

Abstract
Low fidelity Escherichia coli DNA polymerase V (pol V/UmuD′2C) is best characterized for its ability to perform translesion synthesis (TLS). However, in recA730 lexA(Def) strains, the enzyme is expressed under optimal conditions allowing it to compete with the cell’s replicase for access to undamaged chromosomal DNA and leads to a substantial increase in spontaneous mutagenesis. We have recently shown that a Y11A substitution in the “steric gate” residue of UmuC reduces both base and sugar selectivity of pol V, but instead of generating an increased number of spontaneous mutations, strains expressing umuC_Y11A are poorly mutable in vivo. This phenotype is attributed to efficient RNase HII-initiated repair of the misincorporated ribonucleotides that concomitantly removes adjacent misincorporated deoxyribonucleotides. We have utilized the ability of the pol V steric gate mutant to promote incorporation of large numbers of errant ribonucleotides into the E. coli genome to investigate the fundamental mechanisms underlying ribonucleotide excision repair (RER). Here, we demonstrate that RER is normally facilitated by DNA polymerase I (pol I) via classical “nick translation”. In vitro, pol I displaces 1–3 nucleotides of the RNA/DNA hybrid and through its 5′→3′ (exo/endo) nuclease activity releases ribo- and deoxyribonucleotides from DNA. In vivo, umuC_Y11A-dependent mutagenesis changes significantly in polymerase-deficient, or proofreading-deficient polA strains, indicating a pivotal role for pol I in ribonucleotide excision repair (RER). However, there is also considerable redundancy in the RER pathway in E. coli. Pol I’s strand displacement and FLAP- exo/endonuclease activities can be facilitated by alternate enzymes, while the DNA polymerization step can be assumed by high-fidelity pol III. We conclude that RNase HII and pol I normally act to minimize the genomic instability that is generated through errant ribonucleotide incorporation, but that the “nick-translation” activities encoded by the single pol I polypeptide can be undertaken by a variety of back-up enzymes.
Keywords: Y-family DNA polymerase, Steric gate mutant, Ribonucleotide excision repair, UmuC, Spontaneous mutagenesis, RNase H
1. Introduction
Y-family DNA polymerases are best characterized for their ability to traverse lesions in DNA that would otherwise block the cell’s replicase. Structural studies of these polymerases indicate spacious active sites that readily accommodate large bulky adducts within their confines [1]. While this is an extremely important characteristic for efficient lesion bypass, it concomitantly reduces base-selection fidelity when the polymerases replicate undamaged DNA. We recently reported that in addition to low-fidelity DNA synthesis, the E. coli Y-family polymerase pol V (UmuD′2C) also exhibits poor sugar discrimination and readily incorporates ribonucleotides into DNA in vitro [2]. The ability to incorporate ribonucleotides can be modulated by mutations at, or adjacent to, the so-called “steric gate” residue that normally serves to block errant ribonucleotide insertion by DNA polymerases [3]. The steric gate in pol V has been identified as Y11 in the catalytic UmuC subunit of pol V. Not surprisingly, substitution of the large aromatic tyrosine residue by the much smaller aliphatic alanine residue essentially “unlocks” the steric gate and leads to a dramatic increase in the efficiency of pol V-dependent ribonucleotide incorporation [2]. Based upon the tendency of umuC_Y11A to efficiently misincorporate both ribo- and deoxyribonucleotides into DNA, we expected cells expressing pol V steric gate mutant to exhibit a mutator phenotype in vivo. However, to our surprise, both spontaneous and UV-induced umuC_Y11A-dependent mutagenesis were considerably lower than that observed with wild-type pol V [2, 4]. Recently, we have shown that the reduced mutability of the umuC_Y11A expressing strain is caused by efficient repair of the errantly incorporated ribonucleotides with the concomitant removal of adjacent misincorporated deoxyribonucleotides [5]. These conclusions were drawn from the observation that the level of umuC_Y11A spontaneous mutagenesis increases dramatically in ΔrnhB strains lacking endoribonuclease RNase HII [5].
RNase HI cleaves the 3′-O-P bond of the RNA moiety in the RNA/DNA hybrids with more than four sequential rNMPs embedded in a dsDNA strand, while RNase HII can hydrolyze all kinds of hybrids preferring those having a single rNTP over rNTP stretches. Although the role of RNase H enzymes in releasing rNMPs from bacterial DNA has been extensively studied, the precise pathway initiated by these proteins remains to be established. Based on in vitro studies, a general model describing the sequence of events leading to the replacement of a ribonucleotide embedded in DNA with deoxyribonucleotide has been proposed for eukaryotic organisms [6–9]. The ribonucleotide excision repair (RER) pathway in eukaryotes is initiated when RNase HII nicks the phosphodiester bond between the dNMP and rNMP. Then, a second cut 3′ to the rNMP is made by the FLAP endonuclease, FEN-1. Following the release of the cleaved mono-ribonucleotide, pol δ fills the resulting gap by strand displacement DNA synthesis after which DNA ligase seals the nick completing the repair pathway.
As with many other cellular processes, some steps of RER can be accomplished quite effectively by alternative enzymes [9]. For example, a second cut 3′ to rNMP can be made by Exo1 instead of FEN1, while pol ε can substitute for pol δ in the gap-filling step [9]. In contrast, S. cerevisiae RNase HII appears to be essential for the successful processing of rNMPs embedded into DNA since deletion of the RNH201 gene encoding the catalytic subunit of RNase HII leads to replicative stress and genome instability [10]. The instability results from triggering the endoribonuclease activity of topoisomerase 1 (Top1) [11]. In contrast to RNase HII-dependent cleavage, which generates 3′-hydroxyl and 5′-phospho-ribonucleotide ends, Top1-catalyzed cleavage of a bond between ribo- and deoxynucleotides leads to the formation of 2′-3′-cyclic phosphates, which are refractory to religation. As a result, stable ssDNA breaks are formed at the sites of incorporated rNMPs, and when these breaks are located within short repeats, their repair often leads to small deletions and genomic instability [11].
Compared to yeast, bacterial cells appear to be better equipped to withstand errant ribonucleotides inserted into chromosomal DNA. We have shown recently that in the absence of RNase HII, E coli recA730 lexA(Def) ΔdinB strains grow normally and ribonucleotides incorporated by umuC_Y11A can either be removed by nucleotide excision repair (NER), or in a repair pathway initiated by RNase HI [5, 12]. The involvement of RNase HI in RER in this case, is possible because the sugar discrimination of umuC_Y11A is so low that it is able to catalyze incorporation of multiple ribonucleotides consecutively producing RNA/DNA hybrids which could be processed by RNase HI [5]. NER, on the other hand, is able to excise isolated rNMPs as well as RNA fragments although less efficiently than RNase HII-dependent RER [12].
The purpose of the current study is to identify the essential components of the prokaryotic RER pathway. To do so, we have taken advantage of the fact that the Y11A steric gate mutant of E. coli pol V readily incorporates ribonucleotides into DNA and thereby triggers the onset of RER [2]. We have already established that the main repair process is initiated by RNase HII when it nicks DNA 5′ to the incorporated ribonucleotide [5]. But how is the repair process completed? Assuming that there are similarities between the eukaryotic and prokaryotic pathways, repair of ribonucleotides errantly incorporated into the genome would need the participation of a DNA polymerase that can catalyze strand-displacement at the nick and a FLAP endonuclease to remove the displaced ribo- and deoxyribonucleotides.
The unique enzymatic properties of DNA polymerase I make it an ideal candidate for the job [13, 14]. The pol I polypeptide (103 kDa) encoded by the polA gene, consists of two functional domains, a large (68 kDa) C-terminal domain (Klenow fragment) possessing DNA polymerase, strand separation, and 3′→5′ exonuclease activities [15, 16], and a small (35 kDa) N-terminal fragment containing 5′→3′ exo- and FLAP-endonuclease activities [17–20]. The combination of these activities predetermines pol I to be a key component of the E. coli replication machinery by playing an essential role during Okazaki fragment processing [21], and fulfilling functions that in eukaryotes requires the coordinated action of two enzymes, pol δ and FEN1. By means of its 5′→3′ exonuclease activity, pol I removes RNA primers that are used to initiate Okazaki fragment synthesis, while via strand displacement DNA synthesis and 3′→5′ proofreading, it accurately fills the resulting gaps in the lagging strand [22].
Here, we present compelling biochemical and genetic evidence that the same pol I activities are utilized to complete RNase HII-initiated excision of ribonucleotides errantly incorporated into DNA by pol V and as a consequence, it reduces the mutagenic potential of the low-fidelity polymerase. However, similar to eukaryotes, there is also considerable redundancy in the ribonucleotide repair process in E. coli and in the absence of pol I, certain functions are readily assumed by back-up enzymes.
2. Materials and methods
2.1 Bacterial strains, plasmids and oligonucleotides
Bacterial strains used in this study are described in Table 1. Where noted, new strains were generated via generalized transduction using P1vir [23]. The various polA alleles were transduced into the desired background by selecting for the closely linked antibiotic resistance (TetR for polA107; CmR for polA_D424A and polA_SD−; ZeoR for polA_ΔC). In the case of polA107 and polA_ΔC, transductants were screened for sensitivity to methylmethane sulfonate (MMS), whereas the polA_D424A and polA_SD-transductants were screened by colony PCR (see below). Mismatch repair (MMR) defective derivatives (ΔmutL::Kan, ΔmutL::cat or mutL218::Tn10) were usually generated last, so as to limit genomic instability associated with defects in mismatch repair. The exception was the mutD5 zaf13::Tn10 allele that was introduced after the ΔmutL allele.
Table 1.
E. coli strains used in this study
| Strain | Relevant Genotype | Source or Reference |
|---|---|---|
| MG1655 | F- λ − rph- | E. coli Genetic Stock Center |
| MG1655 polA::Zeo |
F- λ − rph- polA_ΔC b | This work |
| MG1655 polA_SD− |
F- λ − rph polA_F769A/F771A ::cat | This work |
| KA797 | polA_D424A ::cat | [22] |
| RW900a | ΔpolA::Kan | LGI stocks |
| KM52 | ΔmutL460::cat | Martin Marinus |
| JW4128 | ΔmutL720::Kan | E. coli Genetic Stock Center |
| ES1484 | mutL218::Tn10 | E. coli Genetic Stock Center |
| JW0178 | ΔrnhB782::Kan | E. coli Genetic Stock Center |
| JW5446 | Δxni725::Kan | E. coli Genetic Stock Center |
| JW0221 | ΔdinB749::Kan | E. coli Genetic Stock Center |
| NR9548 | mutD5 zaf13::Tn10 | [53] |
| EMBL1789 | polA107 | E. coli Genetic Stock Center |
| ET1214 | zih-219::Tn10 | E. coli Genetic Stock Center |
| RW896 | polA107 zih-219::Tn10 | EMBL1789 x P1. ET1214 |
| RW584a | recA730 lexA51(Def) Δ(umuDC)596::ermGT | [5] |
| RW698a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble | [5] |
| RW710a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble mutL215::Tn5 | [34] |
| RW928a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble polA_D424A ::cat | RW698 x P1.KA797 |
| RW1048a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble polA_D424A ::cat ΔmutL720::Kan | RW928 x P1.JW4128 |
| RW932a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble Δxni725::Kan | RW698 x P1 JW5446 |
| RW1050a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble Δxni725c ΔmutL720::Kan | RW932 x P1.JW4128 |
| RW948a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble polA107 zih-219::Tn10 | RW698 x P1. RW896 |
| RW1088a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble polA107 zih-219::Tn10 ΔmutL720::Kan | RW948 x P1. JW4128 |
| RW968a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble polA107 zih-219::Tn10 Δxni725::Kan | RW948 x P1. JW5446 |
| RW1054a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble polA107 zih-219::Tn10 Δxni725::Kan ΔmutL460::cat | RW968 x P1. KM52 |
| RW1030a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble polA_F769A/F771A ::cat | RW698 x P1. MG1655polA_SD− |
| RW1042a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble polA_F769A/F771A ::cat ΔmutL720::Kan | RW1030 x P1. JW4128 |
| RW1060a | recA730 lexA51(Def) Δ(umuDC)596::ermGT polA_ΔC b | RW584 x P1. MG1655polA::Zeo |
| RW1086a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB749::Kan polA_ΔC b | RW1060 x P1. JW0221 |
| RW1098a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB749::Kan polA_ΔC b ΔmutL460::cat | RW1086 x P1. KM52 |
| RW838a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble ΔrnhB782::Kan | [5] |
| RW970a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔrnhB782 ΔdinB61::ble | KanS derivative of RW838c |
| RW1056a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔrnhB782 ΔdinB61::ble ΔmutL720::Kan | RW970 x P1 JW4128 |
| RW1318a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔrnhB782 ΔdinB749::Kan | RW970 x P1. JW0221 |
| RW1326a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔrnhB782 ΔdinB749::Kan polA_ΔC b | RW1318 x MG1655polA::Zeo |
| RW1336a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB749::Kan ΔrnhB782 polA_ΔC b ΔmutL460::cat | RW1326 x KM52 |
| RW1214a | recA730 lexA51(Def) polBex1 Δ(umuDC)596::ermGT ΔdinB749::Kan | LGI lab stocks |
| RW980a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble polA_D424A ::cat ΔrnhB782::Kan | RW928 x P1. JW0178 |
| RW1320 | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble polA_D424A ::cat ΔrnhB782::Kan mutL218::Tn10 | RW980 x P1.ES1418 |
| RW1214a | recA730 lexA51(Def) polBex1 Δ(umuDC)596::ermGT ΔdinB749::Kan | LGI stocks |
| RW1224a | recA730 lexA51(Def) polBex1 Δ(umuDC)596::ermGT ΔdinB749 | KanS derivative of RW1214c |
| RW1226a | recA730 lexA51(Def) polBex1 Δ(umuDC)596::ermGT ΔdinB749 ΔmutL720::Kan | RW1224 x P1.JW4128 |
| RW1242a | recA730 lexA51(Def) polBex1 Δ(umuDC)596::ermGT ΔdinB749 polA_ΔC b | RW1224 x MG1655polA::Zeo |
| RW1324a | recA730 lexA51(Def) polBex1 Δ(umuDC)596::ermGT ΔdinB749 polA_ΔC b ΔmutL460::cat | RW1242 x KM52 |
| RW1218a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB749::Kan polA_ΔC b ΔmutL460::cat mutD5 zaf13::Tn10 | RW1098 x P1. NR9548 |
| RW1250a | recA730 lexA51(Def) Δ(umuDC)596::ermGT ΔdinB61::ble ΔmutL720::Kan mutD5 zaf13::Tn10 | RW1236 x P1. NR9548 |
: thr-1 araD139 Δ(gpt-proA)62 lacY1 tsx-33 glnV44 galK2 hisG4 rpsL31 xyl-5 mtl-1 argE3 thi-1 sulA211
: encodes polA residues 1–768 and expresses 5′→3′ exo/endonuclease and 3′→5′ exonuclease functions of pol I.
: The KanR cassette was “Flp’d” out by transforming the strain with the temperature sensitive plasmid, pCP20 encoding the FLP recombinase, followed by overnight incubation at 43°C [54].
Where noted, the following antibiotics were used for selection; Zeocin (25 μg/ml), Kanamycin (50 μg/ml), Tetracycline (15 μg/ml), Chloramphenicol (20 μg/ml), and Spectinomycin (50 μg/ml).
Plasmids used in this study are listed in supplementary Table 1. Oligonucleotides used in this study are listed in supplementary Table 2.
2.2 Detection of the enzymatic activities of pol I important for ribonucleotide repair pathway
Oligonucleotides were synthesized by Lofstrand Laboratories (Gaithersburg, MD) and gel purified prior to use (Table 3). DNA substrates were prepared byannealing a 49-mer DNA template, XAG49, containing an abasic site 27 nucleotides from the 3′ termini with the 32-mer downstream blocker, FLAPU, with a uracil at its 5′-terminus and the 17-mer (SSP1), or 27-mer (SSP1-27) primer that form a nicked DNA duplex, or a flapped structure, respectively (Fig. 1). In order to separate polymerase and nuclease activities of pol I, either the primer (17-mer or 27-mer), or 32-mer blocker oligonucleotide were 5′-P32 labeled before hybridization. Annealing was performed at a 1.5-fold excess of unlabeled over P32-labeled oligonucleotides by heating in an annealing buffer (50 mM Tris-HCl (pH 8), 5 mM MgCl2, 50 μg/ml BSA, 1.42 mM 2-mercaptoethanol) for 10 min at 100°C, followed by slow cooling to room temperature. Wild type pol I was purified as described earlier [24]. DNA substrates (2 nM) were incubated with pol I (4, 16, 63, 250 pM, or 1 nM) at 37°C in reaction mixtures containing 10 mM Tris-HCl, pH 7.9; 50 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol and 100 μM dNTPs. Reactions were stopped by the addition of an equivalent amount of gel loading buffer containing 50 mM EDTA, 0.1% xylene cyanol and 0.1% bromophenol blue in 90% formamide. Reaction products were heat-denatured and separated in a 20% polyacrylamide gel containing 8 M urea and then visualized and quantified using a Fuji image analyzer FLA-5100.
Fig. 1.
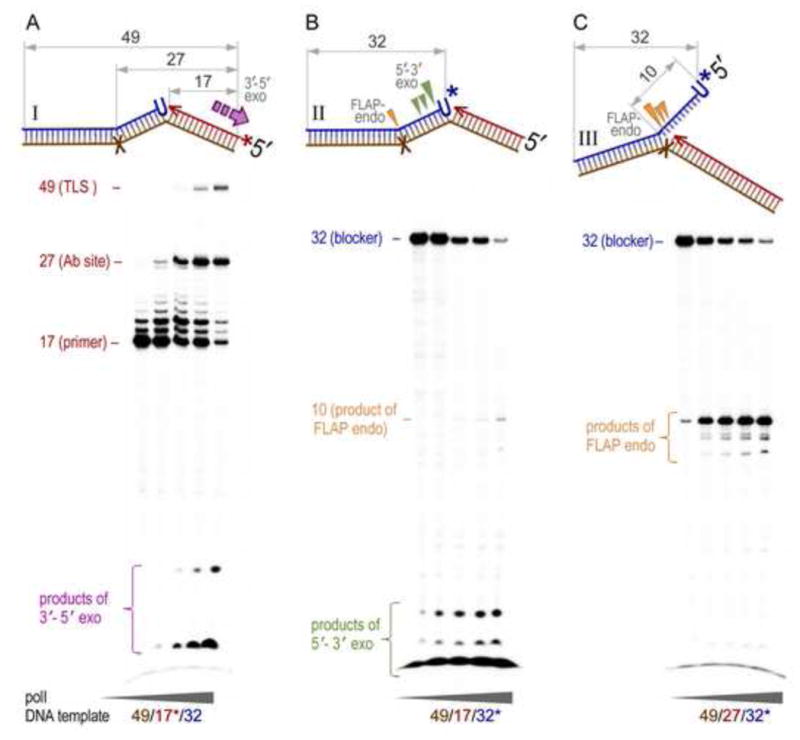
Enzymatic activities of pol I on the FLAP endo-, or exonuclease DNA substrates containing a 5′-terminal monoribonucleotide on the downstream blocking oligonucleotide. The pol I-catalyzed strand displacement synthesis and 3′→5′ exonucleolytic proofreading were assayed using nicked DNA substrate generated by annealing of the 49-mer DNA template containing an abasic site (X) with the P32-labeled 17-mer primer and the 32-mer downstream blocker (A). The nick-specific exonuclease (B) and FLAP-specific endonuclease (C) activities of pol I were visualized using the 17- or 27-mer primers, respectively, hybridized together with the P32-labeled 32-mer blocker to the 49-mer DNA template. Structures of DNA substrates are schematically presented on the top of each gel image (brown – template, red – primer, blue - blocker). In substrate I, the 17mer primer is labeled, while in substrates II and III the 32mer blocking oligonucleotide is labeled (as indicated by, *). The arrows point to the cleavage sites on the DNA made by 3′→5′ exo- (purple ⇪ in I), 5′→3′exo- (green ▼ in II), and flap endonuclease (orange ▼ in II & III) activities of pol I and the corresponding exonucleolytic products, are indicated on the gels. The 27-mer bands seen on the gel in the panel A correspond to the products of primer extension blocked by the abasic site, while the 49-mer band correspond to the primer extensions past the lesion to the full-size product (TLS – translesion synthesis). Reactions were performed using DNA substrate (2 nM), purified pol I (4, 16, 63, 250 pM, or 1 nM), and mixtures of all four dNTPs (100 μM) for 15 min and analyzed by polyacrylamide gel electrophoresis as described in the Materials and Methods section.
2.3 Construction of a polA mutant defective for strand displacement
E. coli MG1655 was aerobically propagated on M9 minimal (M9) medium or on M9 agar plates. Ampicillin (50 μg/ml), Chloramphenicol (15 μg/ml), Tetracycline 3 μg/ml and Zeocin (25 μg/ml) were added as necessary to liquid or solid media for selection. The genome sequence of E. coli MG1655 [25] and Artemis software [26] were used for in silico strain construction.
An ~1.7kb BstAPI-MfeI fragment of the E. coli polA gene was chemically synthesized by Genscript (Piscataway, NJ). The synthesized fragment differed from the wild-type polA sequence at codons Ser769 (AGT→GCA) and F771 (TTC→GCA). These substitutions introduced a novel PstI site into the fragment for ease of subsequent detection and changed the wild-type amino acid to alanine (S769A and F771A respectively). Based upon previous biochemical studies [27] the dual S769A and F77A substitutions should inactivate pol I’s ability to promote strand displacement. For simplicity we will refer to this novel allele as polA_SD−. The synthesized 1.7kb BstAPI-MfeI fragment was sub-cloned into the similarly digested pSKpolAint vector [22] to generate pJM993 (Supplementary Table 1).
Recombineering was performed as described by Gene Bridges (www.genebridges.com) with minor modifications. In brief, 1.4 ml Red/ET proficient E. coli strains were grown at 30°C to an OD600nm of ~0.3. Transient expression of the pRed/ET-encoded (Supplementary Table 1) red genes was induced by adding 50 μl of 10% (w/v) L-arabinose (Sigma) followed by a temperature increase to 37°C. After 40 min incubation, the cells were washed twice with ice cold 10% (v/v) glycerol (AppliChem) and electroporated with 100 ng of DNA in a chilled 1-mm cuvette at 1.35 kV, 10μF, 600ω using an Eppendorf electroporator 2510. Cells were resuspended in 1 ml ice-cold M9 minimal medium and aerobically grown at 37°C for 3h. M9 plates supplemented with appropriate antibiotics were used to select for recombinants.
A two-step Red/ET recombination approach was used to transfer the pJM993 borne polA mutations to the genomic polA gene (2787 bp) of E. coli MG1655 (Supplementary Fig. 1). In the first step, the chromosomal 3′ polA region spanning codons 769 to the ochre codon was replaced by a PCR amplified 664 bpZeo R cassette displaying 50-base pairs homologous to the genomic target region (primer p3 × p4; Supplementary Table 1). Upon the recombineering step, ~550 transformants were selected on M9+zeo plates. No colonies appeared without induction of the Red/ET machinery. Four of the ZeoR colonies were analyzed and one, MG1655 polA_ΔC,that displayed the correct genomic integration of the ZeoR cassette as revealed by sequencing, was chosen for further study (Table. 1). In the second step (Supplementary Fig. 1), ZeoR was replaced by polA_SD−_cat. Therefore, the pJM993 stretch comprising the 3′ region of polA_SD− encompassing the S769A and F771A substitutions (i.e. polA codons 502 to the ochre stop codon) and adjacent cmR marker was PCR amplified using primers cp6 and p2 (Supplementary Table 1). The 2279 bp linear polA_SD−_cat fragment which shares 800 bp (5′) and 50 bp (3′) of terminal sequence homology with the genomic target locus was used to replace the ZeoR cassette from Red/ET proficient MG1655 polA_ΔC by Red/ET recombination. Several independent recombineering approaches yielded two clones that displayed a CmR and ZeoS phenotype characteristic of the successful displacement of the ZeoR marker from the MG1655 polA_ΔC genome. For one clone, the correct insertion of the polA-SD−-cat cassette which introduces the S769A and F771A codons and reconstructs the full-length polA gene in the resulting strain, MG1655 polA_SD− was confirmed by PCR and DNA sequencing (Supplementary Fig. 1). In addition, the MG1655 polA_SD− clones displayed no growth on M9 plates supplemented with Tetracycline or Ampicillin, which confirms the absence of plasmids pRed/ET and pJM993. The polA_SD− allele was subsequently moved to additional strains of E. coli via conventional P1vir transduction [23].
2.4 Colony PCR assay to test for polA_SD− and polA_D424A genotypes
To identify strains carrying the polA_SD− allele, chloramphenicol resistant colonies were subject to colony PCR using primers polA-RBamHI and polA_F2105 (Table 3). Briefly, cells were heated to 95°C for 5 min to lyse cells. An ~700 bp fragment of the polA gene was amplified after 40 PCR cycles (95°C for 30sec, 60°C 45sec and 72°C 2min). The ~700 bp amplicon was then digested with PstI. Strains carrying the polA_SD− allele gave products of 225 bp and 478 bp. The polA_D424A allele was similarly identified by colony PCR as previously described [22], using primers polA-FD424 and polA-RD424 that yields an amplicon of ~1.2kb (Supplementary Table 2). The polA_D424A substitution generates a novel BssHII site in polA. The ~1.2 kb amplicon was therefore digested with BssHII and the polA_D424A allele confirmed by the appearance of 646 bp and 574 bp fragments.
2.5 Quantitative analysis of spontaneous reversion of the hisG4(oc)
Cells transformed with the low-copy-number empty vector plasmid, pGB2, or pGB2-derived plasmid pRW134, or pJM963, expressing wild-type pol V or umuC_Y11A variant respectively (Supplementary Table 1), were grown overnight at 37°C in LB media plus spectinomycin. At least three independent cultures were assayed for each strain. To determine the number of spontaneously arising histidine mutants on the plate, cells were seeded on the Davis and Mingioli minimal agar plates [28] plus glucose (0.4% wt/vol); agar (1.0% wt/vol); proline, threonine, valine, leucine, and isoleucine (all at 100 μg/ml); thiamine (0.25 μg/ml); and either no histidine, 1 μg/ml histidine, or 4μg/ml casamino acids. On the plates lacking histidine, only pre-existing His+ mutants in the culture grew to form colonies. However, on the plates containing 1μg/ml histidine, or 4μg/ml casamino acids, >4 × 107 auxotropic bacteria grew to form a lawn, concomitantly exhausting the low level of histidine after 1–2 days incubation. Spontaneously arising His+ mutants grew up through the lawn and were counted after 4 days incubation at 37°C.
We would like to emphasize that the number of spontaneous revertants arising on the plate is a characteristic of the strain assayed. The number of revertants that arise spontaneously during the 4-day incubation is independent of the initial number of bacterial cells plated (within the range of 105 to 108 cells [29], but is, instead, dependent on the final number of auxotrophs on the plate and that number is a function of the histidine concentration [30, 31]. The actual number of auxotrophic cells (background lawn) on the plate are unimportant, but are nevertheless assumed to be invariant between the strains assayed, since the histidine, or casamino acid concentration is constant and supports the growth of the same absolute number of auxotropic cells per plate, despite any differences in overall growth rates. As a consequence, the extent of spontaneous mutagenesis is expressed as a frequency (mutants per plate) rather than a rate (mutants per cell plated).
The number of His+ mutants spontaneously arising on the plate (at least 3 plates for each culture) was determined by subtracting the number of pre-existing mutants that grew on plates lacking histidine. The values reported in the tables shown in Figs. 2, 3 & 5 represent the average number of His+ mutants per plate from at least 3 independent experiments (± standard error of the mean [SEM]). The relative contribution of umuC_Y11A mutations was calculated by subtracting the number of mutants arising on plates containing the control vector, pGB2 (and lacking pol V) and then expressing the corrected number of umuC_Y11A-dependent His+ mutants (pJM963), as a percentage of the corrected number of wild-type pol V-dependent His+ mutants (pRW134). The data plotted in Figs 2, 3 & 5 represent the mean value for the relative umuC_Y11A-dependent mutagenesis (± standard error of the mean [SEM]). The differences between these values for various strains were assessed for statistical significance using the Student’s t-test (SigmaPlot 11.0; Systat Software Inc., Chicago, IL).
Fig. 2.
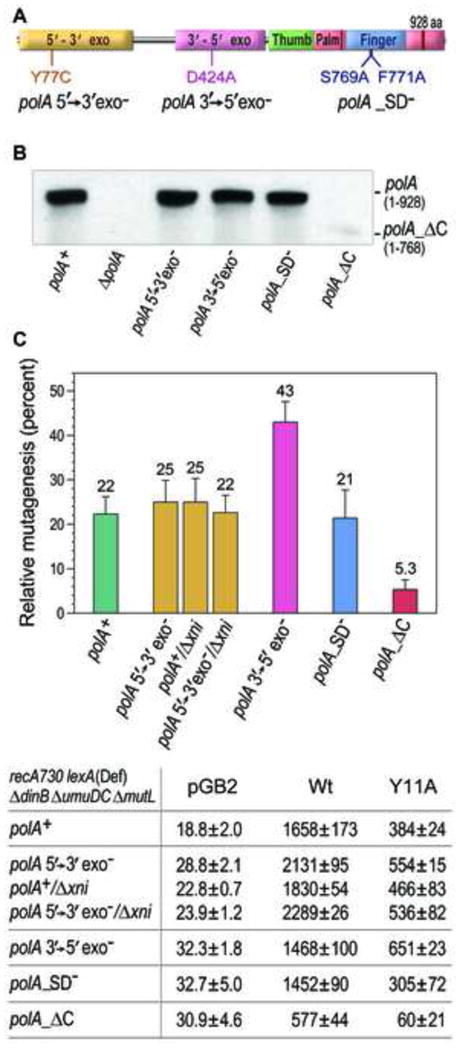
Effect of mutations in the chromosomal polA gene on spontaneous mutagenesis in recA730 lexA(Def) ΔumuDCΔ dinBΔ mutL strains harboring the empty vector, pGB2, or pRW134 expressing wild-type pol V, or JM963 expressing UmuD′ and UmuC_Y11A. A. The location of the amino acid substitutions of each respective missense polA allele are indicated within the structural domains of pol I. The polA_ΔC allele expresses a truncated pol I protein (residues 1–768). B. Western Blot of steady-state levels of pol I in various strains of E. coli. Whole-cell extracts were obtained from the following strains: RW710 (polA+); RW900 (ΔpolA::Kan); (RW1088 (polA107); RW1048 (polA_D424A); RW1042 (polA_F769A/F771A); RW1098 (polA_ΔC) and the steady-state levels of pol I determined as described in Materials and methods. As observed, the steady state levels of pol I encoded by the three missense polA alleles were similar to the level of wild-type pol I. However the steady-state level of the truncated pol I (1–768) protein (encoded by polA_ΔC) was significantly lower compared to the wild-type enzyme and missense pol I mutants and was barely detectable unless the images were greatly overexposed (not shown). C. Spontaneous mutagenesis was measured by assaying reversion of the hisG4 ochre allele (leading to histidine prototroph) as described in Materials and methods. The following mismatch repair-defective strains were used in the assays: RW710 (polA+); RW1088 (polA107); RW1050 (Δxni); RW1054 (polA107/Δxni); RW1048 (polA_D424A); RW1042 (polA_F769A/F771A); RW1098 (polA_ΔC). The average number of His+ revertants per plate ± standard error of the mean (SEM) for the strains lacking pol V, or expressing wild type, or umuC_ Y11A pol V is indicated in the table below the graph. Since the extent of mutagenesis promoted by wild-type pol V differs in the various polA strains, the level of mutagenesis promoted by umuC_ Y11A is expressed as a percentage of wild-type pol V-dependent mutagenesis in the same strain and shown in the graph. Comparison of these values allows us to characterize the effect of mutations in pol I on the excision repair of ribonucleotides incorporated by the umuC_ Y11A allele of pol V. The difference in umuC_ Y11A-dependent mutagenesis between polA+ and either polA 3′→5′ exo− or polA_ΔC strains are statistically significant (p values of 0.02 and 0.015, respectively) as revealed by Student’s t-test analysis.
Fig. 3.
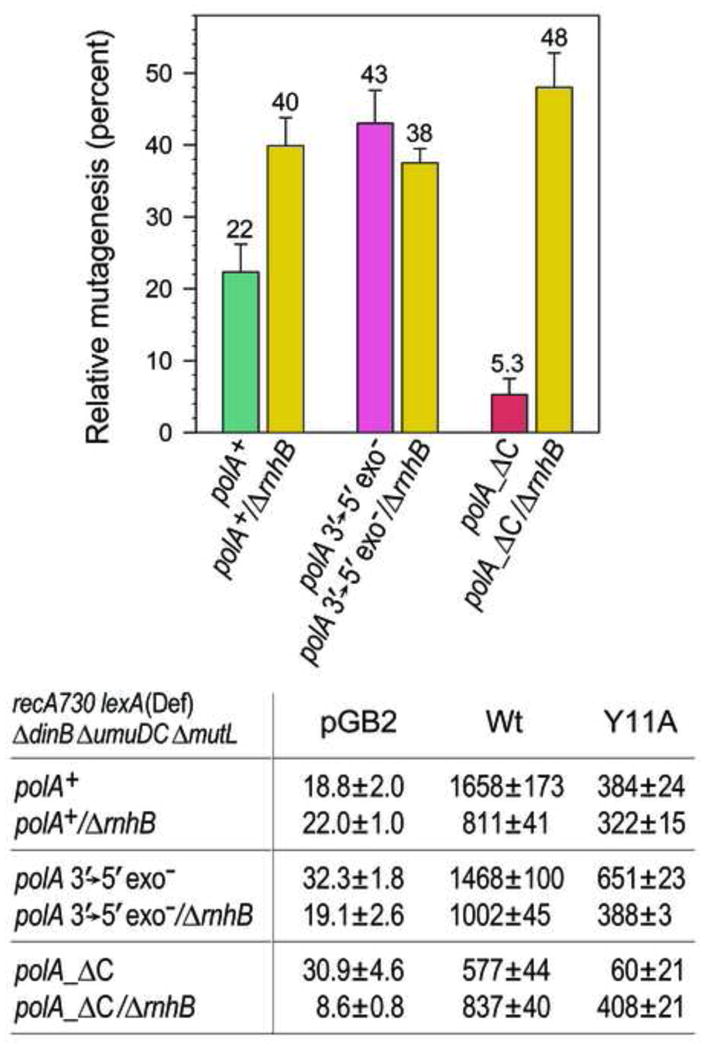
Effect of Rnase HII on the level of spontaneous mutagenesis promoted by the empty vector, pGB2, or pRW134 expressing wild-type pol V, or JM963 expressing UmuD′ and UmuC_Y11A in various polArecA730 lexA (Def) ΔumuDCΔ dinBΔ mutL ± ΔrnhB strains. Spontaneous mutagenesis was measured by assaying reversion of the hisG4 ochre allele (leading to histidine prototrophy) as described in Experimental procedures. The average number of His+ revertants per plate ± standard error of the mean (SEM) for the strains lacking pol V, or expressing wild type, or umuC_ Y11A pol V is indicated in the table below the graph. The average level of mutagenesis promoted by umuC_ Y11A is expressed as a percentage of wild-type pol V-dependent mutagenesis in the same strain and shown in the graph are taken from [14] The following mismatch repair-defective strains were used in the assays: RW710 (polA+); RW1056 (polA+Δ rnhB); RW1048 (polA_D424A); RW1320 (polA_D424A ΔrnhB); RW1098 (polA_ΔC); RW1336 (polA_ΔC ΔrnhB). The data for the polA+, polA 3′→5′ exo− and polA_ΔC strains expressing wild-type RNase HII are taken from Fig. 2 and are shown for direct comparison with the data for the ΔrnhB strains. The difference in umuC_ Y11A-dependent mutagenesis is statistically significant for the polA+ and polA+/ΔrnhB (p=0.02) and for polA_ΔC and polA_ΔC/ΔrnhB (p<0.002) strains.
Fig. 5.
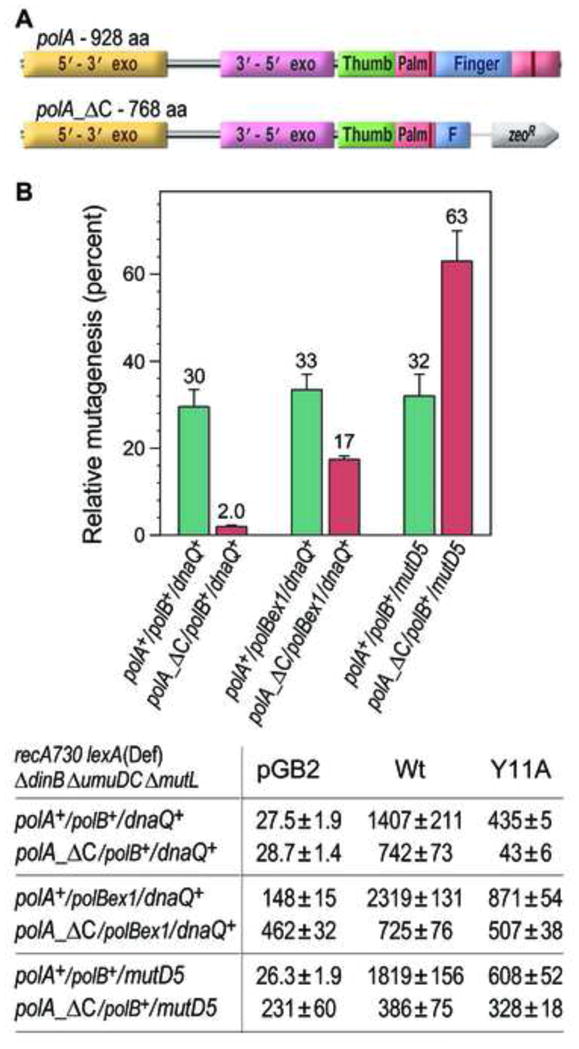
Effect of polBex1 and mutD5 alleles on spontaneous mutagenesis in recA730 lexA(Def) ΔumuDCΔ dinBΔ mutLpolA +/ polA_ΔC strains harboring the empty vector, pGB2, or pRW134 expressing wild-type pol V, or JM963 expressing UmuD′ and UmuC_Y11A. A. Full-length polA encodes a 928 amino acid protein, whereas the polA_ΔC allele expresses a truncated pol I protein (residues 1–768) that lacks key structural domains necessary to catalyze DNA synthesis. B. Because of their low viability on the defined “low histidine” minimal plates, spontaneous mutagenesis was measured by assaying reversion of the hisG4 ochre allele (leading to histidine prototrophy) on minimal agar plates containing 4μg/ml Casamino acids as described in Materials and methods. The following mismatch repair-defective strains were used in the assays: RW1236 (polA+); RW1098 (polA_ΔC); RW1226 (polA+polBex1 ); RW1324 (polA_ΔC polBex1); RW1250 (polA+mutD5 ); RW1218 (polA_ΔC mutD5). The average number of His+ revertants per plate ± standard error of the mean (SEM) for the strains lacking pol V, or expressing wild type, or umuC_ Y11A pol V is indicated in the table below the graph. Since the extent of mutagenesis promoted by wild-type pol V differs in the isogenic strains, the level of mutagenesis promoted by umuC_ Y11A is expressed as a percentage of wild-type pol V-dependent mutagenesis in the same strain and shown in the graph. Comparison of these values allows us to characterize the effect of inactivating the proofreading activity of pol II (polBex1), or proofreading-deficient pol III (mutD5), on the excision repair of ribonucleotides incorporated by the umuC_ Y11A allele of pol V in polA+ or polA_ΔC strains.
2.6. Effect of polA alleles deficient in 3′→5′ exonuclease activity, or DNA polymerase activity on the spectra of spontaneous mutations observed in the rpoB gene of lexA(Def) recA730 ΔdinB ΔumuDC rnhB+/− strains expressing umuC_Y11A
The mutation spectra were generated using the rpoB/RifR mutagenesis assay [32, 33]. E. coli strains RW1048 [relevant genotype: lexA(Def) recA730 ΔdinBΔ umuDCpolA _D424A ΔmutL], RW1320 [relevant genotype: lexA(Def) recA730 ΔdinBΔ umuDCpolA _D424A ΔrnhBΔ mutL], RW1098 [relevant genotype: lexA(Def) recA730 ΔdinBΔ umuDCΔ polA_ΔC ΔmutL], and RW1336 [relevant genotype: lexA(Def) recA730 ΔdinBΔ umuDCΔ polA_ΔC ΔrnhB ΔmutL], expressing the umuC_Y11A plasmid (pJM963) were diluted from frozen stocks and the cultures containing <1000 viable cells were grown in LB for at 37°C. The next day, appropriate dilutions were spread on an LB agar plates containing 100 μg/ml rifampicin. Individual independent RifR colonies were subjected to PCR in a 96-well micro-titer plate using the PCR primers RpoB1: 5′-CAC ACG GCA TCT GGT TGA TAC AG-3′ and RpoF1: 5′-TGG CGA AAT GGC GGA AAA C-3. An <1 kb central region of the rpoB gene was amplified by denaturation at 95°C for 3 min, followed by 30 cycles of 94°C for 30 s, 1 min at 59°C, 2 min at 72°C, with a final extension step done at 72°C for 7 min. Sequencing of the target region of rpoB in each amplicon was determined by Beckman Coulter Genomics (Danvers, MA) using primer WOG923AP01 (5′-CAG TTC CGC GTT GGC CTG-3′). Utilization of a single pair of oligonucleotide primers for PCR amplification and a single primer for DNA sequencing was possible because 88% of all rpoB mutations are localized in the 202 bp region in the middle of the gene [32]. Only base-pair substitutions occurring between positions 1516 and 1717 of the rpoB gene were considered during data analysis. Nucleotide sequences were aligned and analyzed using the ClustalW multiple sequence alignment program (Hinxton, UK). Forward mutations rates in rpoB gene (mutations to rifampicin resistance) were determined as previously described [34].
2.7 Western blot analysis of various polA strains
Cells were grown overnight at 37°C in LB plus appropriate antibiotics. The next morning, cultures were diluted 1:100 in fresh LB and grown with aeration at 37°C until they reached an OD600 of ~0.5. Cells were harvested by centrifugation, resuspended in 1× SDS sample buffer (50 mM Tris-HCl [pH 6.8], 10% glycerol, 2.3% sodium dodecyl sulfate [SDS], 0.1% bromophenol blue, 10 mM dithiothreitol), and immediately frozen in dry ice. Cells were lysed by multiple freeze-thaw cycles and boiled for 5 min. Extracts were immediately applied to a 4–12% SDS-PAGE gel. After separation proteins were transferred to an Immobilon-P membrane (Millipore) using standard Western blot protocols. The membrane was incubated overnight with a 1:5,000 dilution of affinity purified polyclonal rabbit pol I antisera followed by a 1 hour incubation with a 1:10,000 dilution of secondary goat anti-rabbit alkaline phosphatase conjugated antibodies (BioRad). Steady state levels of pol I was determined using the Tropix CPD-star kit (Applied Biosystems) as previously described [35]
3. Results
3.1 Enzymatic properties of pol I required for ribonucleotide excision repair
The previously characterized biochemical properties of pol I make it an ideal candidate as the enzyme that performs strand displacement synthesis at an RNase HII-generated nick and subsequent removal of the resulting 5′-FLAP. To reconstitute the potential pol I-dependent step of ribonucleotide repair pathway, we synthesized oligonucleotides that after annealing generated a model DNA substrate containing a nick adjacent to a unique uracil ribonucleotide located in double stranded DNA (Fig. 1, substrates I and II) and assayed for the ability of pol I to initiate strand displacement and the subsequent exo/endonuclease removal of the resulting displaced product (Fig. 1).
In this model substrate, the template oligonucleotide contains a single abasic DNA lesion 27 nt downstream of the 3′ template terminus. Pol I bypasses an abasic site poorly and as a consequence, the lesion was intended to provide a kinetic barrier to DNA synthesis and thereby ensure ample time for the 5′ FLAP-endonuclease activity of pol I to cleave the displaced strand. As seen in Fig. 1A, labeling of the 17-mer primer (substrate I) reveals dose-dependent primer extension catalyzed by pol I with the majority of products ultimately terminating opposite the abasic site in the template DNA. At the highest concentration of pol I, in addition to a small amount of full length product indicating replication past the abasic site, accumulation of radiolabeled mono- and short polynucleotides could also be detected. These short products are attributed to the 3′→5′ exonucleolytic proofreading of pol I that is activated when elongation of the radiolabeled primer is interrupted by the lesion in the DNA template.
At most concentrations of pol I, DNA synthesis was generally distributive, with primers only elongated by 1–2 nucleotides. When the 32-mer uracil-containing downstream blocker-strand was radiolabeled (Fig. 1B, substrate II), we observed a dose dependent decrease in the full-length oligonucleotide with the concomitant accumulation of products that were predominantly 1 nt long, although products of 2–3 nucleotides were also detected. These products form as a result of the efficient 5′-exo/endonuclease activity of pol I removing a single nucleotide, or the short FLAPs generated by limited distributive strand displacement DNA synthesis observed in Fig. 1A. At the highest enzyme to template ratio, when the first two nucleotides of the downstream blocker are displaced relatively efficiently, FLAP-endonuclease activity becomes visible (marked by the appearance of a faint 10 nt long band), but the majority of the released products are still mono-, di-, and trinucleotides.
To mimic substrates with longer 5′-FLAPs (Fig. 1C, substrate III), we synthesized a 27-mer primer oligonucleotide with its 3-end terminating opposite the abasic site in the template. As a′ consequence, the ten 5′ bases of the radiolabelled uracil-containing downstream blocker oligonucleotide cannot hybridize to the template strand and therefore form a FLAP-structure. As seen in Fig. 1C, under these conditions, the 10-nt FLAP substrate was efficiently hydrolyzed by pol I in a concentration-dependent manner. Based upon our observations presented in Fig. 1B, we suggest that pol I-catalyzed excision of errantly incorporated rNMP from DNA mainly proceeds by limited strand displacement synthesis and efficient 5′→3′ exonucleolytic cleavage. However, under conditions where a longer FLAP-structure is generated, the entire DNA FLAP containing a single 5′ ribonucleotide can be removed by the 5′→3′ endonucleolytic activity of pol I.
3.2 Rationale behind the in vivo assays used to elucidate the mechanism of ribonucleotide excision repair
Although pol V is best characterized for its ability to facilitate translesion DNA synthesis, it has also been shown to contribute to high levels of spontaneous mutagenesis on the E. coli chromosome in recA730 lexA(Def) strains in which the SOS-regulon is fully derepressed and RecA protein is in a constitutively activated form (RecA*) [36]. The mutagenesis is not thought to represent error-prone bypass of cryptic DNA lesions, but rather an ability of the highly activated pol V to compete with E. coli’s four other polymerases for access to undamaged genomic DNA [37]. Furthermore, we have shown that such phenotypes can be enhanced when the highly abundant DNA pol IV (encoded by dinB) is deleted from the chromosome [5]. Even though our genetic system is quite complex, we believe that its utilization as a tool to investigate RER is justified because expression of the pol V steric gate mutant in lexA(Def) recA730 ΔdinB ΔumuDC strains maximizes the amount of ribonucleotides errantly incorporated into chromosomal DNA and thus significantly facilitates the task of identifying individual enzymes, as well as cellular pathways involved in protecting genomic DNA from excessive ribonucleotide misincorporation. Indeed, we have successfully used this approach to elucidate the pivotal role of RNase HII in RER [5] and the important back-up roles for RNase HI and NER in ribonucleotide repair [5, 12].
It should be noted that in these studies, we monitored the extent of umuC_Y11A-dependent mutagenesis relative to that induced by wild-type pol V, rather than the absolute number of His+ revertants promoted by the mutant polymerase in different backgrounds [5, 12]. The reason for such analysis lies in the fact that the level of mutagenesis promoted by pol V differs considerably in the various strain backgrounds and this variability is likely caused by the extent of endogenous single-stranded DNA in the cell, which is an important factor in hyper-activating RecA730 and promoting pol V-induced mutagenesis in vivo [36, 38]. Since the only known biochemical difference between UmuC_Y11A and wild-type pol V is its ability to incorporate riboncleotides [2], by expressing umuC_Y11A-dependent mutagenesis as a percent of the mutagenesis induced by wild-type pol V, we are able to focus only on those mutagenic events that are specifically related to rNMP incorporation and repair.
In previous studies, we reported that in mismatch- and ribonucleotide-repair proficient recA730 lexA(Def) ΔdinB strains, umuC_Y11A-dependent spontaneous mutagenesis is substantially lower (~10 fold) than that observed with wild-type pol V, but it increases significantly when either MMR, or RER is inactivated [5, 12]. By taking advantage of the fact that MMR complex exhibits a strong bias for the repair of transitions made by all DNA polymerases, while pol V, in contrast to other E. coli DNA polymerases, prefers to generate transversions, we demonstrated that the increase in the relative mutagenesis of the strains defective in either MMR, or RER, is determined by unrepaired errors generated by two different polymerases operating in the two different repair pathways [12]. An increase in the relative mutagenesis in the ΔrnhB strain represents persistent umuC_Y11A-generated mutations escaping RER repair. In contrast, in the MMR-defective strains, it reflects misincorporations made by a DNA polymerase (most probably pol I) catalyzing the re-synthesis step of RNase HII-initiated RER, [12]. As a result, for the current study we have inactivated the MMR pathway via mutations in mutL, so as to help identify the E. coli DNA polymerase involved in RER.
3.3 Effect of inactivating FLAP-endonuclease activity on ribonucleotide excision repair in vivo
Our biochemical studies demonstrate that pol I possesses all the necessary activities required for RER. One way to prove that pol I is indeed involved in the repair of rNMPs incorporated by plasmid-encoded pol V in vivo, is to assay for phenotypic changes in umuC_Y11A-dependent mutagenesis in ΔpolA strains devoid of all pol I’s enzymatic activities. Unfortunately, a ΔpolA recA730 strain is inviable [39], thereby precluding the possibility of determining the effects of a ΔpolA allele on umuC_Y11A-dependent mutagenesis. However, certain missense polA mutations are viable in a recA730 strain background. Most relevant, is the polA107 allele [40] that encodes a Y77C mutant of pol I (Fig. 2A) [41] lacking 5′→3′ exo/endonuclease activity [42], which is viable in the recA730 background [39]. To assay the role that the 5′→3′ exo/endonuclease of pol I has in ribonucleotide repair, we transduced the polA107 allele that had previously been linked to zih219::Tn10 into a recA730 lexA51(Def) ΔdinBΔ umuDCΔ mutL strain background (Table 1). We reasoned that if the 5′→3′ exo/endonuclease activity is important for pol I’s role in ribonucleotide excision repair in vivo, then we might observe a difference in the extent of umuC_Y11A-dependent spontaneous mutagenesis relative to that promoted by wild-type pol V, in a manner similar to that observed in ΔrnhB strains lacking RER [5]. However, that turned out not to be the case, since the relative amount of umuC_Y11A-dependent spontaneous mutagenesis remained essentially unchanged in the polA107 strain (Fig. 2C).
We then considered the possibility that similar to the eukaryotic RER pathway, there may be redundancy at various steps of the E. coli RER pathway. Indeed, E. coli possesses another FLAP-endonuclease, ExoIX, that is encoded by the xni gene [43], and which shares considerable homology to the N-terminus (FLAP-domain) of Pol I [44]. As a consequence, we hypothesized that the FLAP-endonuclease activity of ExoIX might substitute for the FLAP endonuclease activity of pol I during ribonucleotide repair. Although ΔpolAΔ xni strains are apparently inviable [45], we had no difficulty in transducing the Δxni725::Kan allele into either polA+ or polA107 recA730 lexA(Def) ΔdinB ΔumuDCΔ mutL strain backgrounds (Table 1). In both strains, umuC_Y11A-dependent mutagenesis was ~25% of that observed with wild-type pol V (Fig. 2C). Thus, inactivation of the 5′-FLAP endonuclease activity of pol I and/or Xni, has no discernible effect of the level of umuC_Y11A-dependent mutagenesis in vivo. As a consequence, we conclude that in the absence of both 5′→3′ exo/endonuclease of pol I and ExoIX, the single-stranded FLAP generated during RER can be processed by one of the numerous E. coli single-stranded nucleases and that the RER does not require a dedicated 5′-FLAP endonuclease.
3.4 Effect of inactivating the 3′→5′ exonuclease activity of pol I on ribonucleotide excision repair in vivo
Since our studies with the polA107 mutant did not delineate an in vivo role for pol I in RER, we assayed the effect of other polA missense alleles on umuC_Y11A-dependent mutagenesis, and in particular, the D424A mutant that inactivates the 3′→5′ exonuclease activity of pol I (Fig. 2A;[22, 46]. E. coli strains with the polA_D424A allele expressed from the chromosome exhibit a modest 1.5–4 fold increase in spontaneous mutation frequencies, which have been attributed to error-prone DNA synthesis during the processing of Okazaki fragments [47]. The polA_D424A allele was transduced into the recA730 lexA(Def) ΔdinB ΔumuDCΔ mutL background and spontaneous His+ mutagenesis was assayed. The relative amount of umuC_Y11A-specific spontaneous mutagenesis increased from ~22% of that observed in the polA+ strain, to ~43% in the strain with defective 3′→5′ exonuclease activity (Fig. 2C). We attribute this phenotype to the reduced fidelity of the exonuclease-deficient pol I enzyme making deoxynucleotide misincorporations during RER of umuC_Y11A-incorporated ribonucleotides. This observation therefore provides the first clear indication that pol I plays an important role in ribonucleotide excision repair in vivo.
3.5 Effect of inactivating the strand-displacement activity of pol I on ribonucleotide excision repair in vivo
In the proposed model for eukaryotic ribonucleotide excision repair, a key step in the process is strand-displacement by pol δ/ε that is initiated at an RNase H2 introduced nick [9]. In E. coli, this step is most likely to depend upon the strand-displacement activity of pol I. We therefore hypothesized that a mutant of pol I unable to promote strand-displacement would exhibit a pronounced umuC_Y11A phenotype.
Previous in vitro studies identified Ser769, Phe771, and Arg841 residues in the fingers subdomain of pol I as being required for strand displacement synthesis [27]. Ser769 and Phe771 are involved in initializing strand separation through the formation of a FLAP-structured DNA, while Arg841 via an interaction with the template strand, ensures optimal strand separation and DNA synthesis [27].
We constructed a polA strain defective in strand displacement by introducing S769(AGT)→A769(GCA) and F771(TTC)→A771(GCA) substitutionsinto the chromosomal polA gene (Supplemental Fig. 1). Briefly, the chromosomal 3′ polA region was first replaced by a ZeoR cassette, and later substituted by a polA_SD− ::cat fragment containing the altered polA codons. The correct insertion of the polA_SD− ::cat cassette was confirmed by PCR, while the precise replacement of the ZeoR cassette and introduction of the altered polA S769A and F771A codons was confirmed by sequencing of the mutant chromosomal polA locus.
The polA_SD− allele was subsequently moved into the recA730 lexA(Def) ΔdinB ΔumuDC ΔmutL background by generalized P1 transduction, and spontaneous His+ mutagenesis assayed. However, the anticipated “pronounced umuC_Y11A phenotype” was not observed. Instead, there was essentially no measurable change in the relative amount of umuC_Y11A-dependent spontaneous mutagenesis compared to wild-type pol V in the polA_SD− mutant versus the wild-type polA strain (Fig. 2C). We attribute the absence of a clear phenotype to the fact that despite exhibiting minimal strand displacement in vitro [27], the polA_SD− mutant may nevertheless possess sufficient strand displacement activity to function in vivo, or that similar to the 5′→3′ exo/endonuclease activity, which can be assumed by a substitute enzyme, the strand-displacement activity of pol I can be provided by an alternate enzyme, such as another DNA polymerase, and/or one of E. coli’s many single-stranded exonucleases.
3.6 Effect of inactivating the DNA polymerase activity of pol I on ribonucleotide excision repair in vivo
During the course of generating the polA_SD− mutant, we made an intermediate construct in which the 3′ end of the polA gene was replaced with a Zeocin resistance cassette (polA_ΔC; Supplemental Fig. 1). This construct is predicted to express a truncated pol I (residues 1–768) that possesses both 5′→3′ and 3′→5′ exonuclease activities, but should be completely defective in polymerase activity, since it lacks key parts of the “finger” and “palm” domains, which are structural features required for DNA synthesis. Joyce and Grindley previously reported that plasmids encoding either the 5′→3′ and 3′→5′ exonuclease activities of pol I enable recA+Δ polA strains to grow to form viable colonies on rich LB-media [19]. We therefore attempted to transduce the polA_ΔC allele into the recA730 lexA(Def) ΔdinB ΔumuDC background and were able to obtain viable colonies on LB plates.
Western blots with antibodies raised to wild-type pol I indicates that the truncated pol I protein is either poorly expressed, or rapidly degraded, compared to either the wild-type or missense polA mutants (Fig. 2B), but nevertheless must be sufficient for cell viability. We assayed spontaneous His+ mutagenesis and found that the relative amount of umuC_Y11A mutagenesis in the polA_ΔC strain decreased significantly (p=0.015) when compared to the wild-type polA strain (5.3% vs. 22%; Fig. 2C). The decrease in mutagenesis in the polA_ΔC polymerase defective strain provides additional evidence for the pivotal role of pol I in RER. We suggest that in the absence of pol I polymerase activity, the DNA synthesis step of RER is performed by an alternate DNA polymerase with higher fidelity than pol I (see below).
3.7 Effect of inactivating rnhB on pol I-dependent ribonucleotide excision repair in vivo
The initial step of RER is Rnase HII-dependent nicking of a DNA duplex 5′ to misincorporated ribonucleotides. In the absence of Rnase HII (as in the case of ΔrnhB strains), there is no nicking and subsequently no pol I-dependent RER. As a consequence, umuC_Y11A-dependent mutations are therefore expected to persist. To confirm that the change in the extent of mutagenesis observed in the rnhB+ 3′→5′ exonuclease-deficient polA and polA_ΔC strains is due to perturbation of pol I’s function in RER, we constructed isogenic ΔrnhB strains and assayed for spontaneous His+ mutagenesis.
Since RER is inactive in the ΔrnhB strains, umuC_Y11A-dependent mutagenesis does not depend on the DNA polymerase performing RER, but rather is determined by persisting mistakes made by the pol V variant. Indeed, the relative extent of umuC_Y11A mutagenesis increased from 22% in the polA+rnhB +strain to 40% in the polA+Δ rnhB strain. Strikingly, umuC_Y11A mutagenesis increased from 5% in the polA_ΔC rnhB+strain to 48% in the polA_ΔC ΔrnhB strain (Fig. 3). In contrast, in the polA_D424Astrain, activation of RNase HII-dependent RER appears to have little effect on the level of relative mutagenesis (43% vs. 38%). At first glance, this might lead one to conclude that the mutagenesis is independent of RNase HII function. However, this is not the case, but is simply due to the fact that the increase in mutagenesis derived from polA_D424A-dependent RER is, by coincidence, in the same range as persisting errors made by umuC_Y11A in the absence of RER.
This assumption was confirmed by determining the spectra of spontaneous rpoB mutations recovered from the polA_D424A rnhB+/−strains. Indeed, the spectrum of the polA_D424A ΔmutLrnhB + strain (Fig. 4A, top; Table 2) is very similar to the spectrum observed by Makiela-Dzbenska et al for the ΔmutL strain expressing proofreading-deficient pol I [47], while the spectrum of the polA_D424A ΔmutLΔ rnhB strain (Fig. 4A, bottom; Table 2) is reminiscent of the umuC_Y11A-dependent spectrum that we previously obtained for the ΔmutLΔ rnhB strain [12]. An even more dramatic effect of RNase HII on the rpoB spectra was observed in the polA_ΔC rnhB+/− strains completely lacking pol I polymerase activity (Fig. 4B; Table 2). Together, we believe that these observations are consistent with the notion that the spectra of mutations manifested in rnhB+/− strains are generated by different DNA polymerases.
Fig 4.
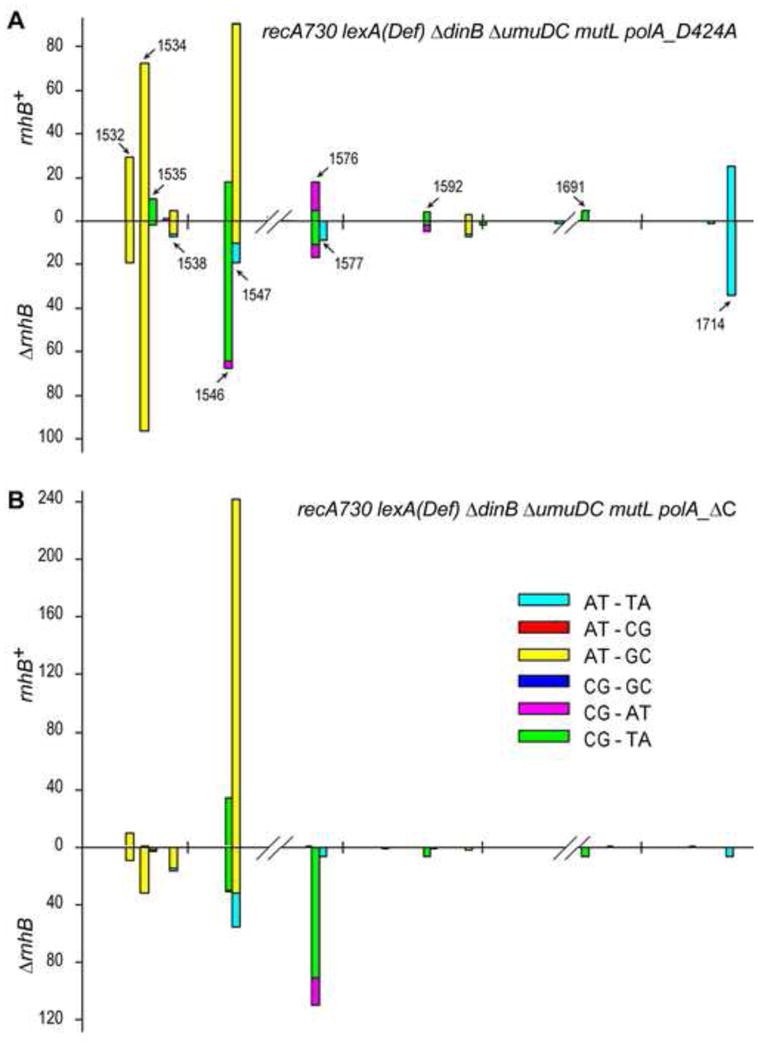
Spectra of spontaneously arising rpoB mutations in recA730 lexA(Def) ΔdinB ΔumuDC ΔmutLrnhB +/− strains expressing umuC_Y11A. A. polA _D424A strains deficient in pol I 3′→5′ exonuclease activity. The arrows indicate mutagenic hot spots within the rpoB. B. polA _ΔC strains deficient in pol Ipolymerase activity. Approximately 300 Rif R mutants were analyzed for each strain (Table 2). The forward mutation rates were assayed by measuring resistance to rifampicin and were in the range of 3–5 × 10−6. The spectra in the rnhB+ and ΔrnhB strains differ in both the types of mutations and locations of mutagenic hot-spots. Because of defects in MMR, all the spectra are dominated by transition mutations. However, since low-fidelity pol V makes a significant number of transversion mutations compared to other E. coli DNA polymerases [34, 37, 52], the number of umuC_Y11A-dependent transversions in the rnhB strains is higher than in the rnhB+ strains (see Table 2). The shift in the types of mutations observed is significantly more pronounced in the polA_ΔC strains. The rnhB+ strains undergo active RER; as a consequence, they have a reduced number of pol V-specific transversions, and the spectrum generated in these strains reflect uncorrected errors made by pol I (panel A) or pol III (panel B) during the DNA re-synthesis step of RER.
Table 2.
Spectrum of spontaneous mutations generated in the rpoB gene of recA730 lexA(Def) ΔdinBΔ umuDCΔ mutLrnhB +/ΔrnhB polA_D424A and polA_ΔC strains in the presence of umuC_Y11Aa.
| Position b | bp change |
rnhB+ polA_D424A |
ΔrnhB polA_D424A |
rnhB+ polA_ΔC |
ΔrnhB polA_ΔC |
|
|---|---|---|---|---|---|---|
| 1532 | AT→GC | 29 | 19 | 10 | 9 | |
| 1534 | AT→GC | 72 | 96 | 1 | 32 | |
|
|
1535 | CG→AT | 1 | |||
| 1535 | CG→TA | 10 | 2 | 2 | ||
| 1537 | CG→AT | 1 | ||||
|
|
1538 | AT→TA | 1 | 1 | ||
| 1538 | AT→GC | 5 | 6 | 15 | ||
|
|
1546 | CG→AT | 3 | 1 | ||
| 1546 | CG→TA | 18 | 64 | 34 | 30 | |
|
|
1547 | AT→TA | 1 | 9 | 23 | |
| 1547 | AT→GC | 90 | 10 | 242 | 32 | |
| 1552 | AT→GC | 2 | 2 | 1 | 1 | |
| 1565 | CG→TA | 1 | ||||
| 1575 | CG→GC | 1 | ||||
|
|
1576 | CG→AT | 13 | 6 | 19 | |
| 1576 | CG→TA | 5 | 11 | 91 | ||
| 1577 | AT→TA | 9 | 6 | |||
| 1586 | CG→TA | 1 | ||||
|
|
1592 | CG→AT | 3 | |||
| 1592 | CG→TA | 4 | 2 | 6 | ||
| 1593 | CG→TA | 1 | ||||
|
|
1598 | AT→TA | 1 | |||
| 1598 | AT→GC | 3 | 6 | 2 | ||
| 1600 | CG→TA | 2 | ||||
| 1611 | CG→TA | 1 | ||||
| 1691 | CG→TA | 5 | 6 | |||
| 1695 | AT→GC | 1 | ||||
| 1708 | CG→TA | 1 | ||||
| 1711 | CG→TA | 1 | ||||
| 1714 | AT→TA | 25 | 34 | 6 | ||
|
| ||||||
| Transitions | 243 (86%) | 221 (77%) | 290 (99%) | 229 (80%) | ||
| Transversions | 40 (14%) | 67 (23%) | 1 (0.3%) | 57 (20%) | ||
| Total | 283 | 288 | 291 | 286 | ||
: The data are the number of mutants found for each type of base substitution at a particular position
: The numbering system originates from Garibyan et al., [32], where the A of the ATG initiation codon is #1.
Furthermore, we believe that because the extent of mutagenesis observed during RER depends upon the polA allele of the strain (increasing ~2-fold in a proofreading-deficient strain and decreasing ~4-fold in a polymerase-deficient strain) it indicates that pol I is the preferred DNA polymerase to facilitate RER in E. coli.
3.8 In the absence of pol I, pol III participates in ribonucleotide excision repair in vivo
Only two DNA polymerases (pol II and pol III) remain in the recA730 lexA(Def) polA_ΔC ΔdinB ΔumuDC ΔmutL strain. To determine which of the two high fidelity polymerases might back up pol I in RER, we constructed a set of isogenic recA730 lexA(Def) polA_ΔC ΔdinB ΔumuDC ΔmutL strains in which the 3′→5′ exonuclease activity of pol II (polBex1), or pol III (mutD5), was inactivated and assayed for spontaneous His+ mutagenesis (Fig. 5). Because these strains lack three of E. coli’s five DNA polymerases and exhibit a strong mutator activity caused by defects in mismatch repair and pol II, or pol III-dependent proofreading, they grew poorly and rapidly acquired mutations. As a result, polBex1 and mutD5 strains were unable to form colonies on the standard minimal “low histidine” plates normally used for the His+ reversion assay. This problem was circumvented by substituting the 1μg/ml histidine with 4μg/ml casamino acids, which not only provides the low levels of histidine necessary for the reversion assay, but also all other essential amino acids sufficient for cell viability on the minimal agar plates. Under these assay conditions, one can clearly observe the mutator activity of the polBex1 and mutD5 alleles by the dramatic increase in the number of polV-independent His+ revertants observed in the mutation assays with the strains containing the control vector, pGB2 (Table in Fig. 5).
The data presented in Fig. 5 support the hypothesis that pol I is the polymerase of choice for RER in E. coli, since no umuC_Y11A mutator effect was observed for either the polBexI or mutD5 exonuclease-deficient alleles in a wild-type polA+ background (relative mutagenesis is ~30% in all these strains). As observed earlier on the low histidine plates (Figs. 2 & 3), the polA_ΔC strain exhibited low levels of umuC_Y11A mutagenesis on the casamino acids plates (Fig. 5) consistent with the hypothesis that a higher fidelity polymerase assumes the DNA synthesis step of RER in the absence of pol I. Interestingly, mutagenesis increased from just ~2% in the polA_ΔC polB+ strain to ~17% in the isogenic polBex1 strain. Even more striking was an increase in umuC_Y11A-dependent mutagenesis in the polA_ΔC mutD5 strain (~63%). We consider such observations as evidence that high-fidelity pol III substitutes for pol I in RER in polA_ΔC strains. The statistically significant (p<0.0001) increase in mutagenesis observed in the polBex1 strain may also be interpreted as an indication of a potential back up role for pol II in RER, but is more likely explained by the fact that pol II is an extrinsic proofreader of pol III errors [48].
4. Discussion
pol V is best characterized by its ability to facilitate translesion DNA synthesis. However, in certain genetic backgrounds, it has been shown to contribute to high levels of spontaneous mutagenesis on the undamaged E. coli chromosome [36, 37]. We have previously taken advantage of this phenotype, along with a steric gate mutant of pol V that readily incorporates ribonucleotides into the chromosome, to investigate the mechanisms of ribonucleotide repair in E. coli [5, 12]. In the present manuscript, we have used the same genetic system to study the potential role of DNA polymerase I in RER. Pol I was chosen as the main focus of the study based upon its previously characterized enzymatic properties that enable it to participate in “nick-translation” [13, 14, 49]. Using model in vitro substrates, we demonstrated that pol I can initiate strand displacement synthesis at a nick with a single 5′ terminal ribonucleotide which is removed during repair synthesis by the 5′→3′ exonuclease or 5′-FLAP endonuclease activities of pol I (Figs. 1 & 6).
Fig. 6.
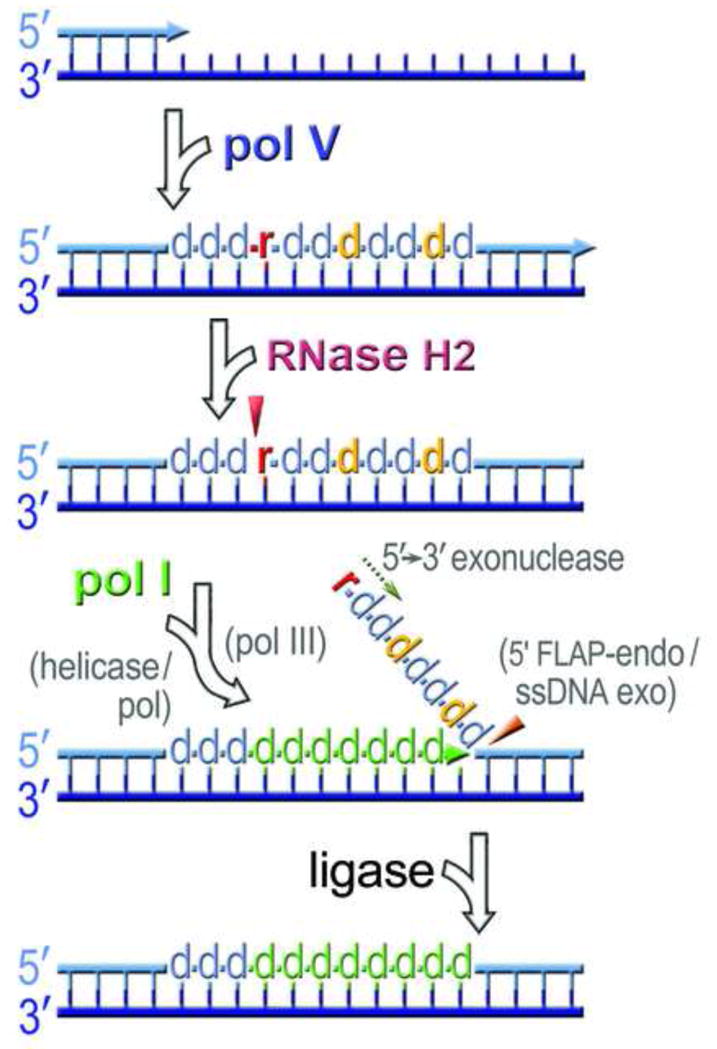
A model for ribonucleotide excision repair in E. coli. Pol V introduces multiple base substitutions (shown as d in orange) when it gains access to undamaged chromosomal DNA. It can also incorporate ribonucleotides (shown as r in red) into the nascent DNA strand. RNase HII incises the bond at the rNMP/dNMP junction 5′ to the ribonucleotide, generating DNA containing a single stranded break with 5′-phospho-ribonucleotide and 3′-hydroxyl ends. Pol I commences DNA synthesis at the nick and promotes strand displacement of the mutagenic pol V tract (shown as d in green). In polA mutants that are unable to promote strand displacement, the unwinding of the nicked intermediate proceeds through the action of an unknown DNA helicase, or polymerase (shown in parentheses). In polA_ΔC strains deficient for DNA synthesis, the 3′-hydroxyl end of the nicked intermediate is extended by high-fidelity pol III (shown in parentheses). The displaced region containing rNMP and misincorporated dNMPs is usually degraded by the 5′→3′ exonuclease or 5′-FLAP endonuclease activity of pol I, but in its absence, the displaced strand may be degraded by the FLAP endonuclease Xni, or another (yet to be identified) single-stranded nuclease (shown in parentheses). Complete repair occurs once the nick is sealed by DNA ligase. Although this model is based upon our present studies with the error-prone umuC_Y11A steric gate mutant of pol V, we envisage that identical processes occur when high-fidelity DNA polymerases inadvertently incorporate ribonucleotides into genomic DNA.
In E. coli strains proficient in RNase H functions, 5′ nicks are generated at sites of errantly incorporated ribonucleotides in vivo. The subsequent step in the repair pathway is initiation of DNA synthesis at the nick and strand-displacement of the ribo-containing DNA downstream from the nick (Fig. 6). We provide several compelling lines of evidence that pol I normally facilitates this step in vivo. First, a polA mutant deficient in 3′→5′ exonucleolytic activity exhibited significantly (p=0.02) higher levels of umuC_Y11A-dependent mutagenesis compared to the isogenic polA+ strain (Fig. 2). We theorize that the increased mutagenesis is caused by error-prone pol I-dependent synthesis during RER of umuC_Y11A incorporated ribonucleotides (Fig. 5). Secondly, in a polA_ΔC strain deficient for pol I DNA synthesis, umuC_Y11A-dependent mutagenesis decreased significantly (p<0.02; Figs. 2, 3 & 5). We believe that this observation can be explained by the fact that in its absence, another higher fidelity polymerase substitutes for pol I in RER. The observed changes in mutagenesis clearly reflect RER, as they are negated when the pathway is blocked by a ΔrnhB allele (Fig. 3) and changes in the spectrum of base substitutions are observed in the rpoB gene in the respective strains (Fig. 4).
Our studies with the mutD5 allele of dnaQ and polBex1 allele of polB provide evidence that it is the cell’s replicase, pol III, that most likely undertakes the role of pol I in its absence (Fig. 5). Importantly, they also support the model in which pol I is the primary DNA polymerase of choice to facilitate RER, as there is no effect of either the mutD5 allele or the polBex1 allele on RER in a polA+ strain.
We observed no significant phenotype of a polA mutant unable to facilitate strand-displacement, but assume that such activity could readily be performed by pol III, or through the actions of a yet to be determined DNA helicase. Similarly, there was minimal effect of the 5′→3′ exo/endonuclease-deficient polA107 mutant, even in the absence of another FLAP endonuclease, ExoIX. However, removal of the 5′-FLAP occurs late in the RER process (immediately prior to ligation), and is simply needed to remove regions of single-stranded DNA generated during strand displacement. This step could potentially be facilitated by any number of E. coli’s single-stranded exonucleases (Fig. 6).
To elucidate the mechanism of RER in E. coli, we have utilized a model system in which a steric gate mutant of pol V with a propensity to incorporate ribonucleotides into DNA has been provided optimal conditions in vivo, so as to ensure access to undamaged DNA, where it acts as a molecular “hypodermic needle” to “inject” errant ribonucleotides into the E. coli genome. Such an approach has recently allowed us to uncover the mechanisms of ribonucleotide repair that were hitherto unknown ([5, 12] and herein). There is no reason to suspect that the mechanisms of RER uncovered utilizing the umuC_Y11A mutant are limited to repair of ribonucleotides specifically incorporated by pol V and given the fact that it has recently been suggested that even high fidelity replicases insert ribonucleotides much more frequently than previously thought [50, 51], we believe that RNase HII-pol I-dependent RER is a global repair pathway that serves as a guardian of the E. coli chromosome and protecting it from the potentially harmful consequences of frequent ribonucleotide incorporation during genome duplication.
Supplementary Material
Mutation Research Highlights.
We have investigated the mechanism of ribonucleotide excision repair (RER) in E. coli.
DNA polymerase I is the primary polymerase of choice for the re-synthesis step of RER.
Back-up enzymes are able to substitute for pol I in the RER pathway in vivo.
Acknowledgments
This work was funded in part by the NICHD/NIH Intramural Research Program to RW, and from the National Institutes of Health (GM21422, ES012259) to MFG. We thank Iwona Fijalkowska for kindly providing the pSKpolAint plasmid vector and the polA 3′→5′ exo− CmR strain, KA796; Martin Marinus for the mutL460::cat strain, KM52; Roel Schaaper for the mutD5 strain, NR9548; and Cathy Joyce for polyclonal rabbit antibodies raised against E. coli pol I.
Abbreviations
- pol
DNA polymerase
- nt
nucleotide
- RER
ribonucleotide excision repair
- Top1
topoisomerase 1
Footnotes
Conflict of interest statement
The authors declare that there is no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol. 2012;13 doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaisman A, Kuban W, McDonald JP, Karata K, Yang W, Goodman MF, Woodgate R. Critical amino acids in Escherichia coli responsible for sugar discrimination and base-substitution fidelity. Nucleic Acids Res. 2012;40:6144–6157. doi: 10.1093/nar/gks233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown JA, Suo Z. Unlocking the sugar “steric gate” of DNA polymerases. Biochemistry. 2011;50:1135–1142. doi: 10.1021/bi101915z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuban W, Vaisman A, McDonald JP, Karata K, Yang W, Goodman MF, Woodgate R. Escherichia coli UmuC active site mutants: effects on translesion DNA synthesis, mutagenesis and cell survival. DNA Repair. 2012;11:726–732. doi: 10.1016/j.dnarep.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDonald JP, Vaisman A, Kuban W, Goodman MF, Woodgate R. Mechanisms employed by Escherichia coli to prevent ribonucleotide incorporation into genomic DNA by pol V. PLoS Genet. 2012;8:e1003030. doi: 10.1371/journal.pgen.1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turchi JJ, Huang L, Murante RS, Kim Y, Bambara RA. Enzymatic completion of mammalian lagging-strand DNA replication. Proc Natl Acad Sci USA. 1994;91:9803–9807. doi: 10.1073/pnas.91.21.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goulian M, Richards SH, Heard CJ, Bigsby BM. Discontinuous DNA synthesis by purified mammalian proteins. J Biol Chem. 1990;265:18461–18471. [PubMed] [Google Scholar]
- 8.Rydberg B, Game J. Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN-1 in cell-free extracts. Proc Natl Acad Sci USA. 2002;99:16654–16659. doi: 10.1073/pnas.262591699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM. RNase H2-Initiated Ribonucleotide Excision Repair. Mol Cell. 2012;47:980–986. doi: 10.1016/j.molcel.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lazzaro F, Novarina D, Amara F, Watt DL, Stone JE, Costanzo V, Burgers PM, Kunkel TA, Plevani P, Muzi-Falconi M. RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol Cell. 2012;45:99–110. doi: 10.1016/j.molcel.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim N, Huang SN, Williams JS, Li YC, Clark AB, Cho JE, Kunkel TA, Pommier Y, Jinks-Robertson S. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science. 2011;332:1561–1564. doi: 10.1126/science.1205016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaisman A, McDonald JP, Huston D, Kuban W, Lu L, Van Houten B, Woodgate R. Removal of misincorporated ribonucleotides from prokaryotic genomes: an unexpected role for nucelotide excision repair. PLoS Genet. 2013:e1003878. doi: 10.1371/journal.pgen.1003878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kornberg A. DNA Replication. Freeman; New York: 1980. [Google Scholar]
- 14.Westergaard O, Brutlag D, Kornberg A. Initiation of deoxyribonucleic acid synthesis. IV. Incorporation of the ribonucleic acid primer into the phage replicative form. J Biol Chem. 1973;248:1361–1364. [PubMed] [Google Scholar]
- 15.Derbyshire V, Grindley ND, Joyce CM. The 3′-5′ exonuclease of DNA polymerase I of Escherichia coli: contribution of each amino acid at the active site to the reaction. EMBO J. 1991;10:17–24. doi: 10.1002/j.1460-2075.1991.tb07916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polesky AH, Steitz TA, Grindley ND, Joyce CM. Identification of residues critical for the polymerase activity of the Klenow fragment of DNA polymerase I from Escherichia coli. J Biol Chem. 1990;265:14579–14591. [PubMed] [Google Scholar]
- 17.Lundquist RC, Olivera BM. Transient generation of displaced single-stranded DNA during nick translation. Cell. 1982;31:53–60. doi: 10.1016/0092-8674(82)90404-4. [DOI] [PubMed] [Google Scholar]
- 18.Lyamichev V, Brow MA, Dahlberg JE. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science. 1993;260:778–783. doi: 10.1126/science.7683443. [DOI] [PubMed] [Google Scholar]
- 19.Joyce CM, Grindley NDF. Method for determining whether a gene of Escherichia coli is essential: application to the polA gene. J Bacteriol. 1984;158:636–643. doi: 10.1128/jb.158.2.636-643.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Derbyshire V, Ng K, Sun XC, Grindley ND, Joyce CM. Biochemical and mutational studies of the 5′-3′ exonuclease of DNA polymerase I of Escherichia coli. J Mol Biol. 1997;268:284–302. doi: 10.1006/jmbi.1997.0967. [DOI] [PubMed] [Google Scholar]
- 21.Okazaki R, Arisawa M, Sugino A. Slow joining of newly replicated DNA chains in DNA polymerase I-deficient Escherichia coli mutants. Proc Natl Acad Sci USA. 1971;68:2954–2957. doi: 10.1073/pnas.68.12.2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makiela-Dzbenska K, Jaszczur M, Banach-Orlowska M, Jonczyk P, Schaaper RM, Fijalkowska IJ. Role of Escherichia coli DNA polymerase I in chromosomal DNA replication fidelity. Mol Microbiol. 2009;74:1114–1127. doi: 10.1111/j.1365-2958.2009.06921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller JH. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1992. [Google Scholar]
- 24.Joyce CM, Derbyshire V. Purification of Escherichia coli DNA polymerase I and Klenow fragment. Methods Enzymol. 1995;262:3–13. doi: 10.1016/0076-6879(95)62003-6. [DOI] [PubMed] [Google Scholar]
- 25.Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 26.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 27.Singh K, Srivastava A, Patel SS, Modak MJ. Participation of the fingers subdomain of Escherichia coli DNA polymerase I in the strand displacement synthesis of DNA. J Biol Chem. 2007;282:10594–10604. doi: 10.1074/jbc.M611242200. [DOI] [PubMed] [Google Scholar]
- 28.Davis BD, Mingioli ES. Mutants of Escherichia coli requiring methionine or vitamin B12. J Bacteriol. 1950;60:17–28. doi: 10.1128/jb.60.1.17-28.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green MHL, Muriel WJ. Mutagen testing using TRP+ reversion in Escherichia coli. Mutat Res. 1976;38:3–32. doi: 10.1016/0165-1161(76)90076-5. [DOI] [PubMed] [Google Scholar]
- 30.Kada T, Brun E, Marcovich H. Comparison of the induction of prototrophic mutants by x-rays and ultraviolet rays in “Escherichia coli” B/r Try. Ann Inst Pasteur (Paris) 1960;99:547–566. [PubMed] [Google Scholar]
- 31.Maron DM, Ames BN. Revised methods for the Salmonella mutagenicity test. Mutat Res. 1983;113:173–215. doi: 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- 32.Garibyan L, Huang T, Kim M, Wolff E, Nguyen A, Nguyen T, Diep A, Hu K, Iverson A, Yang H, Miller JH. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair. 2003;2:593–608. doi: 10.1016/s1568-7864(03)00024-7. [DOI] [PubMed] [Google Scholar]
- 33.Wolff E, Kim M, Hu K, Yang H, Miller JH. Polymerases leave fingerprints: analysis of the mutational spectrum in Escherichia coli rpoB to assess the role of polymerase IV in spontaneous mutation. J Bacteriol. 2004;186:2900–2905. doi: 10.1128/JB.186.9.2900-2905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curti E, McDonald JP, Mead S, Woodgate R. DNA polymerase switching: effects on spontaneous mutagenesis in Escherichia coli. Mol Microbiol. 2009;71:315–331. doi: 10.1111/j.1365-2958.2008.06526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woodgate R, Ennis DG. Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol Gen Genet. 1991;229:10–16. doi: 10.1007/BF00264207. [DOI] [PubMed] [Google Scholar]
- 36.Sweasy JB, Witkin EM, Sinha N, Roegner-Maniscalco V. RecA protein of Escherichia coli has a third essential role in SOS mutator activity. J Bacteriol. 1990;172:3030–3036. doi: 10.1128/jb.172.6.3030-3036.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fijalkowska IJ, Dunn RL, Schaaper RM. Genetic requirements and mutational specificity of the Escherichia coli SOS mutator activity. J Bacteriol. 1997;179:7435–7445. doi: 10.1128/jb.179.23.7435-7445.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vlaši I, Šimatovi A, Kosti KBri. Genetic requirements for high constitutive SOS expression in recA730 mutants of Escherichia coli. J Bacteriol. 2011;193:4643–4651. doi: 10.1128/JB.00368-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fijalkowska I, Jonczyk P, Ciesla Z. Conditional lethality of the recA441 and recA730 mutants of Escherichia coli deficient in DNA polymerase I. Mutat Res. 1989;217:117–122. doi: 10.1016/0921-8777(89)90063-3. [DOI] [PubMed] [Google Scholar]
- 40.Glickman BW, van Sluis CA, Heijneker HL, Rorsch A. A mutant of Escherichia coli K12 deficient in the 5′-3′ exonucleolytic activity of DNA polymerase I. I. General characterization. Mol Gen Genet. 1973;124:69–82. doi: 10.1007/BF00267166. [DOI] [PubMed] [Google Scholar]
- 41.Joyce CM, Fujii DM, Laks HS, Hughes CM, Grindley ND. Genetic mapping and DNA sequence analysis of mutations in the polA gene of Escherichia coli. J Mol Biol. 1985;186:283–293. doi: 10.1016/0022-2836(85)90105-6. [DOI] [PubMed] [Google Scholar]
- 42.Heijneker HL, Ellens DJ, Tjeerde RH, Glickman BW, van Dorp B, Pouwels PH. A mutant of Escherichia coli K12 deficient in the 5′-3′ exonucleolytic activity of DNA polymerase I. II. Purification and properties of the mutant enzyme. Mol Gen Genet. 1973;124:83–96. doi: 10.1007/BF00267167. [DOI] [PubMed] [Google Scholar]
- 43.Allen LM, Hodskinson MR, Sayers JR. Active site substitutions delineate distinct classes of eubacterial flap endonuclease. Biochem J. 2009;418:285–292. doi: 10.1042/BJ20081637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayers JR. Computer aided identification of a potential 5′-3′ exonuclease gene encoded by Escherichia coli. J Theoret Biol. 1994;170:415–421. doi: 10.1006/jtbi.1994.1202. [DOI] [PubMed] [Google Scholar]
- 45.Fukushima S, Itaya M, Kato H, Ogasawara N, Yoshikawa H. Reassessment of the in vivo functions of DNA polymerase I and RNase H in bacterial cell growth. J Bacteriol. 2007;189:8575–8583. doi: 10.1128/JB.00653-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bebenek K, Joyce CM, Fitzgerald MP, Kunkel TA. The fidelity of DNA synthesis catalyzed by derivatives of Escherichia coli DNA polymerase I. J Biol Chem. 1990;265:13878–13887. [PubMed] [Google Scholar]
- 47.Makiela-Dzbenska K, Jonczyk P, Schaaper RM, Fijalkowska IJ. Proofreading deficiency of Pol I increases the levels of spontaneous rpoB mutations in E. coli. Mutat Res. 2011;712:28–32. doi: 10.1016/j.mrfmmm.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banach-Orlowska M, Fijalkowska IJ, Schaaper RM, Jonczyk P. DNA polymerase II as a fidelity factor in chromosomal DNA synthesis in Escherichia coli. Mol Microbiol. 2005;58:61–70. doi: 10.1111/j.1365-2958.2005.04805.x. [DOI] [PubMed] [Google Scholar]
- 49.Hartman CP, Rabussay D. E. coli DNA polymerase I: enzymatic functions and their application in polymer formation, nick translation and DNA sequencing. Gene Amplif Anal. 1981;2:17–39. [PubMed] [Google Scholar]
- 50.Nick McElhinny SA, Watts BE, Kumar D, Watt DL, Lundstrom EB, Burgers PM, Johansson E, Chabes A, Kunkel TA. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc Natl Acad Sci USA. 2010;107:4949–4954. doi: 10.1073/pnas.0914857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, Devenney PS, Sexton D, Grimes G, Holt IJ, Hill RE, Taylor MS, Lawson KA, Dorin JR, Jackson AP. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149:1008–1022. doi: 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watanabe-Akanuma M, Woodgate R, Ohta T. Enhanced generation of A:T->T:A transversions in a recA730 lexA51(Def) mutant of Escherichia coli. Mutat Res. 1997;373:61–66. doi: 10.1016/s0027-5107(96)00189-3. [DOI] [PubMed] [Google Scholar]
- 53.Schaaper RM, Cornacchio R. An Escherichia coli dnaE mutation with suppressor activity toward mutator mutD5. J Bacteriol. 1992;174:1974–1982. doi: 10.1128/jb.174.6.1974-1982.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.