

Abstract
Large-scale transcriptome profiling in clinical studies often involves assaying multiple samples of a patient to monitor disease progression, treatment effect, and host response in multiple tissues. Such profiling is prone to human error, which often results in mislabeled samples. Here, we present a method to detect mislabeled sample outliers using coding single nucleotide polymorphisms (cSNPs) specifically designed on the microarray and demonstrate that the mislabeled samples can be efficiently identified by either simple clustering of allele-specific expression scores or Mahalanobis distance-based outlier detection method. Based on our results, we recommend the incorporation of cSNPs into future transcriptome array designs as intrinsic markers for sample tracking.
Keywords: microarray, transcriptome profiling, coding SNP, outlier detection, sample tracking
Large-scale transcriptome profiling studies of human health and diseases often involve hybridization of multiple samples from the same patient (1–3). For each sample, a series of experimental steps are performed to generate expression data, including sample collection, cell isolation, total RNA extraction, library preparation, chip hybridization, and scanning. The possibility of mislabeling accumulates at each step and is further increased when multiple lab personnel and/or centers are involved. Standard operating procedures (SOPs) reduce such errors, but do not completely eliminate them (4). For example, from our experience with large-scale clinical studies of inflammation and host response to injury (www.gluegrant.org), which generates approximately 1500 arrays every year by one experienced research staff, the mislabeling rate is about 5% even after implementing SOPs. Such accumulated errors may degrade the power to detect gene signatures, or even cause misinterpretation of the etiology (5). It is therefore important to utilize features of the transcriptome to track samples from different subjects, as an internal measure to prevent inadvertent human errors.
One candidate is the coding single nucleotide polymorphism (cSNP). SNP is genetic variation commonly used in association studies to identify individuals (6). Previous studies demonstrated that an individual can typically be identified from global human samples by tens to hundreds of informative SNPs or haplotypes (7). When genes are transcribed, genetic information in cSNPs is passed into mRNAs, allowing samples from different subjects to be distinguishable. Furthermore, allele-specific expression, the imbalance between allele-specific transcripts, was shown to be widespread across human genome (8) and manifest cell type-dependent patterns (9,10).
Therefore, we hypothesized that cSNPs can be used as the intrinsic markers for sample tracking. Here we demonstrated this possibility using the glue grant human transcriptome (GG-H) array, which incorporated cSNPs into the array design: for each of the approximately 89,000 cSNPs in transcribed regions, six probes were designed for each allele at -4, 0, and +4 positions relative to the SNP locus on both strands (11).
For the test experiment, we constructed a data set of 91 samples from five randomly selected subjects in a large-scale study of trauma patients. Each patient was sampled for three different cell types (T cell, monocyte, and neutrophil) and up to seven time points (12 h and day 1, 4, 7, 14, 21, and 28 post-injury). Patient enrollments, blood sampling, and microarray hybridization were described in References 11 and 12.
Using raw CEL files, we quantified an allelic imbalance score (AIS) of each cSNP as follows:
where A and a denote the two alleles, and is the signal intensity of the probe at position i = {-4, 0, +4} of strand j = {‘+’, ‘-’} of allele A. The AIS captures the genotype of a homozygous cSNP or the allele-specific imbalance of a heterozygous cSNP.
First, we tested if mislabeled samples can be detected as outliers. Using 500 cSNPs with the most variable AIS, we performed hierarchical clustering using dChip (13). The clustering pattern clearly showed that all samples are clustered well by subjects, except S89, S90, and S91 (Figure 1, highlighted in orange). Their distinct patterns clearly separate them from the rest, suggesting that these samples might belong to other subjects. This is further verified by reprocessing the three initial samples and showing that their redos (S89_2, S90_2, and S91_2, highlighted in green) cluster correctly with other samples from the same subjects. In addition, each of the outliers was also detected to have significant Mahalanobis distance (14) (calculated by R function mahalanobis) from the center of its mislabeled subject (χ2 test P < 0.05), while none of the redos was significant (P > 0.1). Overall, this analysis showed that the AIS pattern can identify mislabeled samples.
Figure 1. Hierarchical clustering of the AIS scores of the top 500 most variable cSNPs.
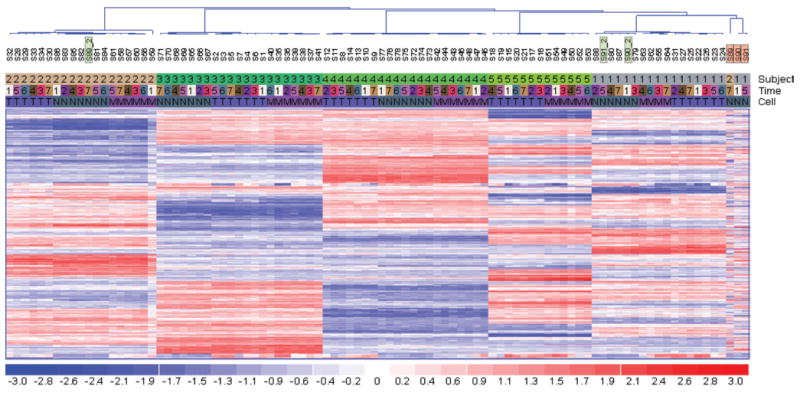
Samples are well clustered by subjects (subject color bar: 1–5 denotes five patients) and cell types (cell color bar: T, T cell; M, monocyte; N, neutrophil), but not time points (time color bar: 1–7 denotes 12 h, day 1, 4, 7, 14, 21, and 28). Three mislabeled samples highlighted in orange (S89, S90, S91) are not clustered with the remaining samples of the same patients, while their redos highlighted in green (S89_2, S90_2, S91_2) are clustered correctly.
Next, we evaluated the number of cSNPs required for outlier detection. A full spectrum eigen-R2 analysis (15) revealed that subject identity is a dominant factor in the AIS variance across the whole range of the number of cSNPs used (Figure 2), with the weight ranging from 93% for 50 cSNPs, to 87% for 500 cSNPs, and 41% for 50,000 cSNPs. We then repeated the hierarchical clustering and Mahalanobis distance methods for the top 50 to 10,000 most variable cSNPs and obtained the same separation as using 500 cSNPs (data not shown), showing that subjects' separation by AIS pattern is a robust method over a broad range of cSNPs.
Figure 2. The percentage of variance explained by the three factors by eigen-R2 analysis (15).
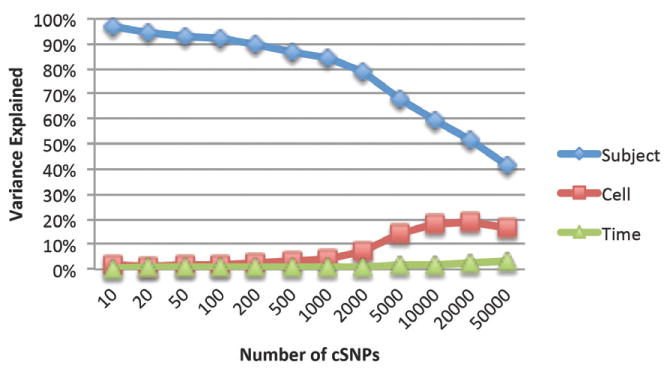
The x-axis indicates the number of cSNPs with most variable AIS included in the analysis.
In addition to the separation by subjects, samples can also be clustered by their cell types within each subject, as shown in Figure 1. However cell type accounts only for less than 4% variance for the top 500 cSNPs (Figure 2), indicating a much larger between-subject variance than the within-subject variance. Further studies are necessary to investigate the extent of potential cell-dependent allele-specific expression (9).
Here we present an approach to select a set of informative cSNPs based on their variance of AIS between individual samples in a given clinical study. Although the selected cSNPs likely include redundant information, this approach is simple to implement without the requirement of additional information of patient genotypes. In addition, since the identities of the cSNPs are not used in this analysis, a randomization can be performed at the array design step to “de-identify” the cSNPs to simplify procedures in patient recruitment and consent. Therefore, we implemented this approach in the simple clustering method with Mahalanobis distance-based outlier detection for sample tracking in our glue grant study.
In terms of whether a different set of cSNPs needs to be used for each project or whether a minimal and sufficient set of cSNPs can be derived that works for any project, we recommend a data-driven approach as described in the paper to identify a set of informative cSNPs for each study and use them for sample tracking in that study. According to Pakstis et al. (7), using DNA samples, ∼100 SNPs should suffice to identify an individual from a set of global samples. However, the detection of sample outliers in mRNA profiling would require the genes of the cSNPs to be expressed in these samples. Since the expression of the vast majority of genes is tissue-specific, the minimal and sufficient set of cSNPs for sample tracking is most likely to be tissue-specific as well. Another potential consideration with typing a predetermined set of cSNPs is the requirement of additional patient's consent.
The current approach is applicable where more than one sample is obtained from each patient. However, if the cSNP genotypes of the patients are available, it will allow the preselection of an optimum set of informative cSNPs to distinguish individuals of the study for the outlier detections, likely without the requirement of multiple samples per patient. The increasing information of personal genomes or exomes provides the possibility in future studies of direct sample tracking using cSNPs as intrinsic markers, as long as the proper patient consent can be acquired.
Acknowledgments
Data presented in this work were generated by the Inflammation and Host Response to Injury Large-Scale Collaborative Research Program. This study was supported by grants from National Institutes of Health (PO1-HG00205, U54-GM62119).
Footnotes
Competing interests: The authors declare no competing interests.
References
- 1.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, et al. A genomic storm in critically injured humans. J Exp Med. 2011;208:2581–2590. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein MB, Silver G, Gamelli RL, Gibran NS, Herndon DN, Hunt JL, Tompkins RG. Inflammation and the host response to injury: an overview of the multicenter study of the genomic and proteomic response to burn injury. J Burn Care Res. 2006;27:448–451. doi: 10.1097/01.BCR.0000227477.33877.E6. [DOI] [PubMed] [Google Scholar]
- 5.Zhang C, Wu C, Blanzieri E, Zhou Y, Wang Y, Du W, Liang Y. Methods for labeling error detection in microarrays based on the effect of data perturbation on the regression model. Bioinformatics. 2009;25:2708–2714. doi: 10.1093/bioinformatics/btp478. [DOI] [PubMed] [Google Scholar]
- 6.Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 7.Pakstis AJ, Speed WC, Fang R, Hyland FC, Furtado MR, Kidd JR, Kidd KK. SNPs for a universal individual identification panel. Hum Genet. 2010;127:315–324. doi: 10.1007/s00439-009-0771-1. [DOI] [PubMed] [Google Scholar]
- 8.Ge B, Pokholok DK, Kwan T, Grundberg E, Morcos L, Verlaan DJ, Le J, Koka V, et al. Global patterns of cis variation in human cells revealed by high-density allelic expression analysis. Nat Genet. 2009;41:1216–1222. doi: 10.1038/ng.473. [DOI] [PubMed] [Google Scholar]
- 9.Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW. Allelic variation in human gene expression. Science. 2002;297:1143. doi: 10.1126/science.1072545. [DOI] [PubMed] [Google Scholar]
- 11.Xu W, Seok J, Mindrinos MN, Schweitzer AC, Jiang H, Wilhelmy J, Clark TA, Kapur K, et al. Human transcriptome array for high-throughput clinical studies. Proc. Natl Acad Sci USA. 2011;108:3707–3712. doi: 10.1073/pnas.1019753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotz KT, Xiao W, Miller-Graziano C, Qian WJ, Russom A, Warner EA, Moldawer LL, De A, et al. Clinical microfluidics for neutrophil genomics and proteomics. Nat Med. 2010;16:1042–1047. doi: 10.1038/nm.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gnanadesikan R, Kettenring JR. Robust estimates, residuals, and outlier detection with multiresponse data. Biometrics. 1972;28:81–124. [Google Scholar]
- 15.Chen LS, Storey JD. Eigen-R2 for dissecting variation in high-dimensional studies. Bioinformatics. 2008;24:2260–2262. doi: 10.1093/bioinformatics/btn411. [DOI] [PMC free article] [PubMed] [Google Scholar]