

Abstract
Effective therapeutic vaccines often require activation of T cell-mediated immunity. Robust T cell activation, including CD8 T cell responses, can be achieved using antibodies or antibody fragments to direct antigens of interest to professional antigen presenting cells. This approach represents an important advance in enhancing vaccine efficacy. Nucleic acid aptamers present a promising alternative to protein-based targeting approaches. We have selected aptamers that specifically bind the murine receptor, DEC205, a C-type lectin expressed predominantly on the surface of CD8α+ dendritic cells (DCs) that has been shown to be efficient at facilitating antigen crosspresentation and subsequent CD8+ T cell activation. Using a minimized aptamer conjugated to the model antigen ovalbumin (OVA), DEC205-targeted antigen crosspresentation was verified in vitro and in vivo by proliferation and cytokine production by primary murine CD8+ T cells expressing a T cell receptor specific for the major histocompatibility complex (MHC) I-restricted OVA257–264 peptide SIINFEKL. Compared with a nonspecific ribonucleic acid (RNA) of similar length, DEC205 aptamer-OVA-mediated antigen delivery stimulated strong proliferation and production of interferon (IFN)-γ and interleukin (IL)-2. The immune responses elicited by aptamer-OVA conjugates were sufficient to inhibit the growth of established OVA-expressing B16 tumor cells. Our results demonstrate a new application of aptamer technology for the development of effective T cell-mediated vaccines.
Introduction
Poor immunogenicity of conventional protein vaccines, in particular an inability to elicit robust T cell-mediated immunity, has limited their use as vaccines targeting diverse diseases including viral infections and cancers. One approach, which has recently been utilized to activate T cell responses, is targeting of antigen to dendritic cells (DCs), a cell type that is pivotal for eliciting T cell activation. Indeed, DC-targeted approaches have recently attracted significant research interest and are rapidly becoming important therapeutic approaches.1,2,3,4 DCs possess the capability of processing self and foreign antigens resulting in presentation of antigen to its cognate T cell receptor. Targeting antigen uptake to DCs via specific DC-enriched receptors has been shown to enhance antigen presentation on major histocompatibility complex (MHC) class I and II molecules by as much as 1,000-fold and 50-fold, respectively.5 Depending on the antigenic stimulus, DCs can induce tolerance or activate the immune system,6 making them important targets in the development of novel therapies for treating autoimmune diseases, viral infections, and cancer.
Targeting antigens to DCs most often involves coupling the antigen of interest to a delivery agent specific for a readily endocytosed cell surface receptor. Typically, targeted antigen delivery has made use of antibodies as the targeting agent. Nucleic acid aptamers, however, due to their unique chemical properties and low immunogenicity, provide a promising alternative to antibody-based antigen delivery.7,8 Aptamers are short ribonucleic acid (RNA) or deoxyribonucleic acid (DNA) sequences generated in vitro by an iterative selection process (systematic evolution of ligands by exponential enrichment (SELEX)) to bind with high affinity and specificity to a given target. Aptamers have demonstrated a wide range of flexibility, finding applications in flow cytometry staining, activating signaling pathways through cell surface receptor ligation, drug or siRNA delivery, blocking protein-protein interactions, and inhibiting enzyme function.7,8,9 Several aptamer-based therapeutics are currently undergoing clinical trials, with one already approved for clinical use.10
With the goal of utilizing aptamers for targeted antigen delivery, we have identified aptamers that recognize the murine receptor DEC205 (mDEC205) and have subsequently adapted them for use as a targeted antigen delivery agent. DEC205 is an excellent target receptor for this proof-of-principle study. This C-type lectin is expressed mainly by CD8α+ DCs, with lower expression by other cells in the hematopoietic lineage (> 1 log less).11 More importantly, studies targeting this receptor with anti-DEC205 antibodies fused to a variety of different antigens have demonstrated that receptor targeting leads to efficient crosspresentation of these molecular cargoes including cancer12,13 and HIV epitopes.14,15 Indeed, a cancer vaccine utilizing anti-human DEC205 antibody is already being tested in the clinic.16
A sequential selection strategy of recombinant mDEC205 protein followed by cells expressing mDEC205 was used to identify nuclease-stabilized RNA aptamers specific for the mDEC205 receptor. Minimized aptamers conjugated to the model chicken antigen ovalbumin (OVA) specifically bound mDEC205-expressing cell lines and primary cells. In the presence of the adjuvant polyinosinic:polycytidylic acid (pIC), incubation of anti-DEC205 aptamer:OVA conjugates with primary DCs in vitro resulted in crosspresentation of antigen, as determined by T cell proliferation assays and cytokine secretion. Fluorescently labeled aptamer localized to DEC205+CD11c+ cells in the spleen following systemic injection but only when displayed multivalently. Administration of multivalent, but not monovalent aptamer:OVA conjugates together with the adjuvant pIC resulted in T cell activation in vivo, as determined by proliferation and production of interferon (IFN)-γ and interleukin (IL)-2 by OVA-specific OT-I cells. Using a flank B16-OVA melanoma model, we found that only injection of multivalent DEC205 aptamer:OVA conjugates, not a multivalent nontargeting control aptamer:OVA conjugate, was able to elicit crosspresentation that was sufficient to cause regression of established B16-OVA tumors. Our results bode well for the future development of aptamer-based vaccines for eliciting immunological responses for the development of immune-targeted therapies such as antiviral and cancer vaccines.
Results
Identification of anti-mDEC205 aptamers
Pool enrichment. In order to identify aptamers that specifically recognized mDEC205 and were readily internalized by cells that naturally express this receptor, we employed a three-stage selection procedure (Figure 1a). Starting with an initial 2′-fluoro-pyrimidine-modified (2′F) RNA library encompassing ~1014 unique sequences, we performed three rounds of selection utilizing a recombinant mDEC205-hIgGFC fusion protein produced in Chinese hamster ovary (CHO) cells. Surprisingly, when we assayed each round of the selection against CHO-mDEC205 cells (a CHO cell line engineered to overexpress mDEC205) by flow cytometry, the Round 3 population already showed marked staining (Figure 1b; CHO-mDEC205). Importantly, no apparent staining was observed when the assay was repeated with the parental CHO cells (Figure 1b; CHO).
Figure 1.

Selection, cloning, and characterization of anti-mDEC205 aptamers. (a) Selection scheme. Three rounds of selection were performed against recombinant mDEC205-hIgG1FC fusion protein, with negative selection against hIgG1FC included in Rounds 2 and 3. Round 4 was performed against the surface of Chinese hamster ovary (CHO)/mDEC205, while Round 5 selected for sequences internalized by mouse bone marrow–derived dendritic cells (BMDCs). See Materials and Methods for full procedure. (b) Binding of selection rounds to surface-expressed mDEC205. Individual selection rounds were hybridized with a biotinylated oligonucleotide complementary to a portion of the aptamer pool's 3′ constant region, incubated with CHO/mDEC205 or CHO cells, counter-stained with SA-PE, and analyzed by flow cytometry. (c) Predicted secondary structure of minimized anti-mDEC205 aptamer, min.2. The conserved seven-base motif appearing in most clones—“UUCAUAA”—is highlighted in red. (d) Apparent binding constant for min.2 against mDEC205+ A20.Kb cells as determined by flow cytometry. Kd app = 23 ± 6 nmol/l. (e) Binding of minimized, Dy649-labeled anti-mDEC205 aptamer min.2 to primary mDEC205+ BMDCs. The identity of each histogram is as indicated in the key adjacent to the plot.
In order to further enrich the population for aptamers that bound to the receptor in the context of the cell surface, for Round 4, we switched to a cell-based selection utilizing CHO-mDEC205 cells. Prior to the selection, we performed a negative selection step on CHO cells to deplete the population of any nonspecific cell binders. Following positive selection on CHO-mDEC205 cells, bound RNA was recovered amplified and assayed by flow cytometry.
The Round 4 RNA showed no improvement in binding and uptake of the aptamer over Round 3 (Figure 1b), which prompted us to turn an additional round with further modification to our selection scheme. For this, we targeted bone marrow–derived DCs (BMDCs), a model for “classical” CD11c+ DCs, which express mDEC205 and are capable of antigen crosspresentation.17,18 Our goal was to ensure that our selected aptamers could bind their target receptor and be efficiently internalized. Therefore, following a 1-hour incubation with the aptamer library in media, the cells were stringently washed with buffer followed by acidic glycine to remove surface-bound molecules. Finally, the cells were trypsinized and then treated with an RNAse cocktail to ensure that only RNAs that had been internalized by the cells were recovered. As shown in Figure 1b, when assayed on CHO/mDEC205 cells, the Round 5 population showed improvement in aptamer binding and uptake over Round 4.
Clone analysis and minimization. Flow cytometric analysis of individual clones identified from Round 5 population revealed that nearly all isolated clones bound CHO-mDEC205, but not CHO cells (Supplementary Figure S1). Sequence analysis revealed a seven base sequence (5′-UUCAUAA-3′) that recurred in several clones (Figure 1c and Supplementary Figure S1b) that often formed a short loop when the constructs were folded. Interestingly, two clones that showed some of the best activity (Clones 1 and 15) possessed identical random regions, with the only difference being that the Clone 1 sequence lacked a significant portion of the 3′ constant region present in Clone 15 and the rest of the library. Using these clones as a starting point, we tested a series of successive truncations aimed at preserving this sequence but minimizing the overall size of the aptamer. This resulted in the identification of a minimized sequence composed of a 42-nucleotide aptamer core, “min.2” which performed better than the bulk Round 5 pool (Figure 1c and Supplementary Figure S2).
The minimized aptamer, min.2, was chemically synthesized bearing a 3′ inverted dT for added serum stability and a 5′ thiol to facilitate subsequent chemical conjugations. Using biotin maleimide, we generated biotinylated minimized aptamers and verified the specificity of this construct for mDEC205 using siRNA to knock down the expression of mDEC205 on CHO/mDEC205 cells by flow cytometry using streptavidin (SA)-phycoerythrin (PE) (Supplementary Figure S3). Specificity for DEC205 was further confirmed using A20.Kb cells, a B cell lymphoma line that naturally expresses mDEC205, as well as A20.Kb-mDEC205 cells, a variant of A20.Kb cells engineered to express higher levels of mDEC205 (Supplementary Figure S4). Using an Alexa Fluor 488-maleimde conjugate of min.2, we measured the apparent dissociation constant of this aptamer for cell-surface mDEC205 on A20.Kb cells (23 ± 6 nmol/l; Figure 1d). Consistent with these results, when we incubated a Dy649-labeled minimized aptamer with CD11c+ BMDCs, significant cell staining was observed (Figure 1e; red). Importantly, a nontargeting control aptamer showed minimal staining under these same conditions (Figure 1e; green).
Using this minimized construct, we performed microscopy studies to confirm that receptor binding was accompanied by internalization. Experiments performed using both CHO-DEC205 cells (Figure 2a) as well as CD11c+ BMDCs (Figure 2b) displayed punctate staining following incubation with the labeled aptamer, but not a nonfunctional sequence of similar size (cntrl.36). To further confirm specificity, we performed colocalization studies using CHO-DEC205 cells. When coincubated with cells, both min.2 and an anti-DEC205 antibody colocalized to the same intracellular compartments (Figure 2c). Consistent with published studies, the DEC205 antibody showed minimal colocalization with early endosomes and more extensive localization with lysosomes,19,20 with min.2 exhibiting similar intracellular localization (Figure 2d,e).
Figure 2.

Min.2, but not cntrl.36, binds to and is internalized by DEC205+ cells. Confocal microscopy images of Chinese hamster ovary (CHO)-mDEC205 cells (a, c, d) or primary mDEC205+ bone marrow–derived dendritic cells (BMDCs) (b) following incubation with fluorescently labeled min.2 or cntrl.36. (a, b) Cells were incubated with 200 nmol/l Dy649-labeled min.2 or cntrl.36 (red) and incubated for 1 hour at 37 °C to allow endocytosis, at which time the cells were stringently washed and the cell surface was stained with (a) DEC205 or (b) CD11c antibodies labeled with AF488 (green). Panel labeled “min.2” (inset) is a magnification of the white boxed region in (b). (c) Colocalization of AF488-labeled min.2 (green) and AF647 labeled anti-DEC205 antibody (clone 205yekta; red)). Colocalization of AF488-labeled min.2 (top) or AF488-labeled anti-DEC205 antibody (bottom) with (d) RFP-labeled Rab5a (early endosome), or (e) RFP-labeled LAMP-1 (lysosome). (f–i) Binding and uptake of large (~300 kDa) molecule cargoes by BMDCs. Flow cytometry was used to assess binding and internalization of aptamer-SA:PE conjugates. Biotinylated min.2 or cntrl.36 were precomplexed with SA:PE and subsequently incubated with BMDCs at the indicated temperatures. Following a 1-hour incubation, cells were washed and then treated with an RNAse cocktail as indicated.
Because antigen delivery requires uptake of cargoes significantly larger than small molecule fluorophores, we generated aptamer-PE conjugates (a ~3 kDa protein) using a biotinylated aptamer and SA-PE and assessed binding and cellular uptake of these large cargoes by BMDCs. The min.2-PE conjugate showed significant staining when incubated with CD11c+ BMDCs at 4 °C, which was reduced to near-background levels when treated with an RNAse cocktail which destroys even 2′F RNA (Figure 2f).21 However, when the same experiment was performed at 37 °C, the bulk of the fluorescence signal remained intact (Figure 2g) indicating that the aptamer and cargo had been internalized and thus protected from digestion. Similar experiments conducted using a nontargeting aptamer control yielded only background fluorescence when cells were incubated at 4 °C (Figure 2h), with slightly higher background levels observed at 37 °C (Figure 2i). Taken as a whole, these data further demonstrate the functionality of min.2 for binding DEC205 and enabling internalization of large molecular cargoes into target cells.
Enhancing crosspresentation with anti-mDEC205 aptamers
Aptamer-OVA conjugation. Antibodies that target DEC205 have been shown to greatly enhance the crosspresentation of antigens.5,6,22 To this end, we chose to utilize the well-characterized OVA system as a model to investigate the ability of min.2 to deliver cargo for crosspresentation.
Using the 5′-thiol-modified min.2, we generated aptamer-OVA conjugates using the heterobifunctional crosslinker N-[γ-maleimidobutyryloxy]sulfosuccinimide ester (sulfo-GMBS; Supplementary Figure S5). As a control to ensure specificity, we also generated OVA conjugates using a nonfunctional control sequence, cntrl.36. Conjugates were subsequently purified by anion exchange high-performance liquid chromatography with the protein conjugate eluting in two distinct peaks (Fraction #1; 1:1 aptamer:OVA and Fraction #2; 2:1 aptamer:OVA; Supplementary Figure S5). Subsequent experiments were performed using the first fraction, which consisted of a single aptamer conjugated to OVA, as determined by the ratio of the UV absorbance at 260 and 280 nm. Finally, to ensure that the aptamer-protein conjugates remained functional, we generated fluorescently labeled conjugates and confirmed binding to CHO/mDEC205 cells (Supplementary Figure S5).
Min.2-OVA is functional for crosspresentation. We initially assessed the ability of our anti-mDEC205 aptamer (min.2) to enhance delivery and crosspresentation of protein antigens in vitro using murine CD11c+ splenocytes, a subset of which are CD8α+DEC205+.22 In short, purified DCs were incubated with the adjuvants lipopolysaccharide and/or pIC and either 10 nmol/l aptamer-OVA conjugate (min.2:OVA or cntrl.36:OVA) or 10 nmol/l antibody-OVA fusion (NLDC145:OVA or GL117:OVA, a rat IgG2a anti-Escherichia coli β-galactosidase antibody which served as an isotype control) for 24 hours at which time the cells were washed and incubated with carboxyfluorescein succinimidyl ester (CFSE)-labeled OT-I cells. T cell activation was assessed 3 days later by flow cytometry. As shown in Figure 3a, targeting antigens with either the mDEC205-specific aptamer-OVA conjugate (min.2:OVA) or antibody-OVA chimera (NLDC145:OVA) induced a strong activation response, with most cells (>90%) dividing after 3 days, whereas incubations performed with the nonspecific RNA conjugate (cntrl.36:OVA) or isotype antibody chimera (rIgG2a:OVA) resulted in minimal T cell division. Importantly, when cells were treated with aptamer alone (data not shown) or a combination of aptamer plus OVA (not conjugated; min.2 + OVA), the observed levels of proliferation were similar to that of the Dulbecco's phosphate-buffered saline (DPBS)-treated control (Figure 3a; DPBS). Consistent with the observed proliferation, only cells which received the min.2:OVA or NLDC145:OVA conjugates upregulated the surface activation marker, CD25 (Figure 3b) and produced the cytokines IFN-γ (Figure 3c) and IL-2 (Figure 3d). Similar levels of activation were observed when cells were treated with min.2:OVA in the presence of pIC only, or following treatment with the SIINFEKL peptide (data not shown). When we assayed IL-2 and IFN-γ in the culture supernatant by enzyme-linked immunosorbent assay, the antibody-targeted and aptamer-targeted OVA displayed slightly different responses. Both treatments induced secretion of IFN-γ and IL-2 by OT-I cells at 24 and 48 hours (Figure 3e,f). However, incubation with min2:OVA resulted in approximately twofold greater level of IFN-γ at 24 hours and significantly lower amounts of IL-2 compared with NLDC145:OVA. When we performed similar experiments to determine the dose response of min.2:OVA, we observed significant crosspresentation at concentrations down to 1 nmol/l (Figure 3g).
Figure 3.

Min.2 is functional for crosspresentation in vitro. (a) Proliferation of carboxyfluorescein succinimidyl ester (CFSE)-labeled OT-I cells following incubation of monovalent aptamer:OVA conjugates or controls with primary dendritic cells (DCs). CD11c+ DCs were isolated from the spleens of C57BL/6 mice. After isolation, 4 × 105 DCs were incubated with pIC plus lipopolysaccharide for 4 hours (except Dulbecco's phosphate-buffered saline labeled), followed by overnight incubation with 10 nmol/l conjugate or controls for 16–20 hours at 37 °C. The next day, a single-cell suspension of 1 × 105 CFSE-labeled OT-I cells was added, and the cells were incubated for an additional 3 days at 37 °C, washed with flow cytometry buffer, stained with 4′,6-diamidino-2-phenylindole and antibodies against CD8α and TCR-β, and analyzed by flow cytometry. See Materials and Methods for full procedure. (b–d) Intracellular cytokine staining of OT-I cells in (a). Cells were treated with 10 μg/ml brefeldin A, incubated for 5 hours at 37 °C, stained with antibodies against CD25, interleukin (IL)-2, and interferon (IFN)-γ, and analyzed by flow cytometry. OT-I responses following incubation with min.2:OVA versus cntrl.36:OVA and NLDC145:OVA versus IgG2a:OVA were compared by student's t-test (****P < 0.0001). Data shown are representative of three experiments. Secretion of cytokines (e) IFN-γ and (f) IL-2 into the media by cells following OT-I proliferation assays performed in the presence of 10 nmol/l aptamer:OVA or antibody:OVA conjugates, free OVA (OVA) or treatment with 100 nmol/l SIINFEKL peptide. (g) Dose–response analysis of cells to aptamer or control treatments. Proliferation of CFSE-labeled OT-I cells was monitored following incubation with primary DCs with a graded dose of aptamer (min.2 or cntrl.36) or antibody (rIgG2a or NLDC145; where rIgG2a is an isotype control) OVA conjugates. The concentration of conjugate used is indicated. OVA, ovalbumin.
To ensure that our aptamers and aptamer-conjugates did not provoke any adverse effects on cells, we monitored DCs treated for 24 hours with our aptamer-OVA conjugates for upregulation of the activation markers CD80, CD86, CD40, and MHC II. Importantly, DCs incubated with the aptamer or control conjugates in the absence of adjuvants were not activated when compared with DCs given DBPS alone (Supplementary Figure S6). Only samples treated with a mixture of lipopolysaccharide and pIC upregulated these activation markers. Similarly, when we monitored cytokine secretion into the media by DCs incubated with aptamer:OVA, IFN-γ, IL-6, IL-10, IL-12, macrophage chemoattractant protein 1, and tumor necrosis factor-α, all remained at background levels (Supplementary Figure S7).
Multivalent aptamer conjugates are required for efficient in vivo targeting. Systemic injection of DEC205-specific antibodies results in uptake by splenic DEC205+CD11c+CD8α+ DCs.23 We therefore used a similar approach to assess the ability of our min.2 aptamer to target this cell subset in vivo. Using a biotinylated variant of min.2 or cntrl.36, we generated aptamer:SA-AF647 conjugates and injected ~20 µg of each construct into mice. Interestingly, when conjugates were generated at a 1:1 aptamer to SA ratio, conjugate uptake was not detectable (data not shown). However, when we saturated SA (a tetrameric protein) with four aptamers, uptake of the multivalent aptamer-conjugates by splenic DEC205+CD11c+CD8α+ DCs was observed (Figure 4a). Importantly, no uptake was seen when similar experiments were performed using a nontargeted aptamer control, ctrl.36.
Figure 4.
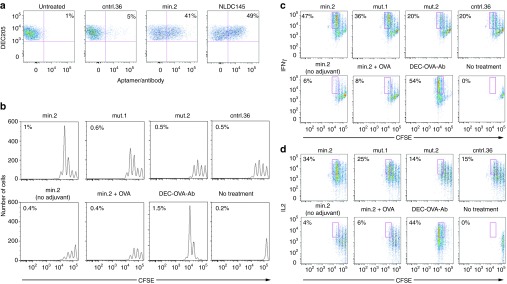
Injection of multivalent min.2 results in uptake by DEC205+ dendritic cells that is functional for antigen crosspresentation. (a) Uptake of multivalent aptamer-SA conjugates by splenic dendritic cells (DCs). C57BL/6 mice were injected with 20 µg fluorescently labeled multimerized cntrl.36, min.2 or NLDC145 antibody. Twenty-four hours later, spleens were harvested, and aptamer uptake by CD11c+DEC205+ cells was determined by flow cytometry. The percentage of CD11c+DEC205+ cells that have taken up fluorescent aptamer/antibody is reported in each plot. (b) Activation of adoptively transferred OT-I cells following treatment with multivalent aptamer:OVA conjugates. 106 OT-I cells were labeled with carboxyfluorescein succinimidyl ester and transferred into congenic B6.SJL-ptprc mice. Twenty-four hours later, mice were injected with 20 µg of aptamer:OVA plus 25 µg pIC. Three days later, OT-I cell proliferation was determined by flow cytometry. The percentage of TCR-β+CD45.2+ cells present in the total splenic population (OT-I cells) is reported on each plot. (c, d) OT-I cells in (b) were analyzed for intracellular expression of interferon (IFN)-γ (c) and interleukin (IL)-2 (d). The percentage of OT-I cells that have undergone three or more divisions is reported in each dot plot. Data shown are representative of three experiments. OVA, ovalbumin.
To better assess the role of valency on binding and uptake, we performed follow-up experiments in vitro using CHO-DEC205 cells. Dye-labeled aptamers or aptamer-SA conjugates generated at a 1:1 or 4:1 aptamer:SA tetramer ratio were incubated at increasing concentrations with CHO-DEC205 cells for 1 hour, after which the cells were washed, and the level of cell staining was determined by flow cytometry. The multivalent conjugates displayed an approximately threefold lower apparent binding constant (8.0 nmol/l) when compared to the aptamer alone or the 1:1 aptamer-SA conjugate (21.5 and 22.7 nmol/l; Supplementary Figure S8). Gel analysis of these SA conjugates suggested that while the 1:1 conjugates were populated by a mixture of species bearing 0, 1, and 2 aptamers per SA (consistent with a Poisson distribution), at a 4:1 ratio, the population was dominated by a mixture of species bearing mostly 3 and 4 aptamers per protein (Supplementary Figure S9). Similar results were observed when experiments were performed on A20 cells (data not shown) or when experiments were performed using a multivalent min.2:OVA conjugate labeled with BOD1PY-FL (Kd app = 15 nmol/l; Supplementary Figure S11).
Multivalent min.2:OVA conjugates are functional for crosspresentation in vivo. Consistent with our in vivo targeting results, injection of 20 µg monovalent min.2:OVA (conjugated at a 1:1 aptamer:OVA ratio) failed to elicit T cell proliferation (data not shown). Drawing from our successful targeting experiments (Figure 4a), we reengineered our aptamer-OVA conjugates utilizing the longer heterobifunctional linker SM(PEG)12 which allowed for the addition of multiple aptamers to each OVA molecule (>4 aptamers per OVA; Supplementary Figure S10). Using this approach, we synthesized and tested conjugates bearing 3–4 aptamers per molecule of OVA. The multivalent conjugates retained the ability to bind DEC205+ cells with an apparent Kd of ~15 nmol/l (Supplementary Figure S11), but were less effective at activating OT-I cells: a ~10-fold higher concentration was needed to achieve similar levels of activation when compared to the monovalent conjugates generated with succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Supplementary Figure S12).
As an additional set of controls, we also generated and tested OVA conjugates of two mutants of min.2 which largely maintained the sequence composition of the parent aptamer but disrupted the predicted folded structure or the order of nucleotides in the conserved heptamer (Supplementary Figure S13). Prior to conjugation to OVA for in vivo experiments, we used biotinylated variants of these additional control aptamers complexed with AF647-labeled SA at a ratio of 4:1 to assess the relative binding affinities of these molecules on CHO-DEC205 cells. As expected, the highest amount of aptamer uptake occurred following incubation with multimerized min.2, with lower levels of uptake (9-, 15-, and 100-fold) observed for the mutants mut.1, mut.2 and the control, cntrl.36, respectively (Supplementary Figure S13).
Using multivalent OVA conjugates bearing either min.2, cntrl.36 or mutant aptamers mut.1 and mut.2, at an average ratio of 3 aptamers per molecule of OVA, we performed in vivo activation experiments in C57BL/6 mice injected with CFSE-labeled OT-I cells. Min.2 targeting elicited approximately two- to sixfold more OT-I proliferation when compared with cntrl.36:OVA or other control treatments (Figure 4b and Supplementary Figure S14). Cytokine production was highest in animals that received min.2:OVA and was similar to the level observed following treatment with NLDC145:OVA (Figure 4c,d). We note that injection with ctrl.36:OVA, mut.1, and mut.2 resulted in some proliferation and cytokine production by OT-I cells. However, the overall level of activation was less than that observed following min.2:OVA injection, and similar to the responses observed following min.2 injection in the absence of adjuvant (Figure 4b–d; min.2 (no adjuvant)). Furthermore, as observed with our in vitro experiments (Figure 3), min.2:OVA must be present as a conjugate to elicit activity—when min.2 was admixed with OVA and coinjected (min.2+OVA), minimal OT-I activation was observed. Other control treatments (pIC alone, Hank's balanced salt solution (HBSS), no injection or OVA alone) did not stimulate responses over background (Supplementary Figure S14 and data not shown).
Finally, to further demonstrate the functionality of the min.2:OVA conjugates, we assessed their ability to activate T cells and induce tumor regression using a B16-flank tumor model. Mice were given B16 cells expressing OVA (M05) on one flank, and palpable tumors developed after 5 days. On day 4, post-tumor injection, OT-I cells were transferred into mice and 1 day later mice were injected with multivalent aptamer:OVA (min.2 or cntrl.36) or NLDC145:OVA plus pIC. As seen in Figure 5, M05 tumor growth was observed in mice that were untreated (OT-I only) or injected with cntrl.36:OVA conjugate. Initial tumor growth was observed in mice given either min.2 aptamer:OVA or NLDC145:OVA. However, for both of these treatment groups, retardation of M05 tumor growth was evident by day 9 (4 days postaptamer/antibody injection), and inhibition of tumor growth was observed through day 15. At this time point, the experiment was terminated due to the large size of the tumors in the control groups. B16-OVA tumors in mice that received cntrl.36:OVA were significantly larger when compared with those in mice injected with min.2:OVA (P < 0.02) or NLDC145:OVA (P < 0.05; analysis by two-way analysis of variance). When similar experiments were performed on mice given parental B16 tumors (no OVA) on one flank, tumor growth was rapid, and progressed with comparable kinetics across all treatment groups (data not shown).
Figure 5.

Injection of min.2 inhibits growth of established M05 flank tumors. Mice were injected s.c. with 106 M05 cells in one flank. Four days later, mice received 106 OT-I cells, and the following day mice were injected with 10 µg aptamer:OVA (min.2 or cntrl.36) or 5 µg NLDC145:OVA plus 25 µg pIC. Tumor growth was monitored and tumor area ± SEM is shown. Statistical significance calculated using two-way analysis of variance. Comparison of cntrl.36:OVA versus min.2:OVA, P < 0.02 (*) and cntrl.36:OVA versus NLDC145:OVA, P < 0.05 (*). Treatment groups were min.2:OVA (green), ctrl.36:OVA (black), NLDC145:OVA (blue), and no treatment (red). Each group contained 5 mice. Data shown are representative of two separate experiments. OVA, ovalbumin.
Discussion
Strategies to target DCs and enhance or control the presentation of antigens by these cells are already finding their way into clinical trials.16,24 Of particular interest are approaches in which antigens are specifically directed to bind DC cell surface receptors using targeting agents. Receptors such as DEC205, the mannose receptor, CD207, DC-SIGN, Clec9a, DCIR2, and others have been shown to be good molecular targets for directing cargoes to intracellular pathways, which can lead to enhanced antigen presentation.22,23,24,25,26 Interestingly, different receptors differ in their ability to stimulate cellular or humoral immunity. This is likely due to factors including cellular expression patterns of the various receptors and to their relative ability to deliver antigenic cargoes to MHC I and II following internalization. For example, in mice, delivery of antigen via the DCIR2 receptor promotes antigen presentation on MHC II, whereas targeting the DEC205 receptor favors MHC I presentation.22 Moreover, costimulatory signals can be used to affect downstream T cell responses. For example, codelivery of adjuvants such as pIC or anti-CD40 antibodies with antigens conjugated to DEC205-specific antibody leads to DC maturation and the production of sustained CD8+ T cell responses.5,23 However, in the absence of such additional signals, CD8+ proliferation is followed by deletion, leading to tolerance.6 Thus, these targeted approaches offer the potential to finely tune the immunological outcome.
Aptamers that are specific for surface receptors have previously been used for targeted delivery of molecular cargoes, including small molecule drugs,27 toxins and other proteins,28,29 as well as nanoparticles.30 And while aptamers have been developed to target some immunologically relevant receptors and modulate immune responses,9,31,32 aptamers have not yet been used for targeted delivery of antigens.
To test the potential to utilize aptamers in this capacity, we generated nuclease-stabilized aptamers that targeted mDEC205. Aptamers were generated using a modification of the traditional SELEX procedure, which combines recombinant protein with a “functional” selection on cells—a strategy we have previously employed for the selection of aptamers which target the human transferrin receptor.33 DEC-specific activity was apparent in the selected population after only three rounds of selection. This was likely due to the sensitivity of flow cytometry when compared to more traditional metrics for monitoring aptamer library function, such as filter binding, which rely on a significant fraction of the population binding to the target to yield a positive signal. While the function of DEC205 remains unknown, it has been reported to be a receptor for apoptotic and necrotic cells34 and phosphorothioated oligonucleotides35 suggesting that DEC205 may simply be a “good” SELEX target.
Using a minimized variant, min.2, which is readily amenable to chemical synthesis, we generated aptamer:OVA conjugates and assessed their ability to specially target CD11C+DEC205+ DCs and enhance crosspresentation to CD8+ OT-I T cells in vitro and in vivo.
In vitro, in the presence of adjuvant, min.2:OVA was efficient at eliciting T cell activation at concentrations as low as 1 nmol/l, with no detectable activation observed when similar experiments were performed with a nontargeting oligonucleotide control (ctrl.36:OVA). T cell proliferation was accompanied by cytokine secretion (IL-2, IFN-γ) as determined by intracellular staining for flow cytometry and enzyme-linked immunosorbent assay. It is interesting to compare our in vitro aptamer-targeted results with those of the anti-DEC205 antibody, NLDC145. In dose–response assays, the antibody performed at least 10-fold better than the aptamer showing almost complete activation of the T cell population at 100 pmol/l. This fact is perhaps not too surprising considering that the observed binding constant of our monovalent aptamer is ~20 nmol/l, and thus appreciable aptamer binding and subsequent uptake would not be expected at concentrations much lower than what we observed. While the binding affinity of NLDC145 has not been reported, comparable antibodies that target human DEC205 have reported dissociation constants of 100 pmol/l to 1 nmol/l, 10–100-fold lower than our aptamer.15 It is also interesting to note that although intracellular staining for flow cytometry experiments showed similar levels of IL-2 or IFN-γ production for aptamer and antibody-mediated delivery (Figure 3b–d), analysis of the culture supernatants by enzyme-linked immunosorbent assay revealed much lower levels of IL-2 for DCs incubated with min2:OVA conjugates (Figure 3e,f). This apparent discrepancy was not due to differences in cell health, as cell viability was similar between aptamer- and antibody-treated samples (data not shown). Altered magnitude or kinetics of IL-2 secretion may be due to differences in potency of T cells in response to antibody- versus aptamer-mediated antigen uptake and/or presentation. IL-2 was detected, albeit at reduced levels, 24 hours following OT-I incubation with DCs cultured with min.2:OVA. IL-2 is utilized as a T cell growth factor, and the lower levels of IL-2 detected in the culture supernatants, when compared with intracellular staining for flow cytometry, coincide with T cell expression of IL-2 receptor.36
Our initial attempts to use min2:OVA conjugates to prime T cell responses in vivo failed to yield positive results (data not shown). This prompted us to assess the localization of our aptamers following systemic injection. Interestingly, although our min.2 aptamer showed robust cell staining in vitro (Figures 1e and 2 and Supplementary Figures S1–S4), when the same dye-labeled aptamers were injected i.v., subsequent ex vivo analysis of DEC205+CD11c+CD8α+ DCs by flow cytometry failed to demonstrate appreciable cell staining (data not shown). Because of their small size, aptamers are rapidly cleared from circulation following injection with a t½ = ~10 minutes.37 Additionally, when compared to an antibody which bears multiple fluorophores (typically 3–4 per Ab), aptamers which bear a single fluorophore yield a corresponding lower signal. To this end, we utilized a biotinylated aptamer to generate aptamer-SA conjugates labeled with three fluorescent dyes (AF647) to increase both size and brightness. At a ratio of 1 aptamer to 1 SA tetramer, this conjugate more closely mimics the size of our aptamer:OVA conjugate (75 kDa for the SA conjugate and 60 kDa for the OVA conjugates). Interestingly, while in vivo uptake of conjugates generated at a 1:1 aptamer to protein ratio by splenic DEC205+CD11c+CD8α+ DCs remained undetectable, multivalent aptamer constructs assembled using a ratio of 4 aptamers to 1 SA tetramer stained cells at a similar level observed for an antibody (NLDC145) control (Figure 4a), although aptamer staining required 10-fold more material than antibody staining suggesting that the in vivo targeting by the aptamer conjugates is less efficient than that for the antibody when delivered i.v. Using an in vitro assay, we assessed the effect of valency on conjugate binding and uptake by flow cytometry. Multivalent conjugates displayed apparent binding constants approximately two- to threefold lower than monomeric conjugates or aptamer only (Figure 1d; Supplementary Figures S8 and S11). Thus, while this increased affinity may account in part for the observed in vivo efficacy, it seems likely that the observed results are due to multiple effects including affinity, clearance time and as yet unknown interactions.
In light of the apparent requirement for multivalency, we developed an alternate approach to generate multivalent aptamer:OVA conjugates. Initial attempts using the heterobifunctional crosslinker GMBS failed to efficiently yield conjugates bearing much more than a single aptamer (data not shown). On the other hand, using the longer linker SM(PEG)12, we found that as many as 4 aptamers could be readily appended to OVA following activation. Using this approach, we generated aptamer:OVA conjugates at a 3:1 aptamer:OVA ratio, which resulted in the generation of conjugates with ~3–4 aptamers per OVA as determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) analysis (Supplementary Figure S10). Although binding of multivalent aptamer conjugates in vitro was enhanced when compared with the monovalent conjugates (Supplementary Figures S8 and S11), our multivalent OVA conjugates were less effective at antigen presentation (Supplementary Figure S12). Why aptamer multimerization negatively impacted crosspresentation is not known but may be the result of alterations in intracellular trafficking following uptake or simply due to the higher degree of surface modification of OVA rendering it more difficult for the cell to process. Regardless, subsequent in vivo experiments performed using multivalent min.2:OVA conjugates in the presence of adjuvant resulted in OT-I activation as determined by CFSE dilution and cytokine production (Figure 4 and Supplementary Figure S14). Importantly, control experiments performed using a nontargeting control conjugate (cntrl.36:OVA) or two different mutants of min.2 (mut.1 and mut.2) that show diminished binding capacity failed to lead to significant T cell proliferation (Figure 4b–d). A lack of response was similarly observed with mice that were treated with min.2 admixed with OVA, confirming the necessity for the aptamer and cargo to be physically linked.
Steinman and coworkers have previously demonstrated that OVA delivery using the anti-DEC205 antibody NLDC145 still resulted in T cell proliferation in the absence of an added adjuvant. However, proliferation was not accompanied by activation—T cells did not produce cytokines and were eventually deleted.6 Interestingly, when we performed experiments using min2:OVA in the absence of the adjuvant pIC, very little proliferation above the nontargeted controls was observed suggesting a potential difference in the pathway or the potency of the immune response when antigen is delivered using the min.2 aptamer versus an NLDC145 antibody (Figure 4b).
Finally, using a M05 flank tumor model, we found that growth of established tumors was slowed following injection of min.2-OVA or NLDC145:OVA plus pIC (Figure 5). This OT-I-mediated response indicated that injection of min.2:OVA resulted in antigen crosspresentation sufficient to elicit tumoricidal CD8+ T cells. The response is equivalent to that observed following treatment with equivalent amounts of NLDC145:OVA. Interestingly, both our in vitro and in vivo data showed that treatment with NLDC145:OVA results in increased proliferation of OT-I cells and higher levels of IL-2 and IFN-γ when compared with min.2:OVA. While min.2:OVA injection elicited reduced proliferation and production of these cytokines, these responses were sufficient to stimulate OT-I cells that were functionally competent for inhibiting tumor growth, similarly to mice given NLDC145:OVA. Therefore, it seems that the quality of the T cell response required for controlling tumor growth was comparable, although a more thorough immunological analysis will be necessary to substantiate this observation. We also note that although our nontargeted control conjugate ctrl36:OVA elicited some OT-I activation (Figures 4 and 5), these responses were not sufficient to inhibit tumor growth. These data suggest that other, as yet unidentified, aspects of T cell function differ significantly when comparing min.2:OVA and ctrl36:OVA in vivo responses.
In summary, our work demonstrates a new approach for targeting antigens to specific DC receptors using nucleic acid aptamers. Our min.2 aptamer targets mDEC205+ cells and is functional for delivering antigenic cargo for crosspriming in vitro and in vivo. We have demonstrated the potential application of this molecule for enhancing T cell-mediated immunity as an antitumor therapy. This DC-targeted aptamer could also be developed as a vaccine platform, or conversely for ameliorating T cell responses by inducing tolerance for treating autoimmunity. When one considers the potential advantages of aptamer technology over protein (antibody) production, aptamers should prove to be a novel powerful tool for use in the clinic.
Materials and Methods
Mice. C57BL/6 and OT-I mice were purchased from Taconic (Hudson, NY). OT-I mice were on a RAG1−/− background. B6.SJL-Ptprc were purchased from Jackson Laboratories (Bar Harbor, ME). All mice used were 6–12 weeks old. All experiments were performed in accordance with Institutional Animal Care and Use Committee regulations and protocols were approved by the Einstein Institutional Animal Care and Use Committee.
Protein expression and purification. CHO cells were cultured in Dulbecco's modified Eagle medium (Life Technologies, Carlsbad, CA) supplemented with 5–10% fetal bovine serum (FBS) or 5% Ultra-Low IgG FBS supplemented with antibiotic-antimycotic, and nonessential amino acids (all from Life Technologies).
CHO cells stably expressing an open reading frame for the full-length mouse DEC205 (CHO/mDEC205) were generated as previously described.15 Similarly, the extracellular domain (residues 1–1,667) of mDEC205 was fused in-frame with the hIgG1 FC domain. The mDEC205/hIgG1FC construct was inserted into the pCMV expression vector (Clontech, Mountain View, CA) and transfected into CHO cells to generate stably expressing CHO/mDEC205/hIgG1FC cells. Culture supernatant from these cells was used to purify the mDEC205/hIgG1FC fusion protein by affinity to Protein A Sepharose (GE Healthcare, Pittsburgh, PA).
Aptamer selection. The sequence of the N50 library used for selection against mDEC205 was: 5′GGGAGGTGAATGGTTCTACGAT-N50-TTACATGCGAGATGACCACGTAATTGAATTAAATGCCCGCCATGACCAG-3′. The single-stranded DNA library was synthesized such that N regions contained an equal probability of containing A, T, G, or C, as previously described.38 Following deprotection, the library was gel purified by denaturing (7 mol/l urea) gel electrophoresis on an 8% polyacrylamide gel. The single-stranded DNA library was amplified by polymerase chain reaction (PCR) to generate a double-stranded DNA bearing a T7 promoter and transcribed in vitro using the Y639F mutant of T7 RNA polymerase39,40 and 2′-fluoro (2′-F) pyrimidines. Following transcription, the RNA was purified on a denaturing (7 mol/l urea) 8% polyacrylamide gel.
For Round 1, we utilized ~3 copies of an RNA library encompassing ~1 × 1014 sequences (~20 µg). The library was diluted in 20 µl HBSS (Life Technologies) and then thermally equilibrated by incubation at 70 °C for 5 minutes, followed by room temperature for 15 minutes prior to addition to the immobilized protein target (mDEC205/hIgG1FC).
Immobilized protein was prepared by incubating 25 pmol mDEC205/hIgG1FC with 25 μl of washed Dynabeads Magnetic Protein G resin in 200 μl of washing/blocking buffer (0.1 mol/l NaPO4 pH 8.2, 0.01% Tween 20). This mixture was incubated 30 minutes at room temperature with rotation and then washed three times with 200 μl DPBS without Ca2+ or Mg2+ (Life Technologies). After the third wash, buffer was removed and replaced with the RNA library in a final volume of 20 µl. The resin was then incubated for an additional 30 minutes at room temperature with rotation, followed by three washes with 200 μl HBSS. After the third wash, protein and RNA were eluted by a 5-minute incubation with 20 μl 200 mmol/l glycine, pH 2.5. The eluent was combined with 400 μl 0.3 mol/l NaOAc, containing 4 μg glycogen and the RNA recovered by ethanol precipitation.
The recovered RNA was reverse transcribed to single-stranded DNA (ssDNA) using Moloney murine leukemia virus reverse transcriptase (M-MLV RT, Life Technologies). The ssDNA was amplified by PCR using Taq DNA polymerase, and PCR product was transcribed into 2′-F-Y-RNA by a modified T7 RNA polymerase (Y639F39 and P266L41 mutations). For subsequent rounds, 1 μg of the previous round's selection product was prepared as above for Round 1.
Prior to Rounds 2 and 3, we used a negative selection step to deplete resin-binding aptamers and aptamers that bound the FC region from the population. The negative selection step was performed by incubating the library with 20 μl Dynabeads Protein G resin prepared as above, but substituting 50 pmol human IgG1 FC region (hIgG1FC) for mDEC205/hIgG1FC fusion protein. Protein G resin loaded with hIgG1FC was then incubated with the prepared RNA for 30 minutes at room temperature with rotation, after which the resin was spun down, and the supernatant was removed and added to 20 μl Dynabeads Protein G resin loaded with 25 pmol mDEC205/hIgG1FC, as above. RNA was recovered, reverse transcribed, amplified, and transcribed as described above.
Round 4 was performed using CHO-mDEC205. Prior to the positive selection, we performed a negative selection on the parental CHO cells. In short, 5 μg of RNA was combined with a twofold molar excess of reverse primer used for amplification (T50R). The mixture was diluted to a final volume of 50 μl in HBSS, denatured for 5 minutes at 70 °C, and allowed to anneal for 15 minutes at room temperature. Reannealed RNA was added to ~1 × 105 CHO cells in a 24-well plate containing 450 μl media and incubated for 30 minutes at 37 °C. Following incubation, the media (containing nonbound RNA) was removed from the CHO cells and transferred to a well containing ~1 × 105 CHO/mDEC205 cells. The RNA was incubated with CHO/mDEC205 for 1 hour at 37 °C, after which cells were washed three times with 1 ml HBSS, and cells were lysed for 5 minutes at room temperature with 500 μl TRIzol. Recovered RNA was reverse transcribed, amplified, and transcribed as described above.
For Round 5, we performed an “internalization selection” in which we targeted BMDCs. BMDCs were prepared from mouse bone marrow by treatment for 5 days with granulocyte-macrophage colony-stimulating factor (GM-CSF) as previously described.17 RNA (5 μg) was combined with a twofold molar excess of reverse primer used for amplification (T50R). The mixture was diluted to a final volume of 50 μl in HBSS, denatured for 5 minutes at 70 °C, and allowed to anneal for 15 minutes at room temperature. The RNA was subsequently added to one well of a 24-well plate containing ~5 × 104 BMDCs in 450 μl advanced Roswell Park Memorial Institute medium (RPMI) 1640 containing 10% FBS and 5% J5 medium (supernatant from J558 cells transfected to express GM-CSF supplemented with single-stranded DNA and transfer RNA (1 mg/ml each).
After a 1-hour incubation at 37 °C, the media was removed, and cells were washed three times with 1 ml HBSS containing 0.1% NaN3 followed by a single wash with 1 ml cold 200 mmol/l glycine, 150 mmol/l NaCl, pH 4. The cells were subsequently washed an additional three times with 1 ml HBSS containing 0.1% NaN3 followed by 1 ml DPBS without Ca2+ or Mg2+ and trypsinized by the addition of 500 μl 0.05% trypsin, 0.53 mmol/l ethylenediaminetetraacetic acid for 15 minutes at 37 °C. Following an additional wash with 1 ml HBSS containing 0.1% NaN3, the cells were pelleted and resuspended in 100 μl HBSS containing 5 μl RiboShredder ribonuclease cocktail (EpiCentre, Madison, WI). The reaction was incubated for 15 minutes at room temperature, after which the cells were washed three times with 1 ml HBSS + 0.1% NaN3 and lysed for 5 minutes at room temperature with 500 μl TRIzol. RNA was recovered, reverse transcribed, amplified, and transcribed as described above.
PCR product from Round 5 was cloned into the pCR2.1-TOPO TA vector by TOPO TA cloning (Life Technologies).
Chemical synthesis of RNA aptamers. Minimized aptamers and controls were synthesized in our laboratory on an Expedite 8909 DNA synthesizer (Applied Biosystems, Carlsbad, CA) using 2′-fluoro-deoxycytidine and 2′-fluoro-deoxyuridine phosphoramidites (Metkinin, Kuusisto, Finland). Unless noted otherwise, all other synthesis reagents were purchased from Glen Research (Sterling, VA). The aptamer was synthesized bearing a 5′ thiol modification using a thiol-modifier C6 S-S phosphoramidite and a 3′ inverted dT residue for added serum stability. The sequences of the minimized aptamer, min.2, and a nonbinding control aptamer, c36, an aptamer-like, nonfunctional sequence which we have previously utilized as a nonbinding control6 were: 5S GGGAGGUGUGUUAGCACACGAUUCAUAAUCAGCUACCCUCCCt and 5SGGCGUAGUGAUUAUGAAUCGUGUGCUAAUACACGCCt, respectively, where “t” is a 3′ inverted dT and “5S” is the 5′ thiol. All aptamers were synthesized with the final 4,4′-dimethoxytrityl protecting group left on. Following deprotection, aptamers were purified by reversed-phase high-performance liquid chromatography on a 10 × 50 mm Xbridge C18 column (Waters, Milford, MA) using a linear gradient of acetonitrile in 0.1 mol/l triethylammonium acetate at pH 7.0.
Aptamer binding by flow cytometry. Aptamer binding was assessed by flow cytometry. Rounds from the selection were first hybridized to a biotinylated labeled oligonucleotide that was complementary to the 3′ end of the library (T50R). In a typical assay, 10 pmol of transcribed RNA pool was added to 11 pmol biotinylated T50R (B-T50R) in 10 μl of DPBS. The RNA was thermally equilibrated by heating to 70 °C for 3 minutes and then allowed to cool on the bench for 15 minutes at room temperature. The RNA was subsequently added to 1 × 105 cells in 100 μl flow cytometry buffer (HBSS containing 1% BSA and 0.1% NaN3) supplemented with 1 mg/ml ssDNA. The RNA and cells were incubated for 15 minutes on ice, pelleted by centrifugation, washed once with 1 ml flow cytometry buffer and then resuspended in 100 μl flow cytometry buffer containing 0.5 μl PhycoLink SA-R-PE (SA-PE), and incubated an additional 15 minutes on ice. Cells were then washed with an additional 1 ml flow cytometry buffer, resuspended with 500 μl flow cytometry buffer, and analyzed by flow cytometry, with exclusion of dead cells by 7-aminoactinomycin D (7-AAD) or 4′,6-diamidino-2-phenylindole (DAPI) staining.
Flow cytometry: fluorophore or biotin-conjugated aptamers. Thiolated aptamers (5S.min2 or 5S.cntrl36) were used to generate the Alexa Fluor 488 (AF488), Dylight 647 (Dy647), or biotin-labeled aptamers used for flow cytometry. Labeling was performed using AF488-C5-malemide (Life Technologies), Dylight 647 maleimide (Pierce, Rockford, IL), or biotin maleimide (Pierce) as follows: thiolated aptamer was reduced using 10 mmol/l tris(2-carboxyethyl)phosphine (TCEP) in 100 μl of 0.1 mol/l triethylammonium acetate. Samples were heated at 70 °C for 3 minutes followed by incubation at room temperature for 1 hour. The reduced aptamers were desalted using a Biospin 6 column (BioRad, Hercules, CA) into phosphate-buffered saline (PBS) supplemented with 50 mmol/l phosphate pH 7.5. Typically, the maleimide in dimethyl sulfoxide was added at a 3–10-fold molar excess over ~10 nmoles of aptamer in 100 μl of 50 mmol/l phosphate, pH 7.5. Following an overnight reaction at 4 °C, the aptamer was desalted an additional time using a Biospin 6 column. Conjugations proceeded to >99% as determined by high-performance liquid chromatography with complete removal of free dye or biotin.
In a typical cytometry assay, 100 pmol of dye/biotin-conjugated RNA was diluted into 100 μl DPBS, denatured for 3 minutes at 70 °C and allowed to cool for 15 minutes at room temperature. Cell staining was performed as described.
Aptamer binding and internalization experiments were performed using BMDCs and CHO-DEC205 cells. To generate BMDCs, bone marrow from the femur and tibia was cultured for 8 days in RPMI complete media plus GM-CSF (10 ng/ml) and MACS sorted for CD11c+ cells (Miltenyi Biotec, San Diego, CA)17 prior to use. For each experiment, 106 cells were resuspended in RPMI complete media containing blocking reagents (tRNA and ssDNA at 1 mg/ml). Biotinylated min.2 or cntrl.36 were thermally equilibrated and mixed at a 1:1 with SA-PE for 15 minutes prior to addition to cells and allowed to incubate for 30 minutes at 4 °C. To determine binding, cells were washed twice and treated with or without Riboshredder for 10 minutess at 37 °C to digest any cell-surface bound aptamer:PE. To determine internalization, following incubation at 4 °C, cells were moved to 37 °C for 1 hour. The cells were then washed twice and incubated with or without Riboshredder for 10 minutes at 37 °C. Cells were then washed and stained with Fc block (anti-mouse CD16; eBioscience, San Diego, CA) and CD11c-APC for 30 minutes at 4 °C. Cells were washed twice, stained with the viability dye DAPI, and analyzed by flow cytometry.
mDEC205 cell surface binding assays and the determination of apparent binding constant. A20.Kb cells were washed with RPMI and resuspended at 1 × 106 cells per milliliter in RPMI supplemented with 1 mg/ml ssDNA. 100 µl cells were incubated with increasing concentrations of thermally equilibrated Alexa Fluor 488-conjugated min.2 or Alexa Fluor 488-conjugated cntrl.36 for 15 minutes at 37 °C. Following incubation, the cells were washed with and resuspended in flow cytometry buffer, and analyzed by flow cytometry. Median Alexa Fluor 488 fluorescence of live (DAPI−) cells was plotted versus min.2 or cntrl.36 concentration using GraphPad Prism, and the dissociation constant (KD) was calculated by fitting a one site binding model to the data.
A20.Kb cells and A20.Kb.mDEC205 cells were generated from A20 cells by viral transduction. The cell lines were generated by superinfection with retroviruses made from pMX-Kb and/or pMX-mDEC205 as described previously.42
Synthesis of aptamer OVA conjugates. Thiolated aptamers (5S.min2 or 5S.cntrl36) were used to generate the aptamer OVA conjugates. Pierce Imject OVA was activated with a 10-fold molar excess of N-[γ-maleimidobutyryloxysulfosuccinimide ester (sulfo-GMBS, Pierce) in 1 mmol/l ethylenediaminetetraacetic acid in DPBS without Ca2+ or Mg2+ according to the manufacturer's protocol, and excess sulfo-GMBS was removed with a Micro Bio-Spin 6 column.
Thiolated aptamers were reduced using 10 mmol/l TCEP and subsequently desalted using a Bio-Spin 6 column into DPBS without Ca2+ or Mg2+. The reduced aptamers were incubated with a fivefold molar excess of sulfo-GMBS-activated OVA. The reaction mixture was incubated for 30 minutes at room temperature or overnight at 4 °C and then quenched by incubation with excess L-cysteine or BODIPY FL L-cysteine (Life Technologies) that had previously been reduced with TCEP.
The reaction mixture was desalted to 20 mmol/l Tris, pH 7 with a Bio-Spin 6 column and then purified on a Mini Q 4.6/50 PE column (GE Healthcare) equilibrated with the same buffer, and the eluted fractions were analyzed by denaturing gel electrophoresis (Supplementary Figure S5). Conjugate function was confirmed by flow cytometry using BODIPY FL L-cysteine quenched conjugates (Supplementary Figure S5).
Mulitmeric aptamer conjugates were generated using a similar approach. In short, OVA was activated with a 50-fold molar excess of the heterobifunctional crosslinker SMPEG12 (Pierce) in place of sulfo-GMBS. The reaction was allowed to proceed for 3 hours at room temperature, after which excess crosslinker was removed by ultrafiltration using an Amicon Ultra, Ultracell 30K filter (Millipore, Billerica, MA). The reduced aptamers we incubated at a 3:1 ratio with activated OVA overnight at 4 °C, after which the reaction was quenched by the addition of 100-fold excess of β-mercaptoethanol. Conjugation efficiency routinely proceeded to >90% as determined by denaturing (7 mol/l urea) gel electrophoresis and SDS–PAGE (Supplementary Figure S8). Multimer:OVA conjugate function was confirmed by flow cytometry on CHO-mDEC205 cells by replacing β-mercaptoethanol in the quench step with BODIPY FL L-cysteine (Life Technologies) that had previously been reduced with TCEP (Supplementary Figure S11).
Confocal microscopy. 2 × 105 CD11c+ BMDCs or CHO-DEC205 cells were plated onto poly-L-lysine coated circular glass coverslips in complete media. CD11c+ BMDCs were treated in 500 µl RPMI complete medium containing 10 ng/ml GM-CSF. CHO-DEC205 cells were treated in 500 µl complete Dulbecco's modified Eagle medium. To visualize aptamer-Dy649 binding, plates were moved to 4 °C and incubated with 200 nmol/l aptamer-Dy649 in presence of blocking agents (1 mg/ml ssDNA and tRNA) for 60 minutes. Cells were then moved to 37 °C for 2 hours to allow for aptamer internalization. Cells were then washed with complete medium to remove unbound aptamer, followed by staining with Fc block and either 10 µg/ml DEC205-biotin (CHO-DEC205) or CD11c-biotin (CD11c+ BMDCs) for 30 minutes at 4 °C. Cells were then washed twice with complete medium and stained with SA-Alexa488 (10 µg/ml) for 30 minutes at 4 °C. Cells were washed twice with cold PBS, fixed with 2% paraformaldehyde, followed by three washes with cold PBS. Coverslips were mounted with 20 µl Vectashield mounting medium containing DAPI. Images were acquired on a Leica SP5 AOBS instrument using the 40x oil immersion objective.
For colocalization studies, 15,000 CHO-DEC205 cells (removed from primary flasks using 5 mmol/l ethylenediaminetetraacetic acid in PBS—not trypsin) were plated in 8-well chamber slides in complete media (advanced Dulbecco's modified Eagle medium, 5% FBS, supplements 2-ME/L-glutamine) and allowed to adhere for 2 hours at 37 °C 5% CO2. 8-well chamber glass bottom slides (LabTek 15547) were pretreated with fibronectin at 25 µg/ml for 30 minutes at room temperature prior to use. Experiments involving coincubation of min.2 and DEC205 antibody were performed using 250 nmol/l min.2-DyLight 650 and 0.2 µg/ml DEC205-Alexa Fluor 488 antibody (clone 205yekta; eBioscience). Experiments involving early endosome or lysosome colocalization studies were performed using CHO-DEC205 cells transduced with a baculovirus vector containing mRFP tagged Rab5a or LAMP1 (Cell Light Reagents BacMam 2.0, Life Technologies). Adherent CHO-DEC205 cells were transduced in complete media at 75 particles/cell. Sixteen hours posttransduction, cells were incubated with 250 nmol/l min2-Alexa Fluor 488 aptamer or 0.2 µg/ml DEC205-Alexa Fluor 488 antibody for 2 hours, after which the cells were washed with complete media and incubated 1 additional hour. Following incubation, cells were washed and then fixed with 4% paraformaldehyde for 20 minutes at room temperature. The fixed cells were again washed and the cell nuclei stained with DAPI. Samples were visualized using a Leica SP5 AOBS confocal microscope using a ×63 oil objective. Captured images were converted to the TIFF format using Leica LAS-AF software (Leica Microsystems, Buffalo Grove, IL). Image processing was performed using NIH ImageJ (National Institutes of Health, Bethesda, MD).
In vitro proliferation assays. A single-cell suspension of splenocytes from C57BL/6 mice, aged 6–12 weeks, was prepared using 400 U/ml collagenase D (Roche, Indianapolis, IN), and CD11c+ cells were isolated using biotinylated antibody clone N418 and MACS SA microbeads, or MACS CD11c+ microbeads alone, according to the manufacturer's protocol (Miltenyi Biotec). CD11c+ splenic DCs were plated in a 96-well U-bottom plate at 4 × 105 in 100 μl Advanced RPMI 1640 (Life Technologies) containing 10% FBS, Penicillin-Streptomycin (Life Technologies), GlutaMAX-I (Life Technologies) and 10 mmol/l hydroxyethyl piperazineethanesulfonic acid (MPBio, Santa Ana, CA) (complete culture medium), 1 μg/ml lipopolysaccharide from Escherichia coli O111:B4 (Sigma-Aldrich, St Louis, MO), and 10 μg/ml polyinosinic-polycytidylic acid (pIC, Sigma-Aldrich). The cells were incubated for 4 hours at 37 °C, after which the media was replaced with media of the same formulation containing additional 1 mg/ml ssDNA and 10 nmol/l of either Imject OVA, rat IgG2a isotype antibody-OVA chimera (Iso:OVA), NLDC145:OVA chimera (NLDC145:OVA), monovalent cntrl.36:OVA, or min.2:OVA (several wells received an equivalent volume of DPBS as a negative control). The cells were incubated for an additional 16–20 hours at 37 °C.
The next day, a single-cell suspension of lymph node cells was prepared from the axillary, brachial, inguinal, popliteal, and mesenteric lymph nodes of 6–12-week-old C57BL/6 RAG−/− OT-I+/+ (OT-I) mice. The OT-I cells were resuspended in DPBS containing 5% FBS, CFSE was added to 5 μmol/l, and the cells were incubated for 5 minutes at 37 °C protected from light. The reaction was quenched by adding 10 volumes of complete culture medium and incubating for 5 minutes at room temperature. Cells were washed three times in complete culture medium and resuspended in complete culture medium at 1 × 106 cells/ml. After their 16–20-hour incubation, the CD11c+ splenocytes were washed three times with DPBS, 1 × 105 CFSE-labeled OT-I cells were added, and the cells were incubated an additional 2–3 days at 37 °C. At the end of this incubation, cells were resuspended, washed with flow cytometry buffer, stained with DAPI and antibodies against CD8α and T cell receptor (TCR)-β, and analyzed by flow cytometry.
Similar experiments were performed to assess the function of our multivalent aptamer:OVA conjugates (Supplementary Figure S12).
In vitro cytokine production. CD11c+ splenic DCs were prepared as described above and treated in triplicate with 10 nmol/l of either of the following: Imject OVA, SIINFEKL (OVA257–264) peptide, rIgG2a:OVA, NLDC145:OVA, cntrl.36:OVA, or min.2:OVA. OT-I cells were prepared as described above, except for CFSE staining. After a 16–20-hour incubation with antigen at 37 °C, the CD11c+ splenic DCs were washed carefully three times with DPBS, and 1 × 105 OT-I cells were added. Following incubation at 37 °C for 2 additional days, media was removed and saved for analysis of secreted IL-2 and IFN-γ by enzyme-linked immunosorbent assay. Fresh media containing 10 μg/ml brefeldin A (Life Technologies) was added, and cells were incubated for 5 hours at 37 °C. Cells were then resuspended and stained for the cell surface molecules CD8α and TCR-β, followed by fixation with 4% paraformaldehyde (Sigma-Aldrich) in pH 7 PBS for 10 minutes at 37 °C. Cells were washed, permeabilized with 0.1% saponin (Sigma-Aldrich) in flow cytometry buffer and stained with antibodies against IL-2 and IFN-γ, and analyzed by flow cytometry.
In vivo uptake of aptamer-SA conjugates. Aptamer:SA conjugates were synthesized at a ratio of 1:1 and 4:1 (aptamer:SA) using biotinylated aptamers (synthesized from thiol modified aptamers as described above) and SA-AF647 (Life Technologies) by simple mixing at the appropriate ratio. The approximate number of aptamers per protein was determined by SDS–PAGE gel electrophoresis. At a mixing ratio of 1:1, conjugates appear as a mixture of species bearing 0, 1, and 2 aptamers, whereas at a ratio of 4:1, conjugates appear as a mixture of species bearing 3 or 4 aptamers (Supplementary Figure S8). Approximately 20 µg aptamer conjugate was injected, i.v. into C57BL/6 mice. One day later, spleens were isolated, digested with 400 units collagenase D (Roche), and following red blood cell lysis, splenocytes were stained with antibodies specific for CD11c and DEC205. Splenocytes were stained with the viability dye Live/Dead Fixable Blue (Life Technologies) and were acquired on an LSRII (BD). Analysis was performed using FlowJo (Treestar, Ashland, OR).
In vivo crosspresentation with aptamer:OVA conjugates. OT-I lymph node cells were labeled with 5 µm CFSE and 106 cells were adoptively transferred into a congenic recipient (B6.SJL-Ptprc) by retroorbital injection. One day later, mice were injected retroorbitally with either 20 µg multivalent aptamer:OVA (min.2, mut.1, mut.2, or ctrl.36) or controls (pIC alone, OVA alone, min.2+OVA admixed, HBSS buffer or 1 µg NLDC145:OVA). Unless stated, all injections included the adjuvant pIC (25 µg per mouse). Three days later, spleens were removed, red blood cells were lysed, and 5 × 106 splenocytes were incubated for 4 hours in the presence of 5 µg/ml brefeldin A with or without 1 µmol/l of the OVA peptide, SIINFEKL. Cells were then stained for cell surface markers, TCR-β and CD45.2, followed by fixation with 4% paraformaldehyde. Intracellular IL-2 and IFN-γ were detected by incubating cells with antibody in the presence of 0.3% saponin. Cells were then acquired on an LSRII and analyzed by FlowJo. Dead cells were excluded using live/dead viability dye.
B16-OVA flank tumor model. C57BL/6 mice were s.c. injected with 106 B16-OVA cells (M05). Four days later, 106 OT-I cells isolated from lymph nodes were injected into the mice i.v. into the retroorbital sinus. The following day, groups of 5–6 mice were injected i.v. with 10 µg cntrl36:OVA, min.2:OVA, or 5 µg NLDC145:OVA plus 25 µg pIC. One group of mice was left untreated. Tumor growth was monitored every 2 days until the tumors reached 1 cm in diameter, at which time mice were euthanized. Experiments were also performed using mice injected with 106 B16-OVA cells (M05) in one flank and 5 × 104 parental B16 cells (no OVA) in the opposite flank. However, rapid growth of the parental B16 across all treatment groups resulted in termination of these experiments prior to observing significant inhibition in min.2:OVA and NLDC145:OVA treated groups.
SUPPLEMENTARY MATERIAL Figure S1. Selection of DEC205-specific aptamers. Figure S2. Analysis of Clone 1 minimizations 1-5. Figure S3. Min.2 specifically binds the DEC205 receptor. Figure S4. Min.2 and NLDC145 bind DEC205+ cells. Figure S5. Characterization and purification of aptamer:OVA conjugates. Figure S6. Aptamer:OVA conjugates do not activate CD11c+ splenic DCs. Figure S7. Cytokine analysis of media from CD11c+ splenocytes treated as described in Figure S6. Figure S8. Cytometric analysis of monovalent and multivalent aptamer:streptavidin conjugates on CHO-DEC205 cells. Figure S9. Analysis of the effects of conjugate valency on binding. Figure S10. Representative analysis of multivalent aptamer:OVA conjugates by gel electrophoresis. Figure S11. Binding analysis of multivalent aptamer:OVA conjugate. Figure S12. Multivalent min.2:OVA conjugates show diminished capacity for antigen presentation in vitro. Figure S13. Mutant forms of min.2 bind DEC205 with lower affinity and are less effective at cross-presentation when compared with min.2. Figure S14. Activation of adoptively transferred OT-I cells following treatment with multivalent aptamer:OVA conjugates.
Acknowledgments
We thank Gregoire Lauvau and Saidi Soudja (AECOM) for helpful discussions and Amy (Amos) Yan for her assistance editing this manuscript. This work was supported in part by a Pilot Project grant from the Albert Einstein Cancer Center P30 CA01330 (M.L.), Stand Up to Cancer and the NIH - R21CA157366-01 (M.L.), R21 AI093539 and R01 AI099567 (D.P.), the NIH Medical Scientist Training Program grant, T32-GM007288 (B.W) and a the Louis and Rachel Rudin Scholarship (B.W).
Supplementary Material
References
- Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- D'Argenio DA, Wilson CB. A decade of vaccines: Integrating immunology and vaccinology for rational vaccine design. Immunity. 2010;33:437–440. doi: 10.1016/j.immuni.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Palucka K, Ueno H, Banchereau J. Recent developments in cancer vaccines. J Immunol. 2011;186:1325–1331. doi: 10.4049/jimmunol.0902539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillman RO. Cancer immunotherapy. Cancer Biother Radiopharm. 2011;26:1–64. doi: 10.1089/cbr.2010.0902. [DOI] [PubMed] [Google Scholar]
- Bonifaz LC, Bonnyay DP, Charalambous A, Darguste DI, Fujii S, Soares H, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199:815–824. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med. 2002;196:1627–1638. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan AC, Levy M. Aptamers and aptamer targeted delivery. RNA Biol. 2009;6:316–320. doi: 10.4161/rna.6.3.8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed MA, Pervaiz S. Advances in aptamers. Oligonucleotides. 2010;20:215–224. doi: 10.1089/oli.2010.0234. [DOI] [PubMed] [Google Scholar]
- Dollins CM, Nair S, Sullenger BA. Aptamers in immunotherapy. Hum Gene Ther. 2008;19:443–450. doi: 10.1089/hum.2008.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Swiggard WJ, Inaba M, Meltzer J, Mirza A, Sasagawa T, et al. Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. I. Expression on dendritic cells and other subsets of mouse leukocytes. Cell Immunol. 1995;163:148–156. doi: 10.1006/cimm.1995.1109. [DOI] [PubMed] [Google Scholar]
- van Broekhoven CL, Parish CR, Demangel C, Britton WJ, Altin JG. Targeting dendritic cells with antigen-containing liposomes: a highly effective procedure for induction of antitumor immunity and for tumor immunotherapy. Cancer Res. 2004;64:4357–4365. doi: 10.1158/0008-5472.CAN-04-0138. [DOI] [PubMed] [Google Scholar]
- Johnson TS, Mahnke K, Storn V, Schönfeld K, Ring S, Nettelbeck DM, et al. Inhibition of melanoma growth by targeting of antigen to dendritic cells via an anti-DEC-205 single-chain fragment variable molecule. Clin Cancer Res. 2008;14:8169–8177. doi: 10.1158/1078-0432.CCR-08-1474. [DOI] [PubMed] [Google Scholar]
- Bozzacco L, Trumpfheller C, Siegal FP, Mehandru S, Markowitz M, Carrington M, et al. DEC-205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+ T cells in a spectrum of human MHC I haplotypes. Proc Natl Acad Sci USA. 2007;104:1289–1294. doi: 10.1073/pnas.0610383104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong C, Choi JH, Vitale L, He LZ, Trumpfheller C, Bozzacco L, et al. Improved cellular and humoral immune responses in vivo following targeting of HIV Gag to dendritic cells within human anti-human DEC205 monoclonal antibody. Blood. 2010;116:3828–3838. doi: 10.1182/blood-2010-06-288068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celldex Therapeutics A Study of CDX-1401 in Patients With Malignancies Known to Express NY-ESO-1. 2009.
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47–64. doi: 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- Chatterjee B, Smed-Sörensen A, Cohn L, Chalouni C, Vandlen R, Lee BC, et al. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood. 2012;120:2011–2020. doi: 10.1182/blood-2012-01-402370. [DOI] [PubMed] [Google Scholar]
- Mahnke K, Guo M, Lee S, Sepulveda H, Swain SL, Nussenzweig M, et al. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J Cell Biol. 2000;151:673–684. doi: 10.1083/jcb.151.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhães ML, Byrom M, Yan A, Kelly L, Li N, Furtado R, et al. A general RNA motif for cellular transfection. Mol Ther. 2012;20:616–624. doi: 10.1038/mt.2011.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- Idoyaga J, Lubkin A, Fiorese C, Lahoud MH, Caminschi I, Huang Y, et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc Natl Acad Sci USA. 2011;108:2384–2389. doi: 10.1073/pnas.1019547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse MA, Chapman R, Powderly J, Blackwell K, Keler T, Green J, et al. Phase I study utilizing a novel antigen-presenting cell-targeted vaccine with Toll-like receptor stimulation to induce immunity to self-antigens in cancer patients. Clin Cancer Res. 2011;17:4844–4853. doi: 10.1158/1078-0432.CCR-11-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell. 2010;143:416–429. doi: 10.1016/j.cell.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keler T, He L, Ramakrishna V, Champion B. Antibody-targeted vaccines. Oncogene. 2007;26:3758–3767. doi: 10.1038/sj.onc.1210375. [DOI] [PubMed] [Google Scholar]
- Huang YF, Shangguan D, Liu H, Phillips JA, Zhang X, Chen Y, et al. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. Chembiochem. 2009;10:862–868. doi: 10.1002/cbic.200800805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu TC, Marks JW, 3rd, Lavery LA, Faulkner S, Rosenblum MG, Ellington AD, et al. Aptamer:toxin conjugates that specifically target prostate tumor cells. Cancer Res. 2006;66:5989–5992. doi: 10.1158/0008-5472.CAN-05-4583. [DOI] [PubMed] [Google Scholar]
- Mallik PK, Nishikawa K, Millis AJ, Shi H. Commandeering a biological pathway using aptamer-derived molecular adaptors. Nucleic Acids Res. 2010;38:e93. doi: 10.1093/nar/gkp1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci USA. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollins CM, Nair S, Boczkowski D, Lee J, Layzer JM, Gilboa E, et al. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem Biol. 2008;15:675–682. doi: 10.1016/j.chembiol.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO, Kolonias D, Pastor F, Mittler RS, Chen L, Giangrande PH, et al. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J Clin Invest. 2008;118:376–386. doi: 10.1172/JCI33365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilner SE, Wengerter B, Maier K, de Lourdes Borba Magalhães M, Del Amo DS, Pai S, et al. An RNA alternative to human transferrin: a new tool for targeting human cells. Mol Ther Nucleic Acids. 2012;1:e21. doi: 10.1038/mtna.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimpton RE, Butler M, Morel AS, Eren E, Hue SS, Ritter MA. CD205 (DEC-205): a recognition receptor for apoptotic and necrotic self. Mol Immunol. 2009;46:1229–1239. doi: 10.1016/j.molimm.2008.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahoud MH, Ahmet F, Zhang JG, Meuter S, Policheni AN, Kitsoulis S, et al. DEC-205 is a cell surface receptor for CpG oligonucleotides. Proc Natl Acad Sci USA. 2012;109:16270–16275. doi: 10.1073/pnas.1208796109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins DP, Luebering BJ, Shaut DM. T-lymphocyte functionality assessed by analysis of cytokine receptor expression, intracellular cytokine expression, and femtomolar detection of cytokine secretion by quantitative flow cytometry. Cytometry. 1998;33:249–255. doi: 10.1002/(sici)1097-0320(19981001)33:2<249::aid-cyto21>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Healy JM, Lewis SD, Kurz M, Boomer RM, Thompson KM, Wilson C, et al. Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm Res. 2004;21:2234–2246. doi: 10.1007/s11095-004-7676-4. [DOI] [PubMed] [Google Scholar]
- Hall B, Micheletti JM, Satya P, Ogle K, Pollard J, Ellington AD. Design, synthesis, and amplification of DNA pools for in vitro selection. Curr Protoc Nucleic Acid Chem. 2009;Chapter 9:Unit 9.2. doi: 10.1002/0471142700.nc0902s39. [DOI] [PubMed] [Google Scholar]
- Sousa R, Padilla R. A mutant T7 RNA polymerase as a DNA polymerase. EMBO J. 1995;14:4609–4621. doi: 10.1002/j.1460-2075.1995.tb00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla R, Sousa R. Efficient synthesis of nucleic acids heavily modified with non-canonical ribose 2'-groups using a mutantT7 RNA polymerase (RNAP) Nucleic Acids Res. 1999;27:1561–1563. doi: 10.1093/nar/27.6.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillerez J, Lopez PJ, Proux F, Launay H, Dreyfus M. A mutation in T7 RNA polymerase that facilitates promoter clearance. Proc Natl Acad Sci USA. 2005;102:5958–5963. doi: 10.1073/pnas.0407141102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CG, Lee SY, Kandala G, Lee SY, Choi Y. A novel gene product that couples TCR signaling to Fas(CD95) expression in activation-induced cell death. Immunity. 1996;4:583–591. doi: 10.1016/s1074-7613(00)80484-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.