

Abstract
Sleep behavior remains one of the most enigmatic areas of life. The unanswered questions range from “why do we sleep?” to “how we can improve sleep in today's society?” Identification of mutations responsible for altered circadian regulation of human sleep lead to unique opportunities for probing these territories. In this review, we summarize causative circadian mutations found from familial genetic studies to date. We also describe how these mutations mechanistically affect circadian function and lead to altered sleep behaviors, including shifted or shortening of sleep patterns. In addition, we discuss how the investigation of mutations can not only expand our understanding of the molecular mechanisms regulating the circadian clock and sleep duration, but also bridge the pathways between clock/sleep and other human physiological conditions and ailments such as metabolic regulation and migraine headaches.
Keywords: Human genetics, mouse model, sleep behavior, sleep duration, sleep schedule
Introduction
Sleep is a fundamental biological phenomenon for most organisms and humans spend, on average, a third to a fourth of their lives in the state of sleep. The lack of sleep and dysregulation of daily sleep rhythms have been linked to a number of ailments and outcomes; cancer (Sahar & Sassone-Corsi, 2009), type two diabetes (Huang et al., 2011), metabolic syndrome (Wolk & Somers, 2007), cellular stress (Hardeland et al., 2003) and most seriously, death (Montagna, 2005). Though great strides have been made in understanding sleep by utilizing model organisms, the regulation of sleep and the functional roles of sleep for humans remain mostly unclear (Frank, 2006). The molecular benefits of sleep have been linked to the pruning and reorganization of dendrites (Benington & Frank, 2003; Maret et al., 2011), and to the modification of gene expression for the synthesis of new biomolecules and metabolites (Tononi & Cirelli, 2006). Theories on the necessity of sleep range from the conservation of energy (Jung et al., 2011) to metabolic repair during sleep (Bonnet & Arand, 1996).
The Earth rotates with a 24-h (circadian) cycle and this dictates that many (if not all) of our physiological functions are subject to circadian regulation (Dunlap, 1999). What we have learnt from past studies is that our endogenous circadian clock regulates many of our body functions including sleep behavior. The most accepted model for describing sleep behavior, the “Two Process Model”, was proposed by Borbély in 1982; Process S represents sleep homeostasis and process C represents circadian rhythm regulation (Borbély & Achermann, 1999). The “Two Process Model” postulates that the interaction between the sleep–wake-dependent “process S” and the circadian “process C” can explain essential aspects of sleep regulation. How both of these processes are changed by human circadian mutations will be discussed throughout this review. Research into the identification of non-circadian genes that regulate sleep homeostasis has been quite fruitful. These non-circadian genes and regulatory networks have been detailed excellently in the following reviews (Andretic et al., 2008; Crocker & Sehgal, 2010; Sehgal & Mignot, 2011).
Investigation of human behavioral traits is inherently challenging since we are known to modify our behaviors by social, cultural and habitual factors. Regardless, the connection between genes and behavioral traits has been established (Plomin et al., 2012). For humans, one of the most compelling evidence for genetic influences on behavior came from the studies done by Thomas Buchard and his colleagues at the Minnesota Center of Twin and Family Research (Bouchard et al., 1990). After decades of study, they concluded that there are deep-seated psychological, behavioral and emotional traits that are determined at birth. Hence, human genetics can be used as a powerful tool to investigate human behaviors. In addition, the wealth of knowledge and information that have been accumulated in the past 20–30 years from studies of model organisms, especially fruit flies (Hardin, 2011) and mice (Lowrey & Takahashi, 2011), has augmented enormously to the understanding of human sleep behaviors. Contributions from these diverse fronts have significantly improved our understanding on the regulatory mechanisms of human sleep schedule in recent years which will be discussed in this review.
Human circadian sleep phenotypes
The majority of people go to bed from 10 pm to midnight and gets up between 6–8 O'clock in the morning. There is a small proportion of people who are either night owls (staying awake until late into the night) or morning larks (demonstrating an extreme early morning awakening) (Jones et al., 1999). Besides environmental influences, rare genetic variants can also directly shift sleep schedules. The first genetic form of human sleep schedule behavior was reported in 1999 (familial advanced sleep phase, FASP) (Jones et al., 1999). FASP individuals demonstrate a forward shift in six different measurements related to sleep (including sleep onset time, sleep offset time, dim light melatonin onset time, core body temperature, and rapid eye movement (REM) sleep onset time) compared to control subjects. Since the discovery of this rare genetic sleep trait, significant progress has been made in our understanding of human circadian rhythm disorders with the characterization of additional sleep phenotypes.
The International Classification of Sleep Disorders includes approximately 60 disorders of human sleep, among them are circadian rhythm sleep disorders (Medicine, n.d.). Circadian rhythm sleep disorders usually exhibit as a social problem in a person's sleep/wake timing, though there are many other sleep disorders demonstrating sleep dysfunction with sleep at the “wrong times”. Circadian rhythm sleep disorders individuals suffer from difficulty initiating or ending sleep at appropriate social times.
Circadian rhythm sleep disorders include the following types according to the American Academy of Sleep Medicine (AASM) (Sack et al., 2007a, b). Advanced sleep phase disorder (ASPD) individuals have earlier sleep/wake times than the average population and their entire sleep–wake cycle is shifted forward several hours. ASPD people are described as “morning larks” that demonstrate an extreme early morning awakening before others are active. However, they also have difficulty in staying awake to satisfy domestic responsibilities in the evening, resulting in significant sleep deprivation if social responsibilities keep them from staying with their internal natural biological clock. It is worth noting, ASPD is more commonly seen in the elderly (Sack et al., 2007b). However, unless patients complain of a problem with waking early, ASPD can be described only as advanced sleep phase (ASP) or with a family history of familial advanced sleep phase (FASP).
In contrast to ASPD, delayed sleep phase disorder (DSPD) is marked by the opposite phenomena where the sleep–wake cycle is shifted later, and DSPD patients feel wide awake, energetic and motivated until late in the night. Although, similar to ASPD, unless patients complain of a problem with waking late, DSPD can be described only as delayed sleep phase (DSP) or with a family history of familial delayed sleep phase (FDSP). DSPD individuals often suffer from sleep deprivation since their sleep onset is delayed by the biological clock and morning waking time is dictated by the alarm clock and social responsibilities. DSPD has an estimated prevalence of 7%–16% in adolescents and young adults (Sack et al., 2007a, b).
Another sleep phenotype is the free-running sleep disorder (FRSD) where the sleep–wake cycle for these individuals is shifted approximately 1 h later everyday. This disorder is linked to retinal blindness and likely precipitates from the lack of light resetting of the circadian clock (Sack, 2007a, b).
Finally, irregular sleep–wake disorder (ISWD) individuals have an undefined sleep–wake cycle where sleep occurs throughout the day. This is often seen in dementia patients who are suffering from neurological dysfunction and can be seen as well in mentally retarded patients. These individuals also suffer from insomnia, as they are unable to experience consolidated sleep (Sack, 2007a, b).
Mammalian molecular clock
Circadian rhythmicity was first noted in the observation that leaf movements (opening and closing) followed a daily rhythm even in the absence of sunlight (de Mairan, 1729). It is now established that the circadian rhythms of human body functions are regulated by an endogenous molecular clock. The mammalian molecular clock is composed of a core set of transcription factors that form functional regulatory feedback loops (Brown et al., 2012; Mohawk et al., 2012; Takahashi et al., 2008). The main feedback loop consists of Period (PER 1/2), Cryptochrome (CRY 1/2), Brain and muscle ARNT-like 1 (BMAL1) and Circadian locomotor output cycles kaput (CLOCK) (Brown et al., 2012; Takahashi et al., 2008). BMAL1 and CLOCK (or NPAS2, a paralog of CLOCK) form a heterodimer and binds to the promoter element described as an enhancer box (E-BOX) of PER and CRY. The transcription and translation of PER and CRY increase protein levels in the cytoplasm, whereby the two proteins then heterodimerize and translocate into the nucleus. In the nucleus, the CRY/PER heterodimer binds to BMAL1/CLOCK to repress transcriptional activation, leading to the termination of their own transcription (Figure 1). PER and CRY are subsequently degraded through the proteasomal pathway, allowing for the reactivation of transcription of PER and CRY by BMAL1/ CLOCK (Brown et al., 2012; Mohawk et al., 2012; Takahashi et al., 2008) (Figure 1). This transcriptional/translational negative feedback loop takes approximately 24 h to complete, thus defining the circadian period, and denotes the proteins involved in the circadian cycle as the “core clock” (Takahashi, 2004). The core circadian loop is further regulated by post-translational regulation summarized later in this review. Circadian regulators that make up additional negative feedback loops or interlocking loops are: activators RAR-related orphan receptor A (RORA), D site of albumin promoter-binding protein (DBP), repressors nuclear receptor subfamily 1, group D, member 1 (NR1D1/REV-ERB) and basic helix-loop-helix family, members e40 and e41 (DEC1, 2 respectively). These activators and repressors act in a circadian manner, providing additional stability and levels of regulation (Ueda et al., 2005). The cycling of these factors operates without requiring extrinsic feedback, and exists in most cell types. Approximately 3%–10% of gene transcripts oscillate in a circadian manner, whose subsets vary according to cell type (Akhtar et al., 2002; Duffield et al., 2002; Hughes et al., 2009; Miller et al., 2007; Panda et al., 2002; Storch et al., 2002). The expression levels of many clock components themselves also demonstrate oscillation. Interestingly, precise rhythmicity of PER2 is necessary for driving cellular circadian oscillations, whereas oscillations of other core clock components such as CRY1, CLOCK and BMAL1 are less critical (Chen et al., 2009).
Figure 1.

Circadian clockwork. The core circadian clock is made up of two interlocking transcriptional and post-translationally regulated feedback loops. BMAL1 and CLOCK heterodimerize to form an activating transcriptional complex that binds to the E-Box on the genes PER, CRY, ROR and REV-ERB. The production of PER and CRY protein builds until PER and CRY are modified post-translationally and then heterodimerize and translocate into the nuclei where they bind to CLOCK and BMAL1 and block the transcriptional activation of E-BOX specific targets. PER, CRY and BMAL1 are degraded by proteasomes. In a second loop, BMAL1 transcription is activated and repressed by ROR and REV-ERB respectively by binding to the RRE DNA element. (see colour version of this figure at www.informahealthcare.com/bmg).
Post-translational modifications in molecular clock
Post-translational regulation gives the core clock its ability to generate precisely tunable timing, since different post-translational modifications can mark the different circadian proteins for different functions at different times over the 24-h clock. Utilizing post-translational modifications the clock is directly tied to the cells’ intermediary metabolism, linking the small molecule metabolites; ATP, Acetyl CoA, NAD+, UDP-GlcNAc, to the core clock's timing. For instance, PER2 functions through heterodimerization with CRY for its translocation and the suppression of CLOCK/ BMAL1 gene activation (Brown et al., 2012; Mohawk et al., 2012; Takahashi et al., 2008), and this heterodimerization is likely modulated by numerous post-translational modifications to ensure the correct and precise timing of transcriptional inhibition. Post-translational modifications likely regulate each of these steps during the cycle of PER2 allowing for the exact control of clock gene regulation and its fine adjustment to outside stimulus. All in all, these various regulatory pathways highlight the intricacy of post-translational regulatory mechanisms of the circadian clock.
Besides PER2, many other circadian clock proteins such as CRY, BMAL1 and CLOCK have been identified as targets of post-translational modifications (Gallego & Virshup, 2007). In fact, an understanding of the importance of complex post-translational modification in regulation of circadian rhythms emerged in recent years. Post-translational modifications are central to the hypothesis that circadian mutations affect sleep schedules through a disruption or change of normal post-translational modifications throughout the circadian period. Here, we first review the known complex network of post-translational modifications and later detail the mutations that affect circadian rhythms through post-translational alterations.
PER2 is the best example illustrating the complexity of post-translational regulation for the molecular clock (Gallego & Virshup, 2007; Vanselow & Kramer, 2007). Phosphorylation has been identified on 21 of 247 serine or threonine residues of mPER2 (Vanselow et al., 2006). Although the mechanistic consequences for most of these phosphorylation sites are unknown, it is hypothesized that phosphorylation localized in the N-terminus of mPER2 can lead to the stabilization and nuclear localization of the protein by CKII (Maier et al., 2009). Also, phosphorylation in the CKI domain increases the half-life of mPER2 (Shanware et al., 2011; Vanselow et al., 2006; Xu et al., 2007). In addition, as described later, the phosphorylation of PER2 at one of the key serine residues (S662) can modulate the period length (Xu et al., 2007). Other clock proteins such as BMAL1 and CRY are also regulated by phosphorylation. Specifically, BMAL1 has been shown to be phosphorylated by c-Jun N- terminal kinase (JNK), and when BMAL1 is hypophosphorylated due to JNK inhibition, the circadian period is lengthened (Yoshitane et al., 2012). Furthermore, BMAL1 is phosphorylated by Protein Kinase Cγ (PKCγ) in response to the food entrainable oscillation. This phosphorylation stabilizes BMAL1 protein by reducing BMAL1 ubiquitination (Zhang et al., 2012). CRY2 is regulated by phosphorylation in a sequential pattern initiated by dual specificity tyrosinephosphorylation-regulated kinase 1 A (DYRK1A) at serine 557 then phosphorylation at serine 553 by glycogen synthase kinase 3β (GSK-3β) leading to CRY2 degradation (Kurabayashi et al., 2010). Conversely, CRY1 is regulated in the liver by adenosine monophosphate-activated protein kinase (AMPK)-mediated phosphorylation leading to CRY1 depletion from the nucleus (Lamia et al., 2009).
Acetylation has gained recent notoriety as another post-translational modification that regulates cellular protein functions (Haigis & Sinclair, 2010; Peng & Seto, 2011). In particular, acetylation and the subsequent deacetylation by Sirtuin-1 (SIRT1) are also implicated in the regulation and degradation of PER2 (Asher et al., 2008). Although the specific residues for acetylation of PER2 and how acetylation affects PER2 biophysically are unknown, SIRT1-mediated deacetylation promotes the degradation of PER2, and loss of SIRT1 increases PER2 protein stability (Asher et al., 2008). Similarly, BMAL1 is regulated by reversible acetylation, specifically acetylated at lysine 537 by the acetyltransferase activity of CLOCK, and deacetylated by SIRT1 (Nakahata et al., 2008). Acetylation of BMAL1 confers circadian timing of BMAL1 by metabolite sensing through nicotinamide adenine dinucleotide (NAD+) (Ramsey et al., 2009).
Moreover, post-translational O-linked N-acetylglucosa-mine modification (O-GlcNAcylation) has recently been shown to exhibit regulatory effects on the circadian clock. O-GlcNAcylation in the heart exhibits diurnal oscillation, which is regulated by glucose, O-GlcNAc transferase (OGT) and O-GlcNAc hydrolase (OGA) levels (Durgan et al., 2011). OGA levels and OGT activities in turn oscillate with daily rhythms. On the other hand, O-GlcNAcylation levels can modulate the clock speed. Higher O-GlcNAcylation leads to longer circadian period and lower O-GlcNAcylation shortens the period (Kaasik et al., 2013; Kim et al., 2012). Like phosphorylation, O-GlcNAcylation regulates the molecular clock through various pathways including modification of transcriptional factor activities, nuclear translocation of clock proteins, and ubiquitin-mediated targeting for degradation of clock proteins (Kaasik et al., 2013; Kim et al., 2012; Li et al., 2013).
Other known post-translational modifications include ADP ribosylation on CLOCK (Asher et al., 2010), sumoylation on BMAL1 (Cardone et al., 2005; Lee et al., 2008), and ubiquitination on CRY (Hirano et al., 2013; Yoo et al., 2013) and BMAL1 (Zhang et al., 2012). Poly ADP-ribosylation of CLOCK enhances CLOCK/BMAL1 complex binding to DNA by poly(ADP-ribose) polymerase 1 (PARP-1) activity initiated in the early light phase of circadian time. Sumoylation of BMAL1 occurs in the liver and promotes the circadian transcriptional activation of the CLOCK/ BMAL1 complex on DNA. This activation occurs in combination with ubiquitination of BMAL1, requiring co- modification for activation. Finally, CRY is ubiquitinated by FBXL21 and FBXL3, F-box-type ubiquitin E3 ligases. These E3 ligases work in the nucleus and cytoplasm, respectively, to activate and time CRY activity for the suppression of circadian gene activation with PER by targeting CRY for proteasomal degradation (Hirano et al., 2013; Yoo et al., 2013).
Mutations causing human circadian phenotypes
Familial advanced sleep phase
PER2-S662G
To determine the genetic basis of FASP, linkage analysis was performed in a large family segregating the FASP trait. The mutation was determined to be on chromosome 2, and further analysis identified PER2 as the gene containing the mutation. In the hPER2 cDNA, position 2106 was changed from A to G which causes the substitution of a serine at amino acid 662 with a glycine (S662G) (Toh et al., 2001). The mutation is localized in the CKI binding domain of PER2, and as the mutation substituted a glycine for a serine residue, the hypothesis that the mutation affected the phosphorylation of PER2 was tested. Initial testing suggested that the mutation of PER2 at Ser662 reduced CKIe phosphorylation (Toh et al., 2001); later data suggested that mutation at Ser662 disrupts the phosphorylation of CKId in vitro (Xu et al., 2007). Because the mutant PER2 protein demonstrated reduced phosphorylation by CKIδ, the functional consequence of a reduction of PER2 phosphorylation in vivo was tested (Xu et al., 2007). BAC transgenic mice carrying this human mutation were generated and then tested in a circadian behavioral assay. These mice recapitulate the human FASP phenotype, including both advanced sleep phase and a shortening of behavioral and cellular circadian period length (Xu et al., 2007). These observed phenotypes are associated with enhanced transcriptional repression, as mRNA levels of both the endogenous copy of mouse Per2 and the introduced human copy of PER2 decreased. The change in repressor activity is caused by a disruption of phosphorylation at Ser662, which normally results in a phosphorylation cascade of four downstream serine sites. Hence, through a still unknown mechanism, the lack of phosphorylation allows PER2 to act as a stronger repressor. Supporting this mechanism, a second transgenic mouse model with a substitution of the Ser662 to aspartic acid (which mimics constitutive phosphorylation) resulted in a lengthened circadian period and decreased repressor activity demonstrated by protein and RNA analysis. In vitro evidence also suggests that mutation of PER2 at S662G enhances PER2 clearance from the nuclei (Vanselow et al., 2006). While enhancing repressor activity would naturally decrease expression levels, another concurrent possibility for protein reduction presented by Vanselow and colleagues is that the changes in PER2 phosphorylation results in deficient nuclear retention, which then promotes PER2 degradation in the cytoplasm (Vanselow et al., 2006).
In the future, it will be necessary to investigate the function of phosphorylation in the CKI domain. Phosphorylation of PER2 has so far been linked to the protein half-life, the ability to interact with other proteins and subcellular localization. Specifically, it is important to investigate how phosphorylation sites work in tandem or in opposition of one another to unravel the mystery of circadian cycling and sleep timing.
CKIγ-T44A
Since the initial identification of a mutation that causes FASP, additional families were identified to have FASP but did not contain the previously identified mutation in PER2. Utilizing a candidate gene approach, a causative mutation for a second FASP family was identified in human CKIγ (Xu et al., 2005). The mutation occurs at Threonine 44 and is substituted with alanine (T44A). This particular amino acid was conserved in the CKI family of proteins in mouse and human as well as in the Drosophila melanogaster CKI homolog Doubletime (homolog of CKIγ and CKIε). Biochemical assays displayed mutation-induced decreases in phosphorylation of alpha-casein, and PER protein substrates by in vitro kinase assays strongly suggesting biological relevance for this genetic variant. To validate the mutated function of CKId, a BAC transgenic mouse model carrying CKIγ-T44A was generated. These mice were tested in wheel-running behavioral assays to determine the functional consequence of the mutant kinase, and their free running period (τ) was shorter than control mice by ~20 min. When the mutant transgenic mice were crossed with CKIγ knock-out mice, an even shorterτ was observed (22.7 h versus 23.4 h) in addition to rescuing the lethal phenotype of the CKIτ knock-out (the mutation does not render the kinase activity null). Intriguingly, expressing the human CKI -T44A mutation in Drosophila generated flies with a significantly longer period than control flies, which highlighted the underlying differences between mammals and invertebrates (Xu et al., 2005).
To address the mechanism of reduced kinase activity, the crystal structure of the mouse homolog CKIδ was analyzed to better understand the functional consequence of the mutating threonine to alanine (Figure 2). The mutation occurs in the loop between the beta sheets that make up the ATP binding domain in the catalytic domain of CKIδ. Specifically, the loss of threonine and replacement with a hydrophobic amino acid (alanine) may cause misfolding or loss of flexibility of the loop, which then restricts the enzyme (Figure 2). Because the mutation causes a reduction of catalytic activity, it is possible that the mutation causes a disruption in chemical catalysis of phosphate transfer from ATP, through restriction of the enzyme's flexibility.
Figure 2.

Molecular modeling of T44A mutation. Utilizing PyMOL, the structure of the human CKIδ with the mutation T44A was modeled from rat CKI (1CKI). The replacement of threonine with the hydrophobic amino acid alanine may cause protein misfolding or loss of flexibility of the loop, which then restricts the enzyme, likely lowering enzymatic activity. (see colour version of this figure at www.informahealthcare.com/bmg).
CKIδ H46R
After identifying CKIδ-T44A, the gene encoding CKIδ in blood samples from over 70 FASP probands were screened, and a second mutation was identified in a single individual that predicted a histidine to arginine change at position 46 (H46R), two amino acids downstream from the T44A substitution (Brennan et al., 2013). Neither T44A nor H46R is present in the 1000 Genomes database and the publicly available CGI 60 Genomes database (1000 Genomes Project Consortium et al., 2012; Drmanac et al., 2010).
As previously discussed, CKIδ-T44A has reduced kinase activity in comparison to wild type (WT) enzyme. Utilizing a CKIδ-H46R isoform, the kinetics of phosphotransfer was analyzed. CKIδ-H46R similarly showed decreased kinase activity on alpha casein and the PER2 peptide (Brennan et al., 2013). The decrease in Vmax suggests that the mutation causes a reduction in the catalytic rate of phosphotransfer. There may be a subtle effect on enzyme recognition of the substrate as the Km of the mutant kinase activity is decreased with PER2 peptide but is not significantly different for casein or ATP. Utilizing molecular modeling, we hypothesize that the reduction of activity can be attributed to the loss of the hydrogen bond that forms between H46 and the glutamine residue at amino acid 48 (Figure 3). Though the function of the hydrogen bond is unknown, it may be to properly fold the loop during catalysis. It is also unclear what the structural consequence of the mutant arginine has on the tertiary structure. However, it is reasonable to hypothesize that the structural integrity in this region is important for the catalytic function of the phosphotransfer.
Figure 3.

Molecular modeling of H46R mutation. Utilizing PyMOL, the structure of the human CKIδ with the mutation H46R was modeled from rat CKI (1CKI). The replacement of histidine with the larger guanidino containing amino acid arginine may cause protein misfolding or loss of flexibility of the loop. Specifically as shown, there is an increased distance between the hydrogen bonding of the mutant arginine with glutamine at position 48, which then restricts the enzyme, likely lowering enzymatic activity. (see colour version of this figure at www.informahealthcare.com/bmg).
Further work is needed to identify the structural ramifications of the H46R and the T44A mutations, which will give insight to the structural function of the loops on CKIδ. It will also be interesting to reveal other possible cellular ramifications of mutating a kinase integral to the Wnt signaling pathway. The function and mechanism of CK1δ phosphorylation may illuminate the role phosphorylation plays in migraine headache (Brennan et al., 2013).
Mutation altering human sleep duration
Familial natural short sleep
A family was found to display natural short-sleep phenotype characterized by the reduction of sleep time and increased daily activity, described as familial natural short sleep (FNSS). Subsequently, a point mutation that changed the amino acid at position 384 from proline to arginine (P384R) in DEC2 was identified (He et al., 2009). This mutation co-segregates with the FNSS phenotype in this family. DEC2 is a negative component of the circadian clock (Butler et al., 2004). It belongs to a basic helix-loop-helix (bHLH) protein family in which members can homodimerize and can affect gene transcription by direct binding to DNA (Fujimoto et al., 2001, 2007). P384 is localized in the C-terminal proline-rich domain and its flanking sequences are highly conserved among mammalian DEC2 orthologs. DEC2-P384R mutation alleviated its transcriptional repressive activity demonstrated by in vitro luciferase reporter assay. BAC transgenic mice carrying this P384R mutation exhibited a shorter total sleep time and longer active time per 24-h period as seen in human mutation carriers. This result was further verified by electroencephalography (EEG) and electromyography (EMG) performed on DEC2-P384R mutant transgenic mice and their littermates. In addition to transgenic mice, transgenic flies carrying the P384R mutation were generated to test whether Drosophila melanogaster would demonstrate a conserved phenomenon with humans and mice. Transgenic flies with mutant Dec2 expressed in their mushroom bodies showed a similar lengthened active duration behavior, suggesting DEC2 plays a significant role in regulating sleep quantity.
The DEC2 mutation is hypothesized to vary sleep quantity by altering the structure of the protein. The mutation of DEC2 incorporates an arginine residue in place of a proline, which can incorporate a locked turn in the protein by the nature of the proline structure. The replacement of the proline with arginine introduces a charged amino acid with the guanidino group of the arginine. In addition, the amino acid substitution is in the glycine and alanine-rich region of the DEC2 protein that has been shown to interact with HDAC1, the protein deacetylase and a known regulator of transcription factors (Fujimoto et al., 2007) (Figure 4). Hence, the mutation may interrupt the interaction between HDAC1 and DEC2, and lead to altered gene transcription, by altering the acetylation status of DEC2 or by affecting HDAC1-dependent deacetylation of other transcription factors. Future work to elucidate the molecular mechanisms of DEC2 on sleep length regulation includes identifying genes that are regulated by DEC2, and specifically how the FNSS mutation affects gene transcription and protein function.
Figure 4.
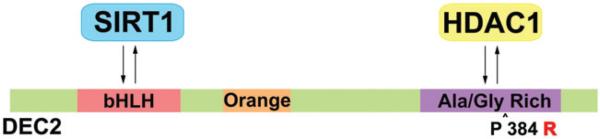
DEC2 interacting domains with HDAC. DEC 2 contains most notably a basic helix-loop-helix domain that has been shown to interact with SIRT1, which may play a significant role in DNA binding regulation. By homology, there is an orange domain. HDAC1 interacts with the Ala/Gly rich region of DEC2, interestingly where the P384R mutation resides. The mutation may disrupt or promote HDAC1 interaction and may be the key to understanding the molecular mechanism of DEC2 regulation of NSS. (see colour version of this figure at www.informahealthcare.com/bmg).
Insight from murine circadian mutations
The initial insight into how circadian rhythm genes affect sleep function was revealed by the tau mutant Syrian hamster (Ralph & Menaker, 1988). This mutation conferred a free-running time in the mutant animals to that of 22 h instead of the wild type 23.5 h. Further characterization of the mutation mapped it to a single nucleotide change in the gene encoding the enzyme CKIε. This mutation has been examined in vitro and hypothesized to decrease enzymatic activity in the general enzyme, but has also been hypothesized to have increased specificity or gain-of-function for PER by increasing PER2 degradation (Lowrey et al., 2000). Interestingly, tau and the mutation in human CKIδ give a similar shortened period, though the mechanisms seem to differ as far as the site and the function of the mutation (gain-of-function versus loss) (Xu et al., 2005). Albeit the exact molecular mechanism of CKIδ regulation needs to be further explored, this reinforces the idea that phosphorylation can have many different effects on a single protein that later can result in the same phenotype.
Another mutation affecting mouse circadian function was identified using a forward genetics screen. Mice were treated with N-ethyl-N-nitrosourea (ENU), and then subjected to free-running activity measurements. Using this method, a semi-dominant mutation was identified in the CLOCK gene (King et al., 1997). This mutation deletes exon 19, which interrupts the bHLH-PAS domain and likely blocks transcriptional regulation of CLOCK. Although to date no human mutations in the CLOCK gene have been identified, mutations in CLOCK could possibly cause the “night owl” phenotype of FDSP by delaying the clock as seen in the mice.
Two mutations, cysteine 358 to serine (Godinho et al., 2007) and isoleucine 364 to threonine (Siepka et al., 2007), in F-box protein 3 (FBXL3) were found independently using ENU mutagenesis. Both mutations lead to long free-running period by affecting the expression of PERs and the stabilization of CRYs. Finally, a new mutation was recently identified in another F-box protein, FBXL21, an E3 ubiquitin ligase specific to CRY1 in the mouse (Yoo et al., 2013). The mutation in FBXL21 conferred a change of amino acid 149 from a glycine to a glutamine. Interestingly, this mutation leads to period shortening and antagonizes the period lengthening effect of the FBXL3 isoleucine 364 threonine mutation. FBXL21 was found to stabilize CRYs and antagonize the destabilizing action of FBXL3 on CRYs (Hirano et al., 2013). The FBXL21-G149E mutation reduces the protective effect of FBXL21. These mutations again point out the importance of post-translational modifications in the precise timing and function of the core circadian oscillators. It is not unreasonable to predict that human FBXL21/3 homologs will play similar roles in regulating human sleep behaviors, highlighting the importance of studying model organisms for understanding human circadian regulatory mechanisms.
How do human circadian mutations help us understand the regulation of human circadian clock and sleep behaviors/beyond finding the mutations?
PER2
The PER2-S662G mutation for FASP was found in the CKI binding region. This region of the PER2 protein is highly conserved among mammalian PERs, and there are four additional serines immediately C-terminal to the mutated serine at amino acid 662 (Xu et al., 2007). In vitro biochemical studies showed that the serine at 662 needs to be phosphorylated by a priming kinase which is followed by phosphorylation of four additional serines by CKIδ (Xu et al., 2007). Transgenic mice that carry the S662G mutation showed a shorter circadian period (22 h) than WT control mice (23.7 h) (Xu et al., 2007). Intriguingly, transgenic mice that carry a serine to aspartic acid change (to mimic a constitutively phosphorylated serine) have a longer period (24.5 h). When serine 662 is phosphorylated, it allows the four additional serines to be phosphorylated; therefore all five serines are phosphorylated. This then leads to weaker repression by PER2 (described above) and animals have a longer period (analogous to a slower clock because it will take longer to finish a cycle). However, when residue S662G cannot be phosphorylated in vitro, the next four serines are not phosphorylated by CKIδ. Then the PER2 protein is a stronger repressor and animals have a shorter period (similar to having a faster clock) (Xu et al., 2007). Studies also revealed that the hypophosphorylated PER2 is less stable and the hyperphosphorylated protein is more stable. Collectively, these results imply that PER2 acts as a circadian repressor and that when S662 phosphorylation is blocked, it adopts a conformation so that it becomes a stronger repressor and is less stable. However, when all five serines in the specific region are phosphorylated, the protein adopts a different conformation, which renders it a weaker repressor and more stable. Therefore, the suicide model for transcriptional factors (Fu, 2008) can be applied to describe PER2 (and potentially other transcription factors of the molecular clock) and offers a possible mechanism linking the transcription repressor activity to its own degradation.
Because the four serines immediately C-terminal to S662 are likely phosphorylated by CKIδ, mouse models were used to test the genetic interactions between CKIδ and PER2. The copy number of CKIδ does not affect the period length of WT mice. However, when PER2-S662G mice were crossed with CKI heterozygous knockout mice, the period was longer (22.33 h) than S662G transgenic mice. In addition, when PER2-S662G mice were crossed with WT CKIδ transgenic mice (carrying four to five copies of CKIδ), the period was shorter (20.83 h) than S662G mice (Xu et al., 2007). These results revealed that there are other sites on PER2 that can be phosphorylated by CKIδ; and when these other sites are phosphorylated, it leads to shorter period (Xu et al., 2007). Therefore, there are multiple sites for CKIδ action on PER2. One of them is the S662 region and when this site is fully phosphorylated, it leads to increased PER2 mRNA and protein levels and a longer period. However, there are other sites on PER2 that when phosphorylated by CKIδ, leads to increased protein degradation, lower protein levels, and a shorter period. Under normal conditions, phosphorylation of different regions maintains a balance, leading to a stable period length. For CKIδ heterozygous knockout, there is lower CKIδ level but it affects both pathways to a similar extent, so period stays the same. Similarly, for WT transgenic mice, the higher CKIδ level will affect both pathways to the same extent and period therefore is unchanged (Xu et al., 2007). For PER2-S662G mutant mice, the pathway for S662 is blocked which then pushes the balance towards the other side and leads to shorter period length (Figure 5). When PER2-S662G is crossed onto CKI-WT transgene mice, the higher CKIδ level will push the balance towards the other side even more and produces an even shorter period (20.83 h) (Xu et al., 2007). When S662G mice is crossed with CKIδ heterozygous knockout, the reduced CKIδ level will decrease the phosphorylation unbalance and therefore gives a less short period of 22.33 h. Overall, these data point out the importance of PER2 phosphorylation in setting the speed of the clock.
Figure 5.

Mechanism of PER2 FASP. Under normal conditions, PER2 is phosphorylated by a specific kinase at the serine 662. This initial phosphorylation “primes” the CKI domain for the subsequent recognition by CKIδ and the phosphorylation of the remaining serine residues S665–S674. The phosphorylation of these residues renders PER2 a weaker repressor and confers a longer period length. Additional phosphorylation site(s) on PER2 by CKIδ increases PER2 degradation and leads to a shorter period. Conversely, in the mutant BAC transgenic animal carrying S662G, the priming kinase cannot recognize or phosphorylate the S662 residue, leaving the protein in the hypophosphorylated state; the lack of priming prevents CKIδ from recognizing S665–S674. However, phosphorylation by CKI on the other site(s) of PER2 is unperturbed and degradation of PER2 continues without counter balance from the S662–S674 region. This then causes a shortened period and advancement of sleep phase. (see colour version of this figure at www.informahealthcare.com/bmg).
Further complicating the molecular scenario, PER2 is O-GlcNAc modified and O-GlcNAcylation further enhances PER2 repressor activity (Kaasik et al., 2013). OGT (O-GlcNAc transferase) does not affect PER2-S662G repressor activity, suggesting that S662 site may be competitively regulated by phosphorylation and O-GlcNAcylation. Interestingly, S662, S668 and S671 of the 5-serine region in PER2 are all O-GlcNAcylation sites. O-GlcNAcylation is dependent on the level of UDP-GlcNAc substrate, and UDPGlcNAc synthesis is dependent on nutrient fluxes including glucose (Hart, 2013). Under low glucose condition, the PER2-S662-S674 region is phosphorylated in the presence of CKId even with OGT and OGA (O-GlcNAcase). However, under conditions of high glucose, phosphorylation in this region is reduced by the presence of OGT or OGA even with the overexpression of CKIδ (Kaasik et al., 2013). As described above, when the first serine is phosphorylated in this 5-serine region, the four subsequent serines are phosphorylated. This then leads to a weaker repressor with longer period. But, phosphorylation in this region is also modulated by O-GlcNAcylation, and under high glucose condition, phosphorylation in this region is blocked by O-GlcNAcylation that then likely leads to a shortened period. Future in vivo investigation will shed light on how sugar can modulate clock speed and further our understanding of the connection between circadian clock and metabolic regulation.
CKI
One interesting observation for the CKIδ-T44A mutation from the very beginning was that human mutation carriers not only have the FASP phenotype but also have asthma and migraine headache (Brennan et al., 2013). One possible explanation for this observation is that the kinase mutation could potentially affect multiple substrates, therefore leading to multiple phenotypes. To gain further insight into this observation, a circadian phosphor-proteomic investigation was initiated for CKIδ (Kategaya et al., 2012). In addition, a parallel study was carried out for CKIε as comparison and control. Intriguingly, CKIδ and ε share some substrates/interacting partners, but also each have their own subset of unique substrates/interacting partners. One significant revelation from this circadian phosphor-proteomic study is that CKIδ and ε both demonstrate interactions with different proteins at different circadian times, pointing out the importance of considering timing while studying kinases, substrates and the pathways in which they are involved (Kategaya et al., 2012).
Because human mutation carriers in families with CKId mutations (T44A and H46R) both experience migraine headaches, the transgenic mouse model of T44A was used to investigate the possibility of migraine phenotype. CKI -T44A mice showed increased sensitivity to a migraine trigger (nitroglycerin), which induced peripheral mechanical and thermal hyperalgesia (Brennan et al., 2013). Migraine-related cortical excitability was examined by measuring cortical spreading depression (CSD) using optical imaging and electrophysiological recording. CKIδ-T44A mice had significantly lower CSD thresholds with increased number of CSD elicited by stimulation, and their cortical surface arteries were more dilated in all phases of CSD than control mice. In addition, astrocytes from CKIδ-T44A mice demonstrated both increased spontaneous and evoked calcium signaling than astrocytes of control mice, suggesting possible astrocytic mechanisms for CKIδ mutations to predispose carriers to migraine. Migraine is common in the general population ~ 12%) and both genetic and environmental factors have been implicated in contributing to this phenotype (Brennan et al., 2013). The association between migraine and two independent CKId mutations, together with the in vitro and in vivo data, suggests that these mutations contribute to the pathogenesis of migraine. Though the relationship between sleep regulation and migraine is unclear, all in all, these studies revealed that CKId plays a diverse yet important role in regulating brain excitability.
Model organisms
Investigating human phenotypes usually has an end goal of understanding their underlying fundamental molecular mechanisms. The approach of finding the mutation and gene in human subjects followed by generating a model animal for further investigation has the advantage of studying a single important gene, which can lead to the most relevant pathways. One critical and powerful feature for studying human conditions, whether in health or disease, is the fact that humans can communicate with researchers about their emotional and mental status. However, the major disadvantage lies in the obvious impossibility of using humans in most experimental procedures. For this reason, model organisms are necessary research tools in pursuing these investigations. On the other hand, not all mutations found in humans will give equivalent (or at times any) phenotypes in model organisms. To establish a “good” or “excellent” model for studying human conditions requires extensive characterization and comparison between the model and human subjects. This also presents challenges since not all comparisons are feasible. With behavioral phenotypes, this challenge is especially augmented and needs additional validations with extra attention from the researchers. To date, the preferred method is to generate transgenic mice that carry human mutations in order to study sleep behavior phenotypes. Mice have the advantage of having closer brain anatomy with humans, and also it is possible to measure EEG for sleep stages in mice. Hence, this model organism has recapitulated the human phenotype beautifully for many mutations as described above. However, there are mutations (or perhaps extremely rare polymorphisms) that were found in humans, but no obvious phenotype could be found in transgenic mice that carry the same mutations (Ptacek & Fu, unpublished results). In addition, by their very nature, mice are nocturnal and display a sleep–wake cycle directly opposite to that of humans. This is further compounded by the animal's natural instinct to mask to light, making sleep and activity measurements at times difficult to interpret (Mrosovsky, 1999). Drosophila also has been used to model human sleep behavior by generating transgenic flies carry human mutations, and in some cases the phenotypes are similar, yet other times different (He et al., 2009; Xu et al., 2005). This further highlights the complexity of choosing the “right” model to assist in unraveling molecular mechanisms involved in regulating human behavioral traits. Yet, Drosophila is a uniquely malleable model organism that researchers can utilize to investigate the possible mechanisms that may shed significant and novel insight as well.
Concluding remarks
The diverse nature of sleep dysfunction and the lack of treatments for many of these conditions accent the potential for a wealth of research and medical opportunities. The fundamental question of how our bodies integrate all information (from environment and internal cues) and then manifests them as certain sleep behaviors remains largely unclear. The search for mutations by directed genetic screens will likely identify numerous new mechanisms and pathways previously thought unrelated to sleep and further enhance our understanding of sleep. In depth studies of these mutations, genes and pathways will reveal novel mechanisms that link sleep to other phenotypes such as obesity, diabetes, mood disorders, cancer and other seemingly unrelated physiological processes. These investigations also underlie the importance of combining the fields of biochemistry, genetics, cellular/ molecular biology and bioinformatics. The complex pheno-type of sleep requires behavioral models in which experimentation can describe the phenotype that can be mechanistically investigated in vitro and in cells. Integrating these interdisciplinary approaches will exponentially increase the possibility of revealing the interwoven nature of human sleep mechanisms that will improve the lives of people in everyday society.
Acknowledgements
The authors thank S.Y. Christin Chong for constructive suggestions and careful editing of this article.
The research in authors’ laboratories is funded by NIH GM079180, HL059596, NS072360. LJP is an investigator of Howard Hughes Medical Institute. W.C.H. is supported by 5 F32HL112598-02 fellowship.
Footnotes
Declaration of interest
The authors have no conflicts of interest, and received no involvement of a pharmaceutical/other company.
References
- 1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar RA, Reddy AB, Maywood ES, et al. Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr Biol. 2002;12:540–50. doi: 10.1016/s0960-9822(02)00759-5. [DOI] [PubMed] [Google Scholar]
- Andretic R, Franken P, Tafti M. Genetics of sleep. Ann Rev Genet. 2008;42:361–88. doi: 10.1146/annurev.genet.42.110807.091541. [DOI] [PubMed] [Google Scholar]
- Asher G, Gatfield D, Stratmann M, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–28. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- Asher G, Reinke H, Altmeyer M, et al. Poly(ADP-Ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell. 2010;142:943–53. doi: 10.1016/j.cell.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Benington JH, Frank MG. Cellular and molecular connections between sleep and synaptic plasticity. Prog Neurobiol. 2003;69:71–101. doi: 10.1016/s0301-0082(03)00018-2. [DOI] [PubMed] [Google Scholar]
- Bonnet MH, Arand DL. Metabolic rate and the restorative function of sleep. Physiol Behav. 1996;59:777–82. doi: 10.1016/0031-9384(95)02093-4. [DOI] [PubMed] [Google Scholar]
- Borbély AA, Achermann P. Sleep homeostasis and models of sleep regulation. J Biol Rhythms. 1999;14:557–68. doi: 10.1177/074873099129000894. [DOI] [PubMed] [Google Scholar]
- Bouchard TJ, Lykken DT, McGue M, et al. Sources of human psychological differences: the Minnesota Study of Twins Reared Apart. Science (New York, N.Y.) 1990;250:223–8. doi: 10.1126/science.2218526. [DOI] [PubMed] [Google Scholar]
- Brennan KC, Bates EA, Shapiro RE, et al. Casein kinase id mutations in familial migraine and advanced sleep phase. Sci Transl Med. 2013;5:183ra56, 1–11. doi: 10.1126/scitranslmed.3005784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Kowalska E, Dallmann R. (Re)inventing the circadian feedback loop. Develop Cell. 2012;22:477–87. doi: 10.1016/j.devcel.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Butler MP, Honma S, Fukumoto T, et al. Dec1 and Dec2 expression is disrupted in the suprachiasmatic nuclei of Clock mutant mice. J Biol Rhythms. 2004;19:126–34. doi: 10.1177/0748730403262870. [DOI] [PubMed] [Google Scholar]
- Cardone L, Hirayama J, Giordano F, et al. Circadian clock control by SUMOylation of BMAL1. Science (New York, N.Y.) 2005;309:1390–4. doi: 10.1126/science.1110689. [DOI] [PubMed] [Google Scholar]
- Chen R, Schirmer A, Lee Y, et al. Rhythmic PER abundance defines a critical nodal point for negative feedback within the circadian clock mechanism. Mol Cell. 2009;36:417–30. doi: 10.1016/j.molcel.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker A, Sehgal A. Genetic analysis of sleep. Genes Dev. 2010;24:1220–35. doi: 10.1101/gad.1913110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mairan JJO. Observation botanique. Histoire de I'Academie Royale des Sciences. 1729:35–6. [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science (New York, N.Y.) 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- Duffield GE, Best JD, Meurers BH, et al. Circadian programs of transcriptional activation, signaling, and protein turnover revealed by microarray analysis of mammalian cells. Curr Biol. 2002;12:551–7. doi: 10.1016/s0960-9822(02)00765-0. [DOI] [PubMed] [Google Scholar]
- Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96:271–90. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- Durgan DJ, Pat BM, Laczy B, et al. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem. 2011;286:44606–19. doi: 10.1074/jbc.M111.278903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG. The mystery of sleep function: current perspectives and future directions. Rev Neurosci. 2006;17:375–92. doi: 10.1515/revneuro.2006.17.4.375. [DOI] [PubMed] [Google Scholar]
- Fu Y-HY. Oscillating percision. PLoS Biol. 2008;6:e192. doi: 10.1371/journal.pbio.0060192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto K, Hamaguchi H, Hashiba T, et al. Molecular cloning and characterization of DEC2, a new member of basic helix-loop-helix proteins. Biochem Biophys Res Commun. 2001;280:164–71. doi: 10.1006/bbrc.2000.4133. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Shen M, Noshiro M, et al. Transcriptional repression by the basic helix-loop-helix protein Dec2: multiple mechanisms through E-box elements. Int J Mol Med. 2007;19:925–32. [PubMed] [Google Scholar]
- Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nature Rev: Mol Cell Biol. 2007;8:139–48. doi: 10.1038/nrm2106. [DOI] [PubMed] [Google Scholar]
- Godinho SIH, Maywood ES, Shaw L, et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science (New York, N.Y.) 2007;316:97–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Ann Rev Pathol. 2010;5:253–95. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardeland R, Coto-Montes A, Poeggeler B. Circadian rhythms, oxidative stress, and antioxidative defense mechanisms. Chronobiol Int. 2003;20:21–62. doi: 10.1081/cbi-120025245. [DOI] [PubMed] [Google Scholar]
- Hardin PE. Molecular genetic analysis of circadian timekeeping in Drosophila. Adv Genet. 2011;74:41–73. doi: 10.1016/B978-0-12-387690-4.00005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW. How sugar tunes your clock. Cell Metab. 2013;17:55–6. doi: 10.1016/j.cmet.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Jones CR, Fujiki N, et al. The transcriptional repressor DEC2 regulates sleep length in mammals. Science (New York, N.Y.) 2009;325:66–70. doi: 10.1126/science.1174443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Yumimoto K, Tsunematsu R, et al. FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell. 2013;152:106–18. doi: 10.1016/j.cell.2013.01.054. [DOI] [PubMed] [Google Scholar]
- Huang W, Ramsey KM, Marcheva B, Bass J. Circadian rhythms, sleep, and metabolism. J Clin Invest. 2011;121:133–41. doi: 10.1172/JCI46043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, DiTacchio L, Hayes KR, et al. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5:1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CR, Campbell SS, Zone SE, et al. Familial advanced sleep-phase syndrome: a short-period circadian rhythm variant in humans. Nature Med. 1999;5:062–5. doi: 10.1038/12502. [DOI] [PubMed] [Google Scholar]
- Jung CM, Melanson EL, Frydendall EJ, et al. Energy expenditure during sleep, sleep deprivation and sleep following sleep deprivation in adult humans. J Physiol. 2011;589:35–44. doi: 10.1113/jphysiol.2010.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik K, Kivimäe S, Allen JJ, et al. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 2013;17:91–302. doi: 10.1016/j.cmet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kategaya LS, Hilliard A, Zhang L, et al. Casein kinase 1 proteomics reveal prohibitin 2 function in molecular clock. PLoS one. 2012;7:31987. doi: 10.1371/journal.pone.0031987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Jeong EH, Park S, et al. A role for O-GlcNAcylation in setting circadian clock speed. Genes Dev. 2012;26:90–502. doi: 10.1101/gad.182378.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DP, Zhao Y, Sangoram AM, et al. Positional cloning of the mouse circadian clock gene. Cell. 1997;89:41–53. doi: 10.1016/s0092-8674(00)80245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurabayashi N, Hirota T, Sakai M, et al. DYRK1A and glycogen synthase kinase 3beta, a dual-kinase mechanism directing proteasomal degradation of CRY2 for circadian timekeeping. Mol Cell Biol. 2010;30:757–68. doi: 10.1128/MCB.01047-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Sachdeva UM, DiTacchio L, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science (New York, N.Y.) 2009;326:37–40. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Lee Y, Lee MJ, et al. Dual modification of BMAL1 by SUMO2/3 and ubiquitin promotes circadian activation of the CLOCK/ BMAL1 complex. Molec Cellular Biol. 2008;28:056–65. doi: 10.1128/MCB.00583-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M-D, Ruan H-B, Hughes ME, et al. O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquiti-nation. Cell Metab. 2013;17:03–10. doi: 10.1016/j.cmet.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Takahashi JS. Genetics of circadian rhythms in Mammalian model organisms. Adv Genet. 2011;74:75–230. doi: 10.1016/B978-0-12-387690-4.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Shimomura K, Antoch MP, et al. Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science (New York, N.Y.) 2000;288:83–92. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B, Wendt S, Vanselow JT, et al. A large-scale functional RNAi screen reveals a role for CK2 in the mammalian circadian clock. Genes Develop. 2009;23:08–18. doi: 10.1101/gad.512209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret S, Faraguna U, Nelson AB, et al. Sleep and waking modulate spine turnover in the adolescent mouse cortex. Nature Neurosci. 2011;14:418–20. doi: 10.1038/nn.2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicine AAOS . International classification of sleep disorders-second edition, ICSD-2. 2nd ed. American Academy of Sleep Medicine; n.d. [Google Scholar]
- Miller BH, McDearmon EL, Panda S, et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci USA. 2007;104:342–7. doi: 10.1073/pnas.0611724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Ann Rev Neurosci. 2012;35:45–62. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna P. Fatal familial insomnia: a model disease in sleep physiopathology. Sleep Med Rev. 2005;9:39–53. doi: 10.1016/j.smrv.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Mrosovsky N. Masking: history, definitions, and measurement. Chronobiol Int. 1999;16:15–29. doi: 10.3109/07420529908998717. [DOI] [PubMed] [Google Scholar]
- Nakahata Y, Kaluzova M, Grimaldi B, et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:29–40. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S, Antoch MP, Miller BH, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:07–20. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Peng L, Seto E. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handbook Exp Pharmacol. 2011;206:9–56. doi: 10.1007/978-3-642-21631-2_3. [DOI] [PubMed] [Google Scholar]
- Plomin R, DeFries JC, Knopik VS, Neiderhiser JM. Behavioral genetics. Worth Publishers; New York: 2012. [Google Scholar]
- Ralph MR, Menaker M. A mutation of the circadian system in golden hamsters. Science (New York, N.Y.) 1988;241:225–7. doi: 10.1126/science.3413487. [DOI] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science (New York, N.Y.) 2009;324:51–4. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack RL, Auckley D, Auger RR, et al. American Academy of Sleep Medicine Circadian rhythm sleep disorders: part I, basic principles, shift work and jet lag disorders. An American Academy of Sleep Medicine review. Sleep. 2007a;30:460–83. doi: 10.1093/sleep/30.11.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack RL, Auckley D, Auger RR, et al. American Academy of Sleep Medicine Circadian rhythm sleep disorders: part II, advanced sleep phase disorder, delayed sleep phase disorder, free-running disorder, and irregular sleep-wake rhythm. An American Academy of Sleep Medicine review. Sleep. 2007b;30:484–501. doi: 10.1093/sleep/30.11.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahar S, Sassone-Corsi P. Metabolism and cancer: the circadian clock connection. Nature Rev: Cancer. 2009;9:886–96. doi: 10.1038/nrc2747. [DOI] [PubMed] [Google Scholar]
- Sehgal A, Mignot E. Genetics of sleep and sleep disorders. Cell. 2011;146:194–207. doi: 10.1016/j.cell.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanware NP, Hutchinson JA, Kim SH, et al. Casein kinase 1-dependent phosphorylation of familial advanced sleep phase syndrome-associated residues controls PERIOD 2 stability. J Biol Chem. 2011;286:12766–74. doi: 10.1074/jbc.M111.224014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepka SM, Yoo S-H, Park J, et al. Circadian mutant overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129:1011–23. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch K-F, Lipan O, Leykin I, et al. Extensive and divergent circadian gene expression in liver and heart. Nature. 2002;417:78–83. doi: 10.1038/nature744. [DOI] [PubMed] [Google Scholar]
- Takahashi JS. Finding new clock components: past and future. J Biol Rhythms. 2004;19:339–47. doi: 10.1177/0748730404269151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi JS, Hong H-K, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nature Rev: Genet. 2008;9:764–75. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh KL, Jones CR, He Y, et al. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science (New York, N.Y.) 2001;291:1040–3. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Ueda HR, Hayashi S, Chen W, et al. System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nature Genet. 2005;37:187–92. doi: 10.1038/ng1504. [DOI] [PubMed] [Google Scholar]
- Vanselow K, Kramer A. Role of phosphorylation in the mammalian circadian clock. Cold Spring Harbor symposia on quantitative biology. 2007;72:167–176. doi: 10.1101/sqb.2007.72.036. [DOI] [PubMed] [Google Scholar]
- Vanselow K, Vanselow JT, Westermark PO, et al. Differential effects of PER2 phosphorylation: molecular basis for the human familial advanced sleep phase syndrome (FASPS). Genes Dev. 2006;20:2660–72. doi: 10.1101/gad.397006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk R, Somers VK. Sleep and the metabolic syndrome. Exp Physiol. 2007;92:67–78. doi: 10.1113/expphysiol.2006.033787. [DOI] [PubMed] [Google Scholar]
- Xu Y, Padiath QS, Shapiro RE, et al. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature. 2005;434:640–4. doi: 10.1038/nature03453. [DOI] [PubMed] [Google Scholar]
- Xu Y, Toh KL, Jones CR, et al. Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell. 2007;128:59–70. doi: 10.1016/j.cell.2006.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S-H, Mohawk JA, Siepka SM, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell. 2013;152:1091–105. doi: 10.1016/j.cell.2013.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitane H, Honma S, Imamura K, et al. JNK regulates the photic response of the mammalian circadian clock. EMBO Reports. 2012;13:455–61. doi: 10.1038/embor.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Abraham D, Lin S-T, et al. PKCg participates in food entrainment by regulating BMAL1. Proc Natl Acad Sci USA. 2012;109:20679–84. doi: 10.1073/pnas.1218699110. [DOI] [PMC free article] [PubMed] [Google Scholar]