

Abstract
In 1953, Stanley Miller reported the production of biomolecules from simple gaseous starting materials, using an apparatus constructed to simulate the primordial Earth's atmosphere-ocean system. Miller introduced 200 ml of water, 100 mmHg of H2, 200 mmHg of CH4, and 200 mmHg of NH3 into the apparatus, then subjected this mixture, under reflux, to an electric discharge for a week, while the water was simultaneously heated. The purpose of this manuscript is to provide the reader with a general experimental protocol that can be used to conduct a Miller-Urey type spark discharge experiment, using a simplified 3 L reaction flask. Since the experiment involves exposing inflammable gases to a high voltage electric discharge, it is worth highlighting important steps that reduce the risk of explosion. The general procedures described in this work can be extrapolated to design and conduct a wide variety of electric discharge experiments simulating primitive planetary environments.
Keywords: Chemistry, Issue 83, Geosciences (General), Exobiology, Miller-Urey, Prebiotic chemistry, amino acids, spark discharge
Introduction
The nature of the origins of life on Earth remains one of the most inscrutable scientific questions. In the 1920s Russian biologist Alexander Oparin and British evolutionary biologist and geneticist John Haldane proposed the concept of a "primordial soup"1,2, describing the primitive terrestrial oceans containing organic compounds that may have facilitated chemical evolution. However, it wasn't until the 1950s when chemists began to conduct deliberate laboratory studies aimed at understanding how organic molecules could have been synthesized from simple starting materials on the early Earth. One of the first reports to this end was the synthesis of formic acid from the irradiation of aqueous CO2 solutions in 19513.
In 1952, Stanley Miller, then a graduate student at the University of Chicago, approached Harold Urey about doing an experiment to evaluate the possibility that organic compounds important for the origin of life may have been formed abiologically on the early Earth. The experiment was conducted using a custom-built glass apparatus (Figure 1A) designed to simulate the primitive Earth. Miller's experiment mimicked lightning by the action of an electric discharge on a mixture of gases representing the early atmosphere, in the presence of a liquid water reservoir, representing the early oceans. The apparatus also simulated evaporation and precipitation through the use of a heating mantle and a condenser, respectively. Specific details about the apparatus Miller used can be found elsewhere4. After a week of sparking, the contents in the flask were visibly transformed. The water turned a turbid, reddish color5 and yellow-brown material accumulated on the electrodes4. This groundbreaking work is considered to be the first deliberate, efficient synthesis of biomolecules under simulated primitive Earth conditions.
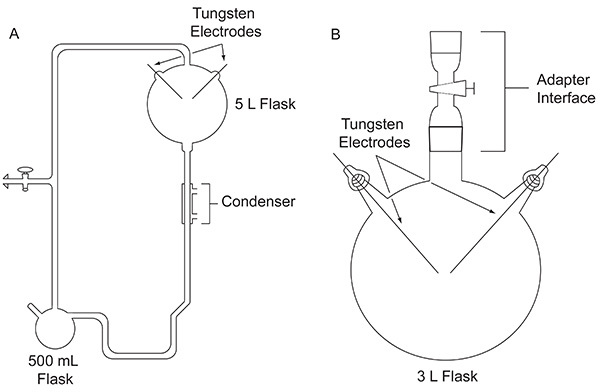 Figure 1. Comparison between the two types of apparatuses discussed in this paper. The classic apparatus used for the original Miller-Urey experiment (A) and the simplified apparatus used in the protocol outlined here (B). Click here to view larger image.
Figure 1. Comparison between the two types of apparatuses discussed in this paper. The classic apparatus used for the original Miller-Urey experiment (A) and the simplified apparatus used in the protocol outlined here (B). Click here to view larger image.
After the 1953 publication of results from Miller's classic experiment, numerous variations of the spark discharge experiment, for example using other gas mixtures, were performed to explore the plausibility of producing organic compounds important for life under a variety of possible early Earth conditions. For example, a CH4/H2O/NH3/H2S gas mixture was tested for its ability to produce the coded sulfur-containing α-amino acids, although these were not detected6. Gas chromatography-mass spectrometry (GC-MS) analysis of a CH4/NH3 mixture subjected to an electric discharge showed the synthesis of α-aminonitriles, which are amino acid precursors7. In 1972, using a simpler apparatus, first introduced by Oró8 (Figure 1B), Miller and colleagues demonstrated the synthesis of all of the coded α-amino acids9 and nonprotein amino acids10 that had been identified in the Murchison meteorite to date, by subjecting CH4, N2, and small amounts of NH3 to an electric discharge. Later, using this same simplified experimental design, gas mixtures containing H2O, N2, and CH4, CO2, or CO were sparked to study the yield of hydrogen cyanide, formaldehyde, and amino acids as a function of the oxidation state of atmospheric carbon species11.
In addition to the exploration of alternative experimental designs over the years, significant analytical advances have occurred since Miller's classic experiment, which recently aided more probing investigations of electric discharge experimental samples archived by Miller, than would have been facilitated by the techniques Miller had access to in the 1950s. Miller's volcanic experiment12, first reported in 19554, and a 1958 H2S-containing experiment13 were shown to have formed a wider variety, and greater abundances, of numerous amino acids and amines than the classic experiment, including many of which that had not been previously identified in spark discharge experiments.
The experiment described in this paper can be conducted using a variety of gas mixtures. Typically, at the very least, such experiments will contain a C-bearing gas, an N-bearing gas, and water. With some planning, almost any mixture of gases can be explored, however, it is important to consider some chemical aspects of the system. For example, the pH of the aqueous phase can have a significant impact on the chemistry that occurs there14.
The method described here has been tailored to instruct researchers how to conduct spark discharge experiments that resemble the Miller-Urey experiment using a simplified 3 L reaction vessel, as described in Miller's 1972 publications9,10. Since this experiment involves a high voltage electric arc acting on inflammable gases, it is crucial to remove O2 from the reaction flask to eliminate the risk of explosion, which can occur upon combustion of reduced carbon-bearing gases such as methane or carbon monoxide, or reaction of H2 with oxygen.
There are additional details that should be kept in mind when preparing to conduct the experiment discussed here. First, whenever working with glass vacuum lines and pressurized gases, there exists the inherent danger of both implosion and over-pressuring. Therefore, safety glasses must be worn at all times. Second, the experiment is typically conducted at less than atmospheric pressure. This minimizes the risk of over-pressuring the manifold and reaction flask. Glassware may be rated at or above atmospheric pressure, however, pressures above 1 atm are not recommended. Pressures may increase in these experiments as water-insoluble H2 is liberated from reduced gases (such as CH4 and NH3). Over-pressuring can lead to seal leakage, which can allow atmospheric O2 to enter the reaction flask, making it possible to induce combustion, resulting in an explosion. Third, it should be borne in mind that modification of this protocol to conduct variations of the experiment requires careful planning to ensure unsafe conditions are not created. Fourth, it is highly recommended that the prospective experimenter read through the entire protocol carefully several times prior to attempting this experiment to be sure he or she is familiar with potential pitfalls and that all necessary hardware is available and in place. Lastly, conducting experiments involving combustible gases require compliance with the experimenter's host institution's Environmental Health and Safety departmental guidelines. Please observe these recommendations before proceeding with any experiments. All steps detailed in the protocol here are in compliance with the authors' host institutional Environmental Health and Safety guidelines.
Protocol
1. Setting Up a Manifold/Vacuum System
- Use a glass manifold to introduce gases into the reaction flask. This manifold can be purchased or constructed by a glass-blowing facility, but must include vacuum-tight ports that can be connected to a vacuum system, gas cylinders, a vacuum gauge, and the reaction vessel.
- Use ground glass joints and glass plugs with valves on the manifold. Ensure that all O-rings on the plugs are capable of making the necessary seals. If using glass joints, a sufficient amount of vacuum grease can be applied to help make a seal, if necessary. Silicon vacuum grease can be used to avoid potential organic contamination.
- Use glass stopcocks on the manifold. Apply the minimum amount of vacuum grease necessary to make a seal.
- Measure the manifold volume. This volume will be used for calculations related to final gas pressures in the 3 L reaction flask and should be known as precisely as possible.
- Unless the manifold has enough connections to accommodate all gas cylinders simultaneously, connect one cylinder at a time to the manifold. Include in this connection, a tap allowing the manifold to be isolated from the ambient atmosphere.
- Use suitable, clean, inert, and chemical and leak resistant tubing and ultratorr vacuum fittings to connect the gas cylinders to the manifold. Ultratorr fittings, where used, are to be finger-tightened.
- Connect to the manifold, a vacuum pump capable of establishing a vacuum of <1 mmHg. The vacuum pump exhaust should be located within the fume hood, or properly vented by other means.
- To ensure rapid attainment of vacuum and to protect the pump, insert a trap between the manifold and the vacuum pump. A liquid nitrogen finger-trap is recommended as it will prevent volatiles such as NH3, CO2, and H2O from entering the pump. Care should be taken, as trapped volatiles, upon warming, may overpressure the manifold and result in glass rupture.
- Connect to the manifold, a manometer or other vacuum gauge capable of 1 mmHg resolution or better. While various devices can be used, a mercury manometer, or MacLeod gauge, is preferable as mercury is fairly nonreactive.
- Measure and record the ambient temperature using a suitable thermometer.
2. Preparation of Reaction Flask
- Heat all glassware at 500 °C for at least 3 hr in air prior to use, to remove organic contaminants.
- Clean the tungsten electrodes by gently washing with clean laboratory wipes and methanol, and drying in air.
- Pour 200 ml of ultrapure water (18.2 MΩ cm, <5 ppb TOC) into the 3 L reaction flask.
- Introduce a precleaned and sterilized magnetic stir bar, which will ensure rapid dissolution of soluble gases and mixing of reactants during the experiment.
Attach the tungsten electrodes to the 3 L reaction flask using a minimal amount of vacuum grease, with tips separated by approximately 1 cm inside the flask. Fasten with clips.
Insert an adapter with a built-in stopcock into the neck of the 3 L reaction flask and secure with a clip.
- Attach the 3 L reaction flask to the gas manifold via the adapter. Use a clip or clamp to help secure the flask.
- Lightly grease all connections to ensure a good vacuum seal.
Open all valves and stopcocks on the manifold, except Valve 6 and Stopcock 1 (Figure 4), and turn on the vacuum pump to evacuate the manifold. Once a stable vacuum reading of <1 mmHg has been attained, close Valve 1 and allow the manifold to sit for ~15 min to check for vacuum leaks. If none are detected, proceed to step 2.8. Otherwise troubleshoot the various connections until the leaks can be identified and fixed.
Apply magnetic stirring to the reaction vessel. Open Valve 1 and Stopcock 1 (Figure 4) to evacuate the headspace of the 3 L reaction flask until the pressure has reached <1 mmHg.
Close Valve 1 (Figure 4) and monitor the pressure inside the 3 L reaction flask. The measured pressure should increase to the vapor pressure of water. To ensure that no leaks exist, wait ~5 min at this stage. If the pressure (as read on the manometer) increases while Valve 1 is closed during this step, check for leaks in Stopcock 1 and the various reaction flask connections. If no leak is found, proceed to the next step.
3. Introduction of Gaseous NH3
Calculate the necessary pressure of gaseous NH3 to introduce into the manifold such that 200 mmHg of NH3 will be introduced into the reaction flask. Details on how to do this are provided in the Discussion section.
Close Valves 1 and 6, and Stopcock 1 (Figure 4) before introducing any gas into the manifold. Leave the other valves and stopcock open.
Introduce NH3 into the manifold until a small pressure (approximately 10 mmHg) is reached and then evacuate the manifold to a pressure of <1 mmHg by opening Valve 1 (Figure 4). Repeat 3x.
Introduce NH3 into the manifold to reach the pressure determined in step 3.1.
Open Stopcock 1 (Figure 4) to introduce 200 mmHg of NH3 into the 3 L reaction flask. The NH3 will dissolve in the water in the reaction flask and the pressure will fall slowly.
Once the pressure stops dropping, close Stopcock 1 (Figure 4) and record the pressure read by the manometer. This value represents the pressure inside the flask and will be used to calculate the pressures for other gases that will be introduced into the manifold later.
Open Valve 1 (Figure 4) to evacuate the manifold to a pressure of <1 mmHg.
Close Valve 2 (Figure 4) and disconnect the NH3 gas cylinder from the manifold.
4. Introduction of CH4
Calculate the necessary pressure of CH4 to be introduced into the manifold such that 200 mmHg of CH4 will be introduced into the 3 L reaction flask. Example calculations are shown in the Discussion section.
Connect the CH4 gas cylinder to the manifold.
Open all valves and stopcocks, except Valve 6 and Stopcock 1 (Figure 4), and evacuate the manifold to a pressure of <1 mmHg.
Close Valve 1 once the manifold has been evacuated (Figure 4).
Introduce CH4 into the manifold until a small pressure (approximately 10 mmHg) is obtained. This purges the line of any contaminant gases from preceding steps. Open Valve 1 (Figure 4) to evacuate the manifold to <1 mmHg. Repeat 2x more.
Introduce CH4 into the manifold until the pressure calculated in step 4.1, is reached.
Open Stopcock 1 (Figure 4) to introduce 200 mmHg of CH4 into the 3 L reaction flask.
Close Stopcock 1 once the intended pressure of CH4 has been introduced into the 3 L reaction flask (Figure 4) and record the pressure measured by the manometer.
Open Valve 1 (Figure 4) to evacuate the manifold to <1 mmHg.
Close Valve 2 (Figure 4) and disconnect the CH4 cylinder from the manifold.
5. Introduction of Further Gases (e.g. N2)
At this point, it is not necessary to introduce additional gases. However, if desired, it is recommended to add 100 mmHg of N2. In this case, calculate the necessary pressure of N2 to be introduced into the manifold such that 100 mmHg of N2 will be introduced into the 3 L reaction flask. Example calculations are shown in the Discussion section.
Connect the N2 gas cylinder to the manifold.
Open all valves and stopcocks, except Valve 6 and Stopcock 1 (Figure 4), and evacuate the manifold to a pressure of <1 mmHg.
Close Valve 1 once the manifold has been evacuated (Figure 4).
Introduce N2 into the manifold until a small pressure (approximately 10 mmHg) is obtained. Open Valve 1 (Figure 4) to evacuate the manifold to <1 mmHg. Repeat 2x more.
Introduce N2 into the manifold until the pressure calculated in step 5.1 is reached.
Open Stopcock 1 (Figure 4) to introduce 100 mmHg of N2 into the reaction flask.
Close Stopcock 1 once the intended pressure of N2 has been introduced into the reaction flask, (Figure 4) and record the pressure using the manometer.
Open Valve 1 (Figure 4) to evacuate the manifold to <1 mmHg.
Close Valve 2 (Figure 4) and disconnect the N2 cylinder from the manifold.
6. Beginning the Experiment
Detach the reaction flask from the manifold by closing Stopcock 1 and Valve 1 (Figure 4) once all gases have been introduced into the reaction flask, so that ambient air may enter the manifold and bring the manifold up to ambient pressure.
After carefully disconnecting the reaction flask from the manifold, set the flask somewhere it will not be disturbed (e.g. inside an empty fume hood).
Disconnect the vacuum pump and carefully remove the cold trap and allow venting inside a fully operational fume hood.
Secure the Tesla coil connected to the high frequency spark generator.
Connect the opposite tungsten electrode to an electrical ground to enable the efficient passage of electrical current across the gap between the two electrodes.
Set the output voltage of the spark generator to approximately 30,000 V, as detailed by documents available from the manufacturer.
Prior to initiating the spark, close the fume hood sash, to serve as a safety shield between the apparatus and the experimenter. Turn the Tesla coil on to start the experiment, and allow sparking to continue for 2 weeks (or other desired period) in 1 hr on/off cycles.
7. End of Experiment
Stop the experiment by turning off the Tesla coil.
Open Stopcock 1 (Figure 4) to slowly introduce ambient air into the reaction flask and facilitate the removal of the adapter and the tungsten electrodes so samples can be collected. If desired, a vacuum can be used to evacuate the reaction flask of noxious reaction gases.
8. Collecting Liquid Sample
- Using a pyrolyzed glass pipette, remove liquid samples from the reaction flask, being careful to minimize exposure to contaminants, such as those that might be introduced by touching the pipette to the vacuum grease or other nonsterile surfaces.
- Transfer the sample to a sterile plastic or glass receptacle. Plastic receptacles are less prone to cracking or breaking upon freezing, compared to glass receptacles.
Seal sample containers and store in a freezer capable of reaching temperatures of -20 °C or lower, as insoluble products may prevent the sample solution from freezing at 0 °C.
9. Cleaning the Apparatus
Use clean laboratory wipes to carefully remove vacuum grease from the neck of the apparatus, the adapter and stopcock, and the glass surrounding the tungsten electrodes.
Thoroughly clean the same surfaces described in step 9.1 with toluene to fully remove organic vacuum grease from the glassware. If using silicon grease, the high vacuum grease may remain on the glassware after pyrolysis, creating future problems, as detailed in the Discussion section.
Thoroughly clean the reaction flask with a brush and the following solvents in order: ultrapure water (18.2 MΩ cm, <5 ppb TOC), ultrapure water (18.2 MΩ cm, <5 ppb TOC) with 5% cleaning detergent, methanol, toluene, methanol, ultrapure water (18.2 MΩ cm, <5 ppb TOC) with 5% cleaning detergent, and finally ultrapure water (18.2 MΩ cm, <5 ppb TOC).
Cover all open orifices of the reaction flask with aluminum foil and wrap the adapter and its components in aluminum foil.
Once all the glassware has been wrapped in aluminum foil, pyrolyze for at least 3 hr in air at 500 °C.
Gently clean electrodes with methanol and let air dry.
10. Sample Analysis
Note: When preparing samples for analysis, the use of an acid hydrolysis protocol such as has been described in detail elsewhere15, is useful for obtaining more amino acids. Hydrolysis of a portion of the recovered sample provides the opportunity to analyze both free amino acids as well as their acid-labile precursors that are synthesized under abiotic conditions.
For amino acid analysis, use a suitable technique (such as liquid chromatography and mass spectrometry-based methods, or other appropriate approaches). Such analytical techniques include high performance liquid chromatography with fluorescence detection (HPLC-FD)14, and ultrahigh performance liquid chromatography with fluorescence detection in parallel with time-of-flight positive electrospray ionization mass spectrometry (UHPLC-FD/ToF-MS)12,13. This manuscript describes analysis using mass spectrometric analyses via a triple quadrupole mass spectrometer (QqQ-MS) in conjunction with HPLC-FD.
Representative Results
The products synthesized in electric discharge experiments can be quite complex, and there are numerous analytical approaches that can be used to study them. Some of the more commonly used techniques in the literature for analyzing amino acids are discussed here. Chromatographic and mass spectrometric methods are highly informative techniques for analyzing the complex chemical mixtures produced by Miller-Urey type spark discharge experiments. Amino acid analyses can be conducted using o-phthaldialdehyde/N-acetyl-L-cysteine (OPA/NAC)16, a chiral reagent pair that tags primary amino groups, yielding fluorescent diastereomer derivatives that can be separated on an achiral stationary phase. Figure 2 shows a chromatogram of an OPA/NAC-derivatized amino acid standard obtained by HPLC coupled to fluorescence detection and QqQ-MS. The amino acids contained in the standard include those typically produced in Miller-Urey type spark discharge experiments. The identities of these amino acids are listed in Table 1. Representative fluorescence traces of a typical sample and analytical blank are shown in Figure 3, demonstrating the molecular complexity of Miller-Urey type electric discharge samples. The sample chromatogram in Figure 3 was produced from a spark discharge experiment using the following starting conditions: 300 mmHg of CH4, 250 mmHg of NH3, and 250 ml of water.
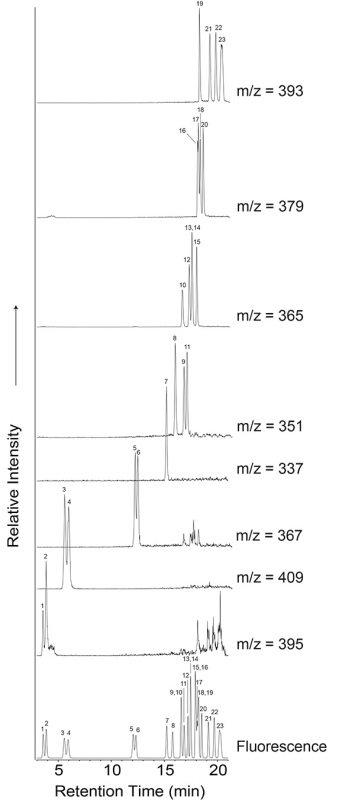 Figure 2. The 3-21 min region of the HPLC-FD/QqQ-MS chromatograms produced from the analysis of an OPA/NAC-derivatized amino acid standard. Amino acid peak identities are listed in Table 1. The fluorescence trace is shown at the bottom and the corresponding extracted mass chromatograms are shown above. The electrospray ionization (ESI) QqQ-MS was operated in positive mode and monitored a mass range of 50-500 m/z. The ESI settings were: desolvation gas (N2) temperature: 350 °C, 650 L/hr; capillary voltage: 3.8 kV; cone voltage: 30 V. The unlabeled peaks in the 367 extracted ion chromatogram are the 13C2 peaks from the 365 extracted ion chromatogram, as a result of the approximately 1% natural abundance of 13C. Click here to view larger image.
Figure 2. The 3-21 min region of the HPLC-FD/QqQ-MS chromatograms produced from the analysis of an OPA/NAC-derivatized amino acid standard. Amino acid peak identities are listed in Table 1. The fluorescence trace is shown at the bottom and the corresponding extracted mass chromatograms are shown above. The electrospray ionization (ESI) QqQ-MS was operated in positive mode and monitored a mass range of 50-500 m/z. The ESI settings were: desolvation gas (N2) temperature: 350 °C, 650 L/hr; capillary voltage: 3.8 kV; cone voltage: 30 V. The unlabeled peaks in the 367 extracted ion chromatogram are the 13C2 peaks from the 365 extracted ion chromatogram, as a result of the approximately 1% natural abundance of 13C. Click here to view larger image.
| Peak | Amino Acid |
| 1 | D-aspartic acid |
| 2 | L-aspartic acid |
| 3 | L-glutamic acid |
| 4 | D-glutamic acid |
| 5 | D-serine |
| 6 | L-serine |
| 7 | Glycine |
| 8 | b-Alanine |
| 9 | D-alanine |
| 10 | g-amino-n-butyric acid (g-ABA) |
| 11 | L-alanine |
| 12 | D-b-amino-n-butyric acid (D-b-ABA) |
| 13 | a-aminoisobutyric acid (a-AIB) |
| 14 | L-b-amino-n-butyric acid (L-b-ABA) |
| 15 | D/L-a-amino-n-butyric acid (D/L-a-ABA) |
| 16 | D-isovaline |
| 17 | L-isovaline |
| 18 | L-valine |
| 19 | e-amino-n-caproic acid (EACA) |
| 20 | D-valine |
| 21 | D-isoleucine |
| 22 | L-isoleucine |
| 23 | D/L-leucine |
Table 1. Peak identities for amino acids detected in the standard and that are typically produced in Miller-Urey type spark discharge experiments.
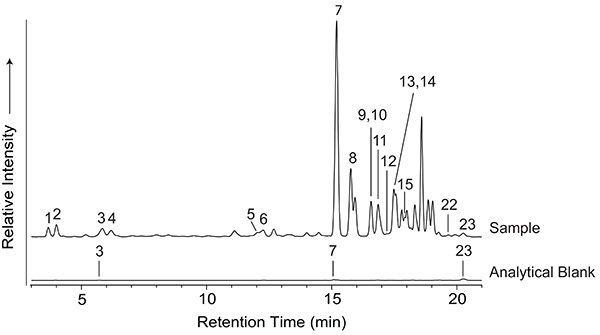 Figure 3. The 3-21 min region of the HPLC-FD chromatograms representative of Miller-Urey type spark discharge experiments. Peaks were identified and quantitated by retention time and mass analysis of target compounds compared to a standard and analytical blank. All target analytes with coeluting fluorescence retention times can be separated and quantitated using mass spectrometry, except for α-AIB and L-β-ABA (peaks 13 and 14), and D/L-norleucine, which coelutes with D/L-leucine (peak 23), under the chromatographic conditions used. D/L-norleucine was added as an internal standard to samples and analytical blanks during sample preparation. Amino acid separation was achieved using a 4.6 mm x 250 mm, 5 μm particle size Phenyl-Hexyl HPLC column. The mobile phase was composed of: A) ultrapure water (18.2 MΩ cm, <5 ppb TOC), B) methanol, and C) 50 mM ammonium formate with 8% methanol, at pH 8. The gradient used was: 0-5 min, 100% C; 5-15 min, 0-83% A, 0-12% B, 100-5% C; 15-22 min, 83-75% A, 12-20% B, 5% C; 22-35 min, 75-35% A, 20-60% B, 5% C; 35-37 min, 35-0% A, 60-100% B, 5-0% C; 37-45 min, 100% B; 45-46 min, 100-0% B, 0-100% C 46-55 min, 100% C. The flow rate was 1 ml/min. Click here to view larger image.
Figure 3. The 3-21 min region of the HPLC-FD chromatograms representative of Miller-Urey type spark discharge experiments. Peaks were identified and quantitated by retention time and mass analysis of target compounds compared to a standard and analytical blank. All target analytes with coeluting fluorescence retention times can be separated and quantitated using mass spectrometry, except for α-AIB and L-β-ABA (peaks 13 and 14), and D/L-norleucine, which coelutes with D/L-leucine (peak 23), under the chromatographic conditions used. D/L-norleucine was added as an internal standard to samples and analytical blanks during sample preparation. Amino acid separation was achieved using a 4.6 mm x 250 mm, 5 μm particle size Phenyl-Hexyl HPLC column. The mobile phase was composed of: A) ultrapure water (18.2 MΩ cm, <5 ppb TOC), B) methanol, and C) 50 mM ammonium formate with 8% methanol, at pH 8. The gradient used was: 0-5 min, 100% C; 5-15 min, 0-83% A, 0-12% B, 100-5% C; 15-22 min, 83-75% A, 12-20% B, 5% C; 22-35 min, 75-35% A, 20-60% B, 5% C; 35-37 min, 35-0% A, 60-100% B, 5-0% C; 37-45 min, 100% B; 45-46 min, 100-0% B, 0-100% C 46-55 min, 100% C. The flow rate was 1 ml/min. Click here to view larger image.
Discussion
Numerous steps in the protocol described here are critical for conducting Miller-Urey type experiments safely and correctly. First, all glassware and sample handling tools that will come in contact with the reaction flask or sample need to be sterilized. Sterilization is achieved by thoroughly rinsing the items in question with ultrapure water (18.2 MΩ cm, <5 ppb TOC) and then wrapping them in aluminum foil, prior to pyrolyzing at 500 °C in air for at least 3 hr. Once the equipment has been pyrolyzed and while preparing samples for analysis, care must be taken to avoid organic contamination. The risk of contamination can be minimized by wearing nitrile gloves, a laboratory coat, and protective eyewear. Be sure to work with samples away from one's body as common sources of contamination include finger prints, skin, hair, and exhaled breath. Avoid contact with wet gloves and do not use any latex or Nylon materials. Second, thorough degassing of the reaction flask prior to gas addition into the reaction flask is critical. The presence of even small amounts of molecular oxygen in the reaction flask poses an explosion risk when the spark is discharged into inflammable gases such as CH4. While degassing the flask, the water inside the flask will boil, which will prevent a stable reading. At this stage there are two options: 1) degas the flask via freeze-thaw cycles (typically 3 are used), or 2) simply degas the liquid solution. In the latter case, some water will be lost, however, the amount will be relatively minor compared to the remaining volume. Third, a well-equipped and efficient setup must be carefully constructed to establish a consistent spark across the electrodes throughout the entirety of the experiment. BD-50E Tesla coils are not designed for prolonged operation, as they are intended for vacuum leak detection. Intermittent cooling of the Tesla coil is thus recommended for extended operational lifetime. There are multiple ways of achieving this. One simple way is to attach a timer in-line between the spark tester and its power supply and program the timer such that it alternates in 1 hour on/off cycles. Cooling the Tesla coil with a commercial fan may also be necessary to prolong the life of the Tesla coil. The Tesla coil tip should be touching or almost touching one of the tungsten electrodes; a distance between the two of approximately 1 mm or less. Additionally, an intense discharge can be achieved using a length of conductive metal wire with a loop in one end draped lightly over the electrode opposite the one touching the Tesla coil to avoid breaking the seal to the contents. It is also recommended to have a second spark generator available in case the primary spark generator fails due to extended use.
There are many additional notes worth keeping in mind when carrying out various steps in the protocol outlined here. When preparing the manifold system for an experiment and using a mercury manometer, it is generally conceded that a precision of 1 mmHg is the best achievable, due to the resolution of the human eye. Some gases may present conductivity problems with resistance-based gauges. Mercury manometers present potential spill hazards, which should be prepared for in advance.
While assembling the 3 L reaction flask, the use of silicon vacuum grease can mitigate potential organic contamination, but care should be taken to remove this thoroughly between runs. Failure to do so will result in the accumulation of silica deposits during high-temperature pyrolysis, which can interfere with vacuum seals. Additionally, the tungsten electrodes are commercially available as 2% thoriated tungsten and should be annealed into half-round ground glass fittings. Do not pyrolyze the glass-fitted tungsten electrodes in an oven. The coefficients of thermal expansion of tungsten and glass are different and heating above 100 °C may weaken the seal around the glass annealed electrodes and introduce leaks to the system. Also, ultrapure water can be introduced into the 3 L reaction flask by pouring, using care to avoid contact with any grease on the port used, or by pipetting, using a prepyrolyzed glass pipette. The aqueous phase in the reaction flask can be buffered, if desired. For example, Miller and colleagues9 buffered the solution to pH ~8.7 with an NH3/NH4Cl buffer. To do this the aqueous phase is made 0.05 M in NH4Cl prior to introducing it into the reaction flask. NH4Cl of 99.5% purity, or greater, should be used. The remainder of the NH3 is then added to the reaction flask as a gas.
In preparation for gas introduction into the 3 L reaction flask, the flask can be secured onto the manifold by placing the flask on a cork ring, set atop a lab jack and gently raising the flask assembly until a snug connection is achieved. When checking for leaks, it is worth noting that likely sources of leaks include poor seals at the junctions of the half-round ground glass joints, which attach the tungsten electrodes to the reaction flask, and the stopcock of the adapter attached to the neck of the 3 L reaction flask. If leaks from these sources are detected, carefully remove the 3 L reaction flask from the manifold, wipe these areas with clean laboratory tissue, reapply a fresh coating of vacuum grease and reattach the flask to the manifold to search for leaks. If no leaks are found, proceed to introduce gases into the reaction flask.
While introducing gases into the apparatus, gas cylinders should be securely fastened to a support. Care should be taken to introduce gases slowly. Valves on gas cylinders should be opened slowly and carefully while monitoring the manometer to avoid over-pressuring the glassware and attached fittings. It is important to note that while adding NH3 into the reaction flask, because NH3 is appreciably soluble in water below the pKa of NH4+ (~9.2), essentially all of the NH3 gas introduced into the manifold will dissolve in the aqueous phase, rendering the final pressure in the flask and manifold as the vapor pressure of water at the ambient temperature. Once this pressure is attained, one may assume the transfer is complete. The following are examples of the calculations that must be executed in order to precisely introduce gases into the reaction flask at their desired pressures:
Introduction of Gaseous NH3
Due to the solubility of NH3, essentially all of it will transfer from the manifold to the reaction flask and dissolve in the aqueous phase as long as the NH3 in the manifold is at a higher pressure than the vapor pressure of water in the reaction flask. Therefore, the ambient temperature should be noted and the vapor pressure of water at that temperature should be referenced prior to introducing NH3 into the manifold. The target pressure of NH3 to be introduced into the reaction flask should be equal to the target pressure of NH3 in the 3 L reaction flask, plus the vapor pressure of water in the reaction flask, at the recorded ambient temperature. For example, at 25 °C, the vapor pressure of water is approximately 24 mmHg. Thus, in order to introduce 200 mmHg of NH3 into the reaction flask, load roughly 225 mmHg of NH3 into the manifold prior to transferring NH3 from the manifold and into the reaction flask. This will result in approximately 200 mmHg of NH3 being introduced into the reaction flask.
Introduction of CH4
After NH3 addition and its dissolution in the aqueous phase, the pressure in the headspace of the reaction flask will be equal to the vapor pressure of water at 25 °C, approximately 24 mmHg. This value will be used, in conjunction with the example manifold shown in Figure 4, to carry out a calculation for how much CH4 to introduce into the manifold such that 200 mmHg of CH4 will be introduced into the reaction flask:
P1 = total pressure desired throughout the entire system, including the reaction flask V1 = total volume of the entire system, including the reaction flask
P2 = pressure of CH4 needed to fill manifold volume prior to introduction into reaction flask V2 = volume of manifold used for gas introduction
P3 = pressure already in the headspace of the reaction flask V3 = volume of the reaction flask
P1 = 200 mmHg of CH4 + 24 mmHg of H2O = 224 mmHg V1 = 3,000 ml + 100 ml + 300 ml + 40 ml + 20 ml + 3,000 ml + 40 ml + 500 ml = 7,000 ml
P2 = pressure of CH4 being calculated V2 = 100 ml + 300 ml + 40 + 20 + 3,000 ml+ 40 ml + 500 ml = 4,000 ml
P3 = 24 mmHg of H2O V3 = 3,000 ml
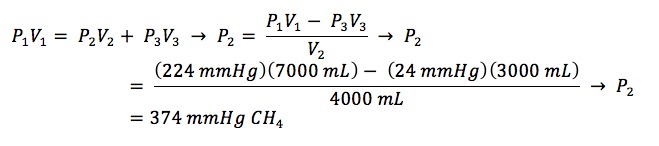
Introduction of N2
After introduction of CH4, the headspace of the reaction flask is occupied by 200 mmHg of CH4 and 24 mmHg of H2O for a total of 224 mmHg. This value will be used, along with the dimensions of the example manifold shown in Figure 4, to calculate the N2 pressure that needs to be introduced into the manifold such that 100 mmHg of N2 will be introduced into the reaction flask:
P1 = total pressure desired throughout the entire system, including the reaction flask V1 = total volume of the entire system, including the reaction flask
P2 = pressure of N2 needed to fill manifold volume prior to introduction into reaction flask V2 = volume of manifold used for gas introduction
P3 = pressure already in the headspace of the reaction flask V3 = volume of the reaction flask
P1 = 24 mmHg of H2O + 200 mmHg of CH4 + 100 mmHg of N2 = 324 mmHg V1 = 3,000 ml + 100 ml > + 300 ml + 40 ml + 20 ml + 3,000 ml + 40 ml + 500 ml = 7,000 ml
P2 = pressure of N2 being calculated V2 = 100 ml + 300 ml + 40 ml + 20 ml + 3,000 ml + 40 ml + 500 ml = 4,000 ml
P3 = 200 mmHg of CH4 + 24 mmHg of H2O = 224 mmHg V3 = 3,000 ml
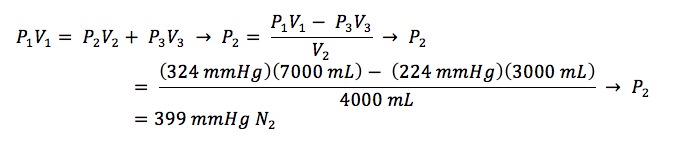
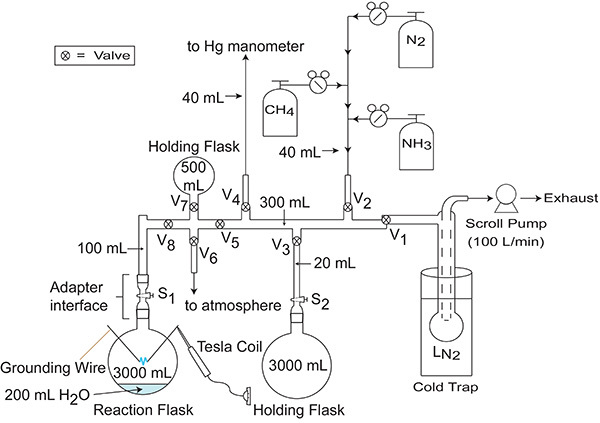 Figure 4. Manifold/vacuum system used to introduce gases into the 3 L reaction flask. Valves controlling gas flow are labeled as V1 - V8, while stopcocks controlling gas flow are labeled as S1 and S2. It is worth noting that while Valves 1, 2, and 6, and Stopcock 1 are referred to explicitly in the protocol, the other valves and stopcock in the manifold shown here are useful for adding or removing volume (i.e. holding flasks) to or from the manifold. For example, when introducing gases into the manifold at relatively high pressures (approximately 500 mmHg or greater), it is advised that the experimenter makes use of all purge flasks attached to the manifold to increase the accessible volume of the manifold and help minimize the risk of over-pressuring the manifold.
Figure 4. Manifold/vacuum system used to introduce gases into the 3 L reaction flask. Valves controlling gas flow are labeled as V1 - V8, while stopcocks controlling gas flow are labeled as S1 and S2. It is worth noting that while Valves 1, 2, and 6, and Stopcock 1 are referred to explicitly in the protocol, the other valves and stopcock in the manifold shown here are useful for adding or removing volume (i.e. holding flasks) to or from the manifold. For example, when introducing gases into the manifold at relatively high pressures (approximately 500 mmHg or greater), it is advised that the experimenter makes use of all purge flasks attached to the manifold to increase the accessible volume of the manifold and help minimize the risk of over-pressuring the manifold.
After initiating the experiment, the system must be checked on regularly to ensure the experiment is running properly. Things to check include: 1) the spark generator is producing a spark, and 2) the spark is being generated across the tungsten electrodes in a continuous manner. If the above conditions are not met, disconnect the Tesla coil from its power supply and replace it with the backup Tesla coil. Meanwhile, repairs to the malfunctioning Tesla coil can be made. Often times, the contact plates inside the spark generator housing can become corroded from extended use and should be polished, or replaced.
Upon completion of the experiment, the gases in the head-space may be irritating to the respiratory system. Harmful gases, such as hydrogen cyanide4 can be produced by the experiment. If the experimenter is not collecting gas samples for analysis, it may be helpful to connect the apparatus to a water aspirator to evacuate volatiles for approximately one hour after completion of the experiment, while the apparatus remains in the fume hood, prior to collecting liquid samples. For safety reasons, it is advised that the apparatus is vented in a fully-operational fume hood. Sample collection should be performed in an operational fume hood and sample handling in a positive-pressure HEPA filtered flow bench is recommended.
Among the numerous types of products formed by spark discharge experiments, amino acids are of significance. Amino acids are synthesized readily via the Strecker synthesis17. The Strecker synthesis of amino acids involves the reaction of aldehydes or ketones and HCN generated by the action of electric discharge on the gases introduced into the reaction apparatus, which upon dissolving in the aqueous phase, may react with ammonia to form α-aminonitriles that undergo hydrolysis to yield amino acids. This is, of course, but one mechanism of synthesis, and others may also be operative, such as direct amination of precursors including acrylonitrile to give β-alanine precursors, or direct hydrolysis of higher molecular weight tholin-like material to give amino acids directly, by-passing the Strecker mechanism.
Amino acid contamination of the samples produced by Miller-Urey experiments can occur if the precautions mentioned earlier are not followed explicitly. During sample analysis, it is important to search for signs of terrestrial contamination that may have originated from sample handling or sample storage. The use of OPA/NAC16 in conjunction with LC-FD techniques allows for the chromatographic separation of D- and L-enantiomers of amino acids with chiral centers and their respective, individual quantitation. Chiral amino acids synthesized by the experiment should be racemic. Acceptable experimental error during the synthesis of amino acids with chiral centers is generally considered to be approximately 10%. Therefore chiral amino acid D/L ratios suggestive of enrichment in one enantiomer by more than 10% is a good metric by which to determine if the sample has been contaminated.
The methods presented here are intended to instruct how to conduct a Miller-Urey type spark discharge experiment; however, there are limitations to the technique described here that should be noted. First, heating the single 3 L reaction flask (Figure 1B), will result in condensation of water vapor onto the tips of the electrodes, dampening the spark, and reducing the generation of radical species that drive much of the chemistry taking place within the experiment. Furthermore, the use of a heating mantle to heat the apparatus is not necessary to synthesize organic compounds, such as amino acids. This differs from Miller's original experiment where he used a more complex, custom-built, dual flask apparatus (Figure 1A)5 and heated the small flask at the bottom of the apparatus, which had water in it (Figure 1A). Heating the apparatus helped with circulation of the starting materials and aimed to mimic evaporation in an early Earth system. Second, the protocol detailed here recommends a 1 hr on/off cycle when using the Tesla coil, which effectively doubles the amount of time an experiment takes to complete, compared to the experiments conducted by Miller, as he continuously discharged electricity into the system4. Third, as spark generators are not intended for long-term use, they are prone to malfunction during prolonged use and must be regularly maintained and sometimes replaced by a back-up unit, if the primary spark generator fails during the course of an experiment. Last, the protocol described here involves the use of glass stopcocks, which require high vacuum grease to make appropriate seals. If desired, polytetrafluoroethylene (PTFE) stopcocks can be used to avoid vacuum grease. However, if examining these stopcocks for potential leaks with a spark leak detector, be cautious to not overexpose the PTFE to the spark as this can compromise the integrity of the PTFE and lead to poor seals being made by these stopcocks.
The significance of the method reported here with respect to existing techniques, lies within its simplicity. It uses a commercially available 3 L flask, which is also considerably less fragile and easier to clean between experiments than the original design used by Miller5. Because the apparatus is less cumbersome, it is small enough to carry out an experiment inside a fume hood.
Once the technique outlined here has been mastered, it can be modified in a variety of ways to simulate numerous types of primitive terrestrial environments. For example, more oxidized gas mixtures can be used14,18,19. Furthermore, using modifications of the apparatus, the energy source can be changed, for example, by using a silent discharge4, ultraviolet light20, simulating volcanic systems4,12,21, imitating radioactivity from Earth's crust22, and mimicking energy produced by shockwaves from meteoritic impacts23, and also cosmic radiation18,19.
The classic Miller-Urey experiment demonstrated that amino acids, important building blocks of biological proteins, can be synthesized using simple starting materials under simulated prebiotic terrestrial conditions. The excitation of gaseous molecules by electric discharge leads to the production of organic compounds, including amino acids, under such conditions. While amino acids are important for contemporary biology, the Miller-Urey experiment only provides one possible mechanism for their abiotic synthesis, and does not explain the origin of life, as the processes that give rise to living organisms were likely more complex than the formation of simple organic molecules.
Disclosures
The authors declare no competing financial interests.
Acknowledgments
This work was jointly supported by the NSF and NASA Astrobiology Program, under the NSF Center for Chemical Evolution, CHE-1004570, and the Goddard Center for Astrobiology. E.T.P. would like to acknowledge additional funding provided by the NASA Planetary Biology Internship Program. The authors also want to thank Dr. Asiri Galhena for invaluable help in setting up the initial laboratory facilities.
References
- Oparin AI. The Origin of Life. Izd. Moskovshii Rabochii; 1924. [Google Scholar]
- Haldane JB. The origin of life. Rationalist Annu. 1929;148:3–10. [Google Scholar]
- Garrison WM, Morrison DC, Hamilton JG, Benson AA, Calvin M. Reduction of Carbon Dioxide in Aqueous Solutions by Ionizing Radiation. Science. 1951;114:416–418. doi: 10.1126/science.114.2964.416. [DOI] [PubMed] [Google Scholar]
- Miller SL. Production of Some Organic Compounds under Possible Primitive Earth Conditions. J. Am. Chem. Soc. 1955;77:2351–2361. [Google Scholar]
- Miller SL. A Production of Amino Acids Under Possible Primitive Earth Conditions. Science. 1953;117:528–529. doi: 10.1126/science.117.3046.528. [DOI] [PubMed] [Google Scholar]
- Heyns HK, Walter W, Meyer E. Model experiments on the formation of organic compounds in the atmosphere of simple gases by electrical discharges (Translated from German) Die Naturwissenschaften. 1957;44:385–389. [Google Scholar]
- Ponnamperuma C, Woeller F. α-Aminonitriles formed by an electric discharge through a mixture of anhydrous methane and ammonia. Biosystems. 1967;1:156–158. doi: 10.1016/0303-2647(67)90031-7. [DOI] [PubMed] [Google Scholar]
- Oró J. Synthesis of Organic Compounds by Electric Discharges. Nature. 1963;197:862–867. [Google Scholar]
- Ring D, Wolman Y, Friedmann N, Miller SL. Prebiotic Synthesis of Hydrophobic and Protein Amino Acids. Proc. Natl. Acad. Sci. U.S.A. 1972;69:765–768. doi: 10.1073/pnas.69.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolman Y, Haverland WJ, Miller SL. Nonprotein Amino Acids from Spark Discharges and Their Comparison with the Murchison Meteorite Amino Acids. Proc. Natl. Acad. Sci. U.S.A. 1972;69:809–811. doi: 10.1073/pnas.69.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscoe S, Miller SL. Energy Yields for Hydrogen Cyanide and Formaldehyde Syntheses: The HCN and Amino Acid Concentrations in the Primitive Ocean. Orig. Life. 1987;17:261–273. doi: 10.1007/BF02386466. [DOI] [PubMed] [Google Scholar]
- Johnson AP, et al. The Miller Volcanic Spark Discharge Experiment. Science. 2008;322 doi: 10.1126/science.1161527. [DOI] [PubMed] [Google Scholar]
- Parker ET, et al. Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment. Proc. Natl. Acad. Sci. U.S.A. 2011;108:5526–5531. doi: 10.1073/pnas.1019191108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaves HJ, Chalmers JH, Lazcano A, Miller SL, Bada JL. A reassessment of prebiotic organic synthesis in neutral planetary atmospheres. Orig. Life Evol. Biosph. 2008;38:105–115. doi: 10.1007/s11084-007-9120-3. [DOI] [PubMed] [Google Scholar]
- Glavin DP, et al. Amino acid analyses of Antarctic CM2 meteorites using liquid chromatography-time of flight-mass spectrometry. Meteorit. Planet. Sci. 2006;41:889–902. [Google Scholar]
- Zhao M, Bada JL. Determination of α-dialkylamino acids and their enantiomers in geologic samples by high-performance liquid chromatography after a derivatization with a chiral adduct of o-phthaldialdehyde. J. Chromatogr. A. 1995;690:55–63. doi: 10.1016/0021-9673(94)00927-2. [DOI] [PubMed] [Google Scholar]
- Strecker A. About the artificial formation of lactic acid and a new Glycocoll the homologous body Justus Liebigs Annalen der Chemie. 1850;75:27–45. [Google Scholar]
- Miyakawa S, Yamanashi H, Kobayashi K, Cleaves HJ, Miller SL. Prebiotic synthesis from CO atmospheres: implications for the origins of life. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14628–14631. doi: 10.1073/pnas.192568299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Kaneko T, Saito T, Oshima T. Amino Acid Formation in Gas Mixtures by Particle Irradiation. Orig. Life Evol. Biosph. 1998;28:155–165. doi: 10.1023/a:1006561217063. [DOI] [PubMed] [Google Scholar]
- Sagan C, Khare BN. Long-Wavelength Ultraviolet Photoproduction of Amino Acids on the Primitive Earth. Science. 1971;173:417–420. doi: 10.1126/science.173.3995.417. [DOI] [PubMed] [Google Scholar]
- Harada K, Fox SW. Thermal Synthesis of Natural Amino-Acids from a Postulated Primitive Terrestrial Atmosphere. Nature. 1964;201:335–336. doi: 10.1038/201335a0. [DOI] [PubMed] [Google Scholar]
- Ponnamperuma C, Lemmon RM, Mariner R, Calvin M. Formation of Adenine by Electron Irradiation of Methane Ammonia, and Water. Proc. Natl. Acad. Sci. USA. 1963;49:737–740. doi: 10.1073/pnas.49.5.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Nun A, Bar-Nun N, Bauer SH, Sagan C. Shock Synthesis of Amino Acids in Simulated Primitive Environments. Science. 1970;168:470–473. doi: 10.1126/science.168.3930.470. [DOI] [PubMed] [Google Scholar]