodulin-dependent protein kinase II expression and signalling in skeletal muscle of humans. 2007;583:785–795. doi: 10.1113/jphysiol.2007.138529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AJ, Kiens B, Richter EA. Ca2+-calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise. J Physiol. 2006;574:889–903. doi: 10.1113/jphysiol.2006.111757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi AE, Dirksen RT. Sarcoplasmic reticulum: the dynamic calcium governor of muscle. Muscle Nerve. 2006;33:715–731. doi: 10.1002/mus.20512. [DOI] [PubMed] [Google Scholar]
- Rubinstein NA, Holtzer H. Fast and slow muscles in tissue culture synthesise only fast myosin. Nature. 1979;280:323–325. doi: 10.1038/280323a0. [DOI] [PubMed] [Google Scholar]
- Simmerman HK, Collins JH, Theibert JL, Wegener AD, Jones LR. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. 1986;261:13333–13341. [PubMed] [Google Scholar]
- Slack JP, Grupp IL, Ferguson DG, Rosenthal N, Kranias EG. Ectopic expression of phospholamban in fast-twitch skeletal muscle alters sarcoplasmic reticulum Ca2+ transport and muscle relaxation. J Biol Chem. 1997;272:18862–18868. doi: 10.1074/jbc.272.30.18862. [DOI] [PubMed] [Google Scholar]
- Spencer RF, Porter JD. Biological organization of the extraocular muscles. Prog Brain Res. 2005;151:43–80. doi: 10.1016/S0079-6123(05)51002-1. [DOI] [PubMed] [Google Scholar]
- Traaseth NJ, Ha KN, Verardi R, Shi L, Buffy JJ, Masterson LR, Veglia G. Structural and dynamic basis of phospholamban and sarcolipin inhibition of Ca(2+)-ATPase. Biochemistry. 2008;47:3–13. doi: 10.1021/bi701668v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wescombe L, Lahooti H, Gopinath B, Wall JR. The cardiac calsequestrin gene (CASQ2) is up-regulated in the thyroid in patients with Graves’ ophthalmopathy - support for a role of autoimmunity against calsequestrin as the triggering event. Clin Endocrinol (Oxf) 2009 doi: 10.1111/j.1365-2265.2009.03753.x. in press. [DOI] [PubMed] [Google Scholar]
- Wirtschafter JD, Ferrington DA, McLoon LK. Continuous remodeling of adult extraocular muscles as an explanation for selective craniofacial vulnerability in oculopharyngeal muscular dystrophy. J Neuroophthalmol. 2004;24:62–67. doi: 10.1097/00041327-200403000-00013. [DOI] [PubMed] [Google Scholar]
- Zeiger U, Khurana TS. Distinctive Patterns of MicroRNA Expression in Extraocular Muscles. Physiol Genomics. 2010 doi: 10.1152/physiolgenomics.00169.2009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang M, Laties AM, Mitchell CH. Stimulation of P2X7 receptors elevates Ca2+ and kills retinal ganglion cells. Invest Ophthalmol Vis Sci. 2005;46:2183–2191. doi: 10.1167/iovs.05-0052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used for SYBR Green qPCR.
Ca2+ peak and cell size did not correlate when plotted for EOM and TA myotubes.
To identify the 14kDa band detected by the Pln-phospho-Ser16 antibody (A), the blot was stripped and reprobed with anti-phospholemman (B).