

Abstract
Objectives
We examined the role of brain-derived neurotrophic factor (BDNF) in the pathogenesis of pain in an experimental model of chronic pancreatitis (CP).
Methods
Pancreatitis was induced by retrograde infusion of trinitrobenzene sulfonic acid into the pancreatic duct of adult rats. Twenty-one days after injection, BDNF expression was examined in pancreas-specific dorsal root ganglia (DRGs) by immunohistochemistry, and protein levels were quantified from DRGs and spinal cord extracts. The effects of intrathecal infusion of a neutralizing antibody to BDNF on pancreatic hyperalgesia were assessed by the sensitivity of the abdominal wall to filament probing as well as the nocifensive behavior to electrical stimulation of the pancreas.
Results
Levels of BDNF in DRGs and spinal cords (T9-13) were significantly higher in trinitrobenzene sulfonic acid rats compared with controls, accompanied by an increase in the number of pancreas-specific neurons expressing BDNF immunoreactivity. Brain-derived neurotrophic factor antagonism suppressed phospho–tropomyosin-related kinase B receptor levels in the spinal cord and significantly reduced behavioral responses in rats with CP.
Conclusions
Brain-derived neurotrophic factor is upregulated in pancreas-specific primary afferent neurons in rats with CP, and BDNF antagonism is associated with a reduction of pain-related behavior in these animals, suggesting an important role for this neurotransmitter in the nociception of CP.
Keywords: chronic pancreatitis, visceral pain, sensory neurons, BDNF, TrkB
Pain is a cardinal symptom in chronic pancreatitis (CP), present in approximately 75% of patients with alcoholic CP, 50% of patients with late-onset idiopathic CP, and 100% of patients with early-onset idiopathic CP.1 The management of pain in these patients is a major therapeutic challenge. Treatment is empirical and often highly invasive with long-term results that are often unsatisfactory. More rational and effective therapies will require the discovery of novel targets based on further understanding the neurobiology of pain in this condition.2,3
Ongoing tissue damage results in heightened responsiveness or sensitization of the nociceptive system, both peripherally and centrally. Pain signaling begins in the periphery with transduction of local injury into an electrical signal in primary afferent neurons or nociceptors. Once generated at the periphery, action potentials are conducted centripetally to the spinal terminals of the nociceptors, where they initiate neurotransmitter release and thereby relay nociceptive information to second-order neurons. The early response to relatively mild stimuli is transmitted via glutamate, but if the stimulus is sustained or intense enough, it causes release of peptide neurotransmitters such as substance P (SP), calcitonin gene–related peptide (CGRP), and brain-derived neurotrophic factor (BDNF) from nerve growth factor (NGF)–responsive small-diameter sensory neurons that serve to augment the response to glutamate and participate in other ways in nociceptive sensitization.
The development of valid rodent models has facilitated research in the pathogenesis of pancreatic pain. Thus, we have previously shown that classic neurotransmitters such as SP and CGRP and the transient receptor potential cation channel, subfamily V, member 1 (TRPV1) are upregulated in pancreatic sensory neurons in a rat model of CP.4,5 However, the role of other neurotransmitters and channels has yet to be carefully examined in this condition. Brain-derived neurotrophic factor is a powerful modulator of short-term and long-term synaptic efficiencies and is thought to exert its effect on the tropomyosin-related kinase B receptor (TrkB) expressed by second-order neurons in the spinal cord.6 Brain-derived neurotrophic factor has mainly been studied in somatic pain models and remains incompletely understood and somewhat contradictory with the neurotransmitter, playing a pronociceptive role in most inflammatory states and an antinociceptive role in neuropathic models.6 Pancreatic BDNF expression has also been noted in specimens from patients with CP and found to correlate with pain scores.7 The overall aim of this study was therefore to examine the role of BDNF in the pathogenesis of pain in an experimental model of CP. Our results prove that BDNF is upregulated in pancreas-specific primary afferent neurons in rats with CP and that BDNF antagonism is associated with a reduction of pain-related behavior in these animals, suggesting an important role for this neurotransmitter in the nociception of CP.
MATERIALS AND METHODS
Induction of CP, Cell Labeling, and Implantation of Electrodes
Chronic pancreatitis was induced in adult, male Sprague-Dawley rats by retrograde infusion of 0.5 mL of 1% trinitrobenzene sulfonic acid (TNBS) in 10% ethanol in PBS (pH 8.0) into the pancreatic duct. The common bile duct was temporarily occluded with a clamp to avoid TNBS entry into the liver before a 30-gauge needle connected to the PE-10 tubing was inserted into the duodenum and guided through the papilla into the pancreatic duct to allow for the slow injection of the TNBS/ saline solution as described previously.4 For experiments involving immunohistochemistry, rat pancreas was injected with the lipid-soluble fluorescence dye, DiI (1,1′-dioleyl-3,3,3′,3-tetramethylindocarbocyanine methanesulfonate; obtained in crystal form from Molecular Probes, Eugene, Ore), 25 mg in 0.5 mL of methanol, at 8 to 10 sites on the exposed pancreas in 2-μL volumes, before TNBS infusion. For experiments involving electrical stimulation testing, a pair of electrodes (Myo-Wires; A&E Medical, Farmingdale, NJ) was sutured into the pancreas soon after the TNBS infusion, and the open ends were subcutaneously tunneled and externalized at the dorsal neck region.
BDNF Enzyme-Linked Immunosorbent Assay
Three weeks after TNBS injection, animals were killed under ketamine/xylazine anesthesia, and cervical and thoracic dorsal root ganglia (DRGs; T9-13) were rapidly harvested. Corresponding cervical, thoracic, and lumbar spinal cord segments were also collected. The tissue was pulverized in liquid nitrogen using mortar and pestle and then thawed in buffer after which it was homogenized using a rotor-driven Teflon homogenizer. After a further incubation on ice for 30 minutes, it was centrifuged 14,000g 10 minutes at 4°C, and the supernatant was transferred to a fresh tube for enzyme-linked immunosorbent assay (ELISA) and protein determinations. The composition of the buffer was as follows: 20 mM Tris-HCl pH 7.4, 100 mM NaCl, 40 mM NaF, 1 mM EDTA, 1 mM EGTA, 1 mM Na orthovanadate, 25 mM β-glycerophosphate, 5 mM pyrophosphate, and 10% glycerol. Just before use, a protease inhibitor cocktail (1:100, P8340; Sigma, St Louis, Mo) was added, and then BDNF was quantified from the tissue extracts using an ELISA kit (Millipore, Billerica, Mass). Results were normalized to the total protein concentration and expressed as picograms of BDNF per microgram of total protein. Protein was measured by the BCA method (Pierce, Rockford, Ill).
Immunohistochemistry
Three weeks after TNBS injection, rats were anesthetized with ketamine/xylazine and transcardially perfused with 200 mL of 0.9% NaCl solution followed by 500 mL of cold 4% paraformaldehyde in PBS (pH 7.4). Pancreas-specific DRGs (T9-T13) were harvested and fixed overnight in 4% paraformaldehyde and then cryoprotected in 30% sucrose. Dorsal root ganglia were embedded in optimal cutting temperature medium, and 10-μm-thick frozen cryosections were prepared. To ensure that a neuron was counted only once, serial sections were placed on consecutive slides with at least 50 μm between sections on the same slide. Sections were blocked and permeabilized for 30 minutes in 5% normal goat serum with 0.3% Triton X-100 in PBS, then incubated overnight at 4°C in rabbit anti-BDNF primary antibody (Millipore; 1:100), rinsed in PBS, and finally incubated with a fluorescent goat antirabbit secondary antibody (Alexa Fluor 488; Invitrogen, Carlsbad, Calif; 1:300). Slides were then visualized on a Nikon Eclipse E600 (Nikon Instruments Inc, Melville, NY) microscope equipped with filter cubes appropriate for DiI (rhodamine filter) and appropriate band-pass and long-pass filter for Alexa 488 dye. Results were expressed as the percentage of pancreas-specific (DiI-labeled) neurons that were positively stained for BDNF.
Intrathecal Placement of Catheters and Subcutaneous Implantation of Osmotic Pumps for Continuous Delivery of Anti-BDNF
Three weeks after TNBS treatment, under ketamine/ xylazine anesthesia, an intrathecal catheter (6 cm long) was inserted through the atlanto-occipital membrane to spinal cord level T9-10. The tubing was secured to the muscle, and the Osmotic mini pump (Alzet model no. 2001; Durect Corporation, Cupertino, Calif) filled with 200 μL of 500 μg/mL BDNF-neutralizing antibody or, as control, an equal concentration of purified immunoglobulin, both from Innovative Research (Novi, Mich), was implanted subcutaneously and connected to the intrathecal tubing. The pump was designed to constantly deliver the antibody at 1 μL/h for 7 days after an initial activation period of 4 hours inside the animal.
Western Blotting
Immunoblotting for phosphorylated TrkB and β-tubulin were performed as follows. Fresh-frozen, rat spinal cord segments were homogenized on ice in RIPA buffer (150 mM NaCl, 50 mM Tris, pH 8, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) containing phosphatase and protease inhibitors (P5726 and P8340, respectively; Sigma), 2.5 mM phenylmethylsulfonylfluoride, 1 mM sodium orthovanadate, 5 mM EDTA, and 50 mM sodium fluoride. The homogenates were centrifuged at 14,000 rpm for 30 minutes, and the protein concentration in each sample was determined by the BCA assay (Thermo Scientific, Rockford, Ill). Fifty micrograms of total cellular proteins was resolved by 4% to 12% NuPage Bis-Tris gels (Invitrogen, Carlsbad, Calif) and then electroblotted onto Hybond polyvinylidene difluoride membranes (GE Healthcare, Piscataway, NJ). After blocking, membranes were probed with specific primary antibodies (phospho-TrkB [Ab81288; Abcam, Cambridge, Mass] or β-tubulin [Ab6046; Abcam]). The blots were processed by SuperSignal West Pico chemiluminescent Western blotting detection system (34080; Thermo Scientific) according to the manufacturer’s instruction. Densitometric readings of phospho-TrkB were normalized against the corresponding β-tubulin measurements, and the results were expressed as relative units to the mean of the values from rats without CP.
Von Frey Filament Testing
Von Frey filament (VFF) testing was performed as described previously.4 Before testing, the belly was shaved, and areas designated for stimulation were marked in relation to fore limbs and hind limbs. Rats were placed in a plastic cage with a mesh floor and were given 30 minutes to adapt before testing. Von Frey filaments (Stoelting, Wood Dale, Ill) of various caliber/ strengths were applied to the shaved belly of the animals in ascending order to the upper part of the midabdomen close to the rib cage 10 times each for 1 to 2 seconds with a 10-second interval between applications. A positive response consisted of lifting the belly and/or scratching and licking the abdomen. The data were expressed as number of responses during the 10 applications of the filaments. Once a level of 10 was reached in a given animal, further testing was not done, and for analysis purposes, it was assumed that higher filament strengths would also result in the same score. All tests were performed in a blinded manner.
Electrical Stimulation of the Pancreas and Measurement of Nocifensive Responses
After infusion of the pancreatic duct with normal saline or TNBS, a pair of electrodes was attached to the pancreas and externalized behind the head. The animals received successive applications of current at 2, 5, and 10 mA through the pair of electrodes connected to the electrical stimulator for 5 minutes with 20-minute rest between stimulations. The number of nocifensive behaviors were counted during each 5-minute stimulation period. These behaviors consisted of stretching, licking of the abdomen, contraction of abdominal wall muscles, and extension of the hind limbs, as previously described.4
Data Analysis
Data were presented as mean ± SEM. Behavior data from electrical stimulation were analyzed by repeated-measures analysis of variance (ANOVA) with treatment as the between-group factor and stimulation current (mA) as the within-group factor. Statistical differences between other data from control and TNBS-treated rats were determined by the Student t test, Fisher exact test, or χ2 test. P < 0.05 was considered statistically significant.
RESULTS
CP Enhances BDNF Protein Expression in DRG and Spinal Cord Segments Innervating the Pancreas
We determined whether TNBS-induced CP altered the protein expression of BDNF. Total proteins from pancreas-responsive DRG and spinal cord segments from control and TNBS-treated animals were extracted and analyzed for BDNF using an ELISA kit. As shown in Figure 1, the expression of BDNF proteins was significantly elevated approximately 1.4-fold and 1.7-fold in T9-13 segments of DRG and spinal cord, respectively (n = 6 rats in each group). By contrast, BDNF protein levels were not significantly different between control and CP in DRG and spinal cord samples from cervical or lumbar segments (results not shown). In parallel immunofluorescent studies, we also counted the number of DiI-labeled neurons expressing BDNF immunoreactivity in thoracic T9-13 segments (Fig. 1). Rats with CP had a higher proportion of DiI-labeled cells that stained positive for BDNF compared with controls (43.2% vs 50.9%, P < 0.01; n = 6 rats in each group).
FIGURE 1.

Chronic pancreatitis results in up-regulation of BDNF in sensory nerves. A, Cryosections of thoracic DRGs were fluorescently stained for BDNF presence. Representative picture of BDNF-positive neurons in a DRG section (top panel) and DiI-labeled pancreas-specific neurons (middle panel). The bottom panel shows a merged picture identifying both pancreas specific BDNF expressing neurons. Trinitrobenzene sulfonic acid–treated animals had a significantly higher percentage of BDNF-expressing pancreas-specific neurons compared to controls (50.9% vs 43.2%, P < 0.05). Scale bar, 50 μm. B, Levels of BDNF protein as measured by ELISA in thoracic T9-13 DRGs (top panel) and spinal cord (bottom panel) were also significantly higher in TNBS rats compared with controls (P < 0.006 vs P < 0.05, respectively).
BDNF Antagonism Suppresses TrkB Phosphorylation
One week after anti-BDNF treatment, phosphorylated TrkB levels in the thoracic spinal cord of rats with CP (n = 9) were similar to rats without CP (“normal,” n = 5; Fig. 2). By contrast, rats treated with control IgG (n = 10) had significantly higher levels than both normal rats and rats treated with anti-BDNF antibody (P = 0.03 by ANOVA).
FIGURE 2.
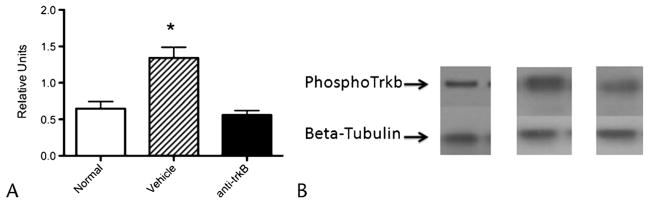
Phosphorylated TrkB levels in spinal cord are upregulated in CP and normalize with intrathecal anti-BDNF. A, Phosphorylated TrkB levels quantified by Western blot and expressed as a ratio of β-tubulin expression normalized to rats without pancreatitis.
B, Representative examples of the actual Western blots (normal= rats without CP).
BDNF Antagonism Diminishes Referred Somatic Sensitization in CP
We next determined whether pancreatic nocifensive behavior observed in TNBS-treated rats was mediated by BDNF by using a neutralizing antibody, as previously reported.8 We first measured the sensitivity of the abdomen to mechanical stimulation (an assay for referred somatic hyperalgesia, a characteristic of painful visceral conditions) before and after application of anti-BDNF or a control immunoglobulin in TNBS-treated rats. Overall, the response frequencies of rats (n = 6) treated with anti-BDNF 3 weeks after TNBS infusion were significantly lower compared with pretreatment baseline, with the stimulus-response curve shifting to the right (Fig. 3B; 2-way ANOVA: stimulus effect, P < 0.0001; treatment effect, P < 0.0001). In contrast, control rats (n = 7) treated with immunoglobulin showed an increase in the response frequency in rats 3 weeks after TNBS infusion (Fig. 3A; 2-way ANOVA: stimulus effect, P < 0.0001; treatment effect, P < 0.0001). Thus, BDNF antagonism produced a marked reduction in sensitivity to mechanical probing of the abdomen in TNBS-treated rats.
FIGURE 3.

Intrathecal neutralization of BDNF attenuates behavioral responses to noxious stimulation in CP. A, Von Frey filament response from before and after intrathecal infusion of control IgG in TNBS-treated rats. Plot of mean number of responses (of 10 applications per filament). These data show increase in responses after control IgG treatment (P < 0.001) in animals with CP. B, Von Frey filament response from before and after intrathecal infusion of anti-BDNF in TNBS-treated rats. Intrathecal infusion of anti-BDNF significantly reduced the response activity of animals with CP during VFF testing (P < 0.001). C, Response to graded electrical stimulation before and after intrathecal infusion of control IgG in TNBS-treated rats. Number of nocifensive responses during 5 minutes of electrical stimulation of pancreas before and after infusion of control IgG. Although a robust response was seen in electrical stimulation in all animals, no significant differences in response numbers were observed after infusion compared with experiments before infusion. D, Response to graded electrical stimulation from before and after intrathecal infusion of anti-BDNF in TNBS-treated rats. Number of nocifensive responses during 5 minutes of electrical stimulation of pancreas before and after infusion of anti-BDNF. Animals had significantly fewer responses to electrical stimulation after intrathecal infusion of anti-BDNF (P < 0.01).
BDNF Antagonism Diminishes Pancreatic Hyperalgesia in CP
Somatic responses to pressure on the upper abdominal wall are an indirect marker of visceral sensitization and do not necessarily implicate the pancreas. To test pancreatic sensitization directly, we have previously established a model using electrical stimulation of the organ.4 Although electrical stimulation is not a natural visceral stimulus, it has been shown to cause pain in both animals and humans; specifically, electrical stimulation of the pancreas produces pain in human subjects in a region-specific manner.9 Our results suggest that, overall, the response curve to graded electrical stimulation was significantly lower after intrathecal infusion of anti-BDNF in rats (n = 8) 3 weeks after TNBS treatment compared with pretreatment responses (Fig. 3D; 2-way ANOVA: stimulus effect, P < 0.01; treatment effect, P < 0.0001). This effect is most marked at 2 mA of stimulation and disappears at the highest level of stimulation used in this study (10 mA). On the contrary, control IgG infusion had no effect on pain behaviors at any level of stimulation in TNBS-treated rats (n = 9; Fig. 3C).
DISCUSSION
Sensitization of the nociceptive system is a hallmark of local injury or inflammation such as CP. Indeed, striking changes in the morphology of pancreatic nerves with associated foci of inflammatory cells have been noted in specimens from humans with CP.10–16 Given the inability to study the function of these nerves in humans, research in this area has been facilitated by the availability of a suitable animal model that mimics the human condition morphologically as well as with reproducible pain behavior in response to pancreas-specific noxious stimulation.4 In this study, we have used this model to study the role of the neurotransmitter, BDNF, in the pathogenesis of pain in CP.
Neuronal BDNF is coexpressed and colocalized with other neurotransmitters such as SP and CGRP in the same population of primary sensory neurons, although it may be released differentially, depending on the pattern of afferent stimulation.17 In general, BDNF acts in a proexcitatory manner by augmenting both presynaptic release of glutamate and other peptides in an autocrine fashion,18,19 enhancing postsynaptic responses of the N-methyl-D-aspartate receptor, probably by TrkB-induced phosphorylation.20–23 These may include activation of intracellular signaling molecules such as ERK1/2 in the spinal cord, as has been shown in rat models of colitis as well as urinary cystitis.24,25 In addition, BDNF may modulate GABA release and chloride channels in complex ways.26,27 The most compelling evidence for a role for BDNF comes from antagonist studies, using pharmacological (antibodies and sequestrating proteins) as well as molecular and genetic (antisense nucleotides and conditional knockouts) approaches. As reviewed recently, these studies suggest that BDNF is a prominent mediator of inflammatory pain, contributing significantly to hyperalgesia in these models.6 This seems to be also true for visceral pain syndromes. There is at least 1 study of visceral pain in a model of TNBS-induced colitis in rats, where antagonism of BDNF using a systemically delivered antibody was able to reverse the hyperalgesic response to colorectal distention, although it had no effect in control rats.28
Inflammation can result in an up-regulation of BDNF at multiple levels: peripheral nerves, their target tissues, as well as other structures in the spinal cord. We have previously shown that BDNF in the parenchyma participates in what seems to be a generalized neurotrophin up-regulation in acute pancreatitis.29 In another model of visceral pain, urinary cystitis results in an increase in bladder BDNF expression.25 Of greater relevance to our study, a peripheral up-regulation is also seen in humans with chronic pancreatitis, along with increased expression in perineural and neural structures, with BDNF levels appearing to correlate with pain intensity and frequency.7 This may reflect a role for BDNF in driving peripheral neural sensitization, distinct from its participation as a neurotransmitter in the spinal cord. An increase in BDNF in nociceptive nerves has also been shown in a variety of inflammatory models of somatic pain27 as well as in experimental colitis.24 Finally, nociceptor neurons are not the only cells producing BDNF; among others, microglia can also be an important source of BDNF in pain states, particularly those due to neuropathy.30–33 Thus, inflammatory but not neuropathic pain is attenuated in a nociceptor-specific BDNF knockout model.34 In our study, we focused on the role of neural BDNF and showed that CP is associated with an increase in BDNF in DRGs and corresponding spinal cord regions in pancreas-relevant segments. Further, the results of the retrograde labeling experiments indicate that at least a proportion of this increase is due to increased expression in pancreas-specific neurons and not simply other spinal structures such as microglia. Taken together, our results suggest that CP results in an up-regulation of BDNF in primary afferent neurons and their spinal projections.
We did not specifically examine the factors responsible for up-regulation of BDNF in our study, although NGF is a likely mediator. Nerve growth factor modulates the chemosensitivity of peptidergic nociceptors and is one of the most important “sensitizers” of primary afferent neurons in inflammatory states. In this context, it acts mainly via its specific receptor TrkA activating multiple mechanisms including induction of gene plasticity, involving ion channels and neurotransmitters including SP and CGRP. We have shown previously that TNBS-induced CP is associated with increased and persistent NGF immunoreactivity in the pancreatic parenchyma,4,5 similar to what has been observed in human CP.35 Because BDNF is expressed in the same population of neurons as the other sensory neuropeptides in DRG, it is not surprising that BDNF mRNA and protein also seem to be upregulated by an NGF-dependent mechanism.36,37
We next studied the role of BDNF in mediating sensitization to pancreatic noxious stimuli in our model using a neutralizing anti-BDNF antibody. Most studies with these antagonists in pain models have assessed the short-term effects of single administrations. These results may not predict the effects of long-term blockade in chronically sensitized animals. By giving anti-BDNF “chronically” during a 1-week period by continuous intrathecal infusion, we wished to eliminate fluctuations in drug delivery and also avoid repetitive handling of the animals, which by itself can create confounding problems associated with a classic conditioning model.38 Using this approach, we have showed that antagonism of BDNF suppresses TrkB phosphorylation to the same levels as that seen in rats without pancreatitis. This was important because the major effects of BDNF in nociception seem to be mediated via its high-affinity receptor, full-length TrkB with intrinsic tyrosine kinase activity.
Brain-derived neurotrophic factor antagonism also significantly ameliorates the referred somatic hypersensitivity (as measured by VFF testing) associated with CP as well as pancreatic hyperalgesia (as measured by the response to electrical stimulation). The degree of analgesia produced by BDNF antagonism was most apparent at lower intensities of stimulation. At higher intensities, the effect disappeared, suggesting that the hyperalgesic effects of BDNF are modest at best. This is, however, not completely surprising considering that many other factors are operative at high intensities of noxious stimulation including the release of other neuropeptides such as SP and CGRP. It is possible that the lack of effects at the higher stimulus were due to inadequate dosing; however, the near-complete suppression of TrkB phosphorylation to normal levels argues against this and suggests that BDNF is an important but not exclusive mediator of nociceptive sensitization in CP. In this regard, CP is similar to an inflammatory (where BDNF is clearly pronociceptive) rather than a neuropathic pain model (where both antinociceptive and pronociceptive have been shown by different investigators). Although our results are consistent with neuronally produced BDNF participating in nociceptive signaling in the spinal cord, they do not rule out a possible role for pancreatic BDNF (acting on the peripheral ends of primary neurons)28,39 or glial BDNF in the spinal cord.
In conclusion, we have demonstrated that CP is associated with an up-regulation of BDNF expression in pancreas specific DRG neurons and that BDNF antagonism can reverse the characteristic behavioral changes in response to noxious pancreatic stimulation in this model. This study also validates BDNF signaling as a rational target for treating pain in patients with CP.
Acknowledgments
This study was supported by a grant from the National Institutes of Health (R01 DK073558 to P.J.P.) and P30 DK56339 (Stanford Digestive Disease Center).
Footnotes
None of the authors have any conflict of interest with respect to this article.
References
- 1.DiMagno EP. Toward understanding (and management) of painful chronic pancreatitis. Gastroenterology. 1999;116(5):1252–1257. doi: 10.1016/s0016-5085(99)70031-4. [DOI] [PubMed] [Google Scholar]
- 2.Lieb JG, 2nd, Forsmark CE. Pain and chronic pancreatitis. Aliment Pharmacol Ther. 2009;29(7):706–719. doi: 10.1111/j.1365-2036.2009.03931.x. [DOI] [PubMed] [Google Scholar]
- 3.Anaparthy R, Pasricha PJ. Pain and chronic pancreatitis: is it the plumbing or the wiring? Curr Gastroenterol Rep. 2008;10(2):101–106. doi: 10.1007/s11894-008-0029-4. [DOI] [PubMed] [Google Scholar]
- 4.Winston JH, He ZJ, Shenoy M, et al. Molecular and behavioral changes in nociception in a novel rat model of chronic pancreatitis for the study of pain. Pain. 2005;117(1–2):214–222. doi: 10.1016/j.pain.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 5.Xu GY, Winston JH, Shenoy M, et al. Transient receptor potential vanilloid 1 mediates hyperalgesia and is up-regulated in rats with chronic pancreatitis. Gastroenterology. 2007;133(4):1282–1292. doi: 10.1053/j.gastro.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 6.Merighi A, Salio C, Ghirri A, et al. BDNF as a pain modulator. Prog Neurobiol. 2008;85(3):297–317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Zhu ZW, Friess H, Wang L, et al. Brain-derived neurotrophic factor (BDNF) is upregulated and associated with pain in chronic pancreatitis. Dig Dis Sci. 2001;46(8):1633–1639. doi: 10.1023/a:1010684916863. [DOI] [PubMed] [Google Scholar]
- 8.Yajima Y, Narita M, Matsumoto N, et al. Involvement of a spinal brain-derived neurotrophic factor/full-length TrkB pathway in the development of nerve injury–induced thermal hyperalgesia in mice. Brain Res. 2002;958(2):338–346. doi: 10.1016/s0006-8993(02)03666-1. [DOI] [PubMed] [Google Scholar]
- 9.Bliss WR, Burch B, Martin MM, et al. Localization of referred pancreatic pain induced by electric stimulation. Gastroenterology. 1950;16(2):317–323. [PubMed] [Google Scholar]
- 10.Ceyhan GO, Deucker S, Demir IE, et al. Neural fractalkine expression is closely linked to pain and pancreatic neuritis in human chronic pancreatitis. Lab Invest. 2009;89(3):347–361. doi: 10.1038/labinvest.2008.170. [DOI] [PubMed] [Google Scholar]
- 11.Ceyhan GO, Bergmann F, Kadihasanoglu M, et al. The neurotrophic factor artemin influences the extent of neural damage and growth in chronic pancreatitis. Gut. 2007;56(4):534–544. doi: 10.1136/gut.2006.105528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Sebastiano P, di Mola FF, Buchler MW, et al. Pathogenesis of pain in chronic pancreatitis. Dig Dis. 2004;22(3):267–272. doi: 10.1159/000082798. [DOI] [PubMed] [Google Scholar]
- 13.Di Sebastiano P, di Mola FF, Bockman DE, et al. Chronic pancreatitis: the perspective of pain generation by neuroimmune interaction. Gut. 2003;52(6):907–911. doi: 10.1136/gut.52.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friess H, Shrikhande S, Shrikhande M, et al. Neural alterations in surgical stage chronic pancreatitis are independent of the underlying aetiology. Gut. 2002;50(5):682–686. doi: 10.1136/gut.50.5.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Sebastiano P, Fink T, Weihe E, et al. Immune cell infiltration and growth-associated protein 43 expression correlate with pain in chronic pancreatitis. Gastroenterology. 1997;112(5):1648–1655. doi: 10.1016/s0016-5085(97)70047-7. [DOI] [PubMed] [Google Scholar]
- 16.Bockman DE, Buchler M, Malfertheiner P, et al. Analysis of nerves in chronic pancreatitis. Gastroenterology. 1988;94(6):1459–1469. doi: 10.1016/0016-5085(88)90687-7. [DOI] [PubMed] [Google Scholar]
- 17.Lever IJ, Bradbury EJ, Cunningham JR, et al. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci. 2001;21(12):4469–4477. doi: 10.1523/JNEUROSCI.21-12-04469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matayoshi S, Jiang N, Katafuchi T, et al. Actions of brain-derived neurotrophic factor on spinal nociceptive transmission during inflammation in the rat. J Physiol. 2005;569(Pt 2):685–695. doi: 10.1113/jphysiol.2005.095331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merighi A, Carmignoto G, Gobbo S, et al. Neurotrophins in spinal cord nociceptive pathways. Prog Brain Res. 2004;146:291–321. doi: 10.1016/s0079-6123(03)46019-6. [DOI] [PubMed] [Google Scholar]
- 20.Groth R, Aanonsen L. Spinal brain-derived neurotrophic factor (BDNF) produces hyperalgesia in normal mice while antisense directed against either BDNF or trkB, prevent inflammation-induced hyperalgesia. Pain. 2002;100(1–2):171–181. doi: 10.1016/s0304-3959(02)00264-6. [DOI] [PubMed] [Google Scholar]
- 21.Slack SE, Thompson SW. Brain-derived neurotrophic factor induces NMDA receptor 1 phosphorylation in rat spinal cord. Neuroreport. 2002;13(15):1967–1970. doi: 10.1097/00001756-200210280-00027. [DOI] [PubMed] [Google Scholar]
- 22.Slack SE, Pezet S, McMahon SB, et al. Brain-derived neurotrophic factor induces NMDA receptor subunit one phosphorylation via ERK and PKC in the rat spinal cord. Eur J Neurosci. 2004;20(7):1769–1778. doi: 10.1111/j.1460-9568.2004.03656.x. [DOI] [PubMed] [Google Scholar]
- 23.Slack SE, Grist J, Mac Q, et al. TrkB expression and phospho-ERK activation by brain-derived neurotrophic factor in rat spinothalamic tract neurons. J Comp Neurol. 2005;489(1):59–68. doi: 10.1002/cne.20606. [DOI] [PubMed] [Google Scholar]
- 24.Qiao LY, Gulick MA, Bowers J, et al. Differential changes in brain-derived neurotrophic factor and extracellular signal–regulated kinase in rat primary afferent pathways with colitis. Neurogastroenterol Motil. 2008;20(8):928–938. doi: 10.1111/j.1365-2982.2008.01119.x. [DOI] [PubMed] [Google Scholar]
- 25.Pinto R, Frias B, Allen S, et al. Sequestration of brain derived nerve factor by intravenous delivery of TrkB-Ig2 reduces bladder overactivity and noxious input in animals with chronic cystitis. Neuroscience. 2010;166(3):907–916. doi: 10.1016/j.neuroscience.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 26.Bardoni R, Ghirri A, Salio C, et al. BDNF-mediated modulation of GABA and glycine release in dorsal horn lamina II from postnatal rats. Dev Neurobiol. 2007;67(7):960–975. doi: 10.1002/dneu.20401. [DOI] [PubMed] [Google Scholar]
- 27.Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- 28.Delafoy L, Gelot A, Ardid D, et al. Interactive involvement of brain derived neurotrophic factor, nerve growth factor, and calcitonin gene related peptide in colonic hypersensitivity in the rat. Gut. 2006;55(7):940–945. doi: 10.1136/gut.2005.064063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toma H, Winston JH, Micci MA, et al. Characterization of the neurotrophic response to acute pancreatitis. Pancreas. 2002;25(1):31–38. doi: 10.1097/00006676-200207000-00009. [DOI] [PubMed] [Google Scholar]
- 30.Coull JA, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438(7070):1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 31.Hayward P. Microglial role in neuropathic pain. Lancet Neurol. 2006;5(2):118–119. doi: 10.1016/s1474-4422(06)70342-8. [DOI] [PubMed] [Google Scholar]
- 32.Lu VB, Biggs JE, Stebbing MJ, et al. Brain-derived neurotrophic factor drives the changes in excitatory synaptic transmission in the rat superficial dorsal horn that follow sciatic nerve injury. J Physiol. 2009;587(Pt 5):1013–1032. doi: 10.1113/jphysiol.2008.166306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suter MR, Wen YR, Decosterd I, et al. Do glial cells control pain? Neuron Glia Biol. 2007;3(3):255–268. doi: 10.1017/S1740925X08000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao J, Seereeram A, Nassar MA, et al. Nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not neuropathic pain. Mol Cell Neurosci. 2006;31(3):539–548. doi: 10.1016/j.mcn.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 35.Friess H, Zhu ZW, di Mola FF, et al. Nerve growth factor and its high-affinity receptor in chronic pancreatitis. Ann Surg. 1999;230(5):615–624. doi: 10.1097/00000658-199911000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pezet S, Malcangio M, Lever IJ, et al. Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Mol Cell Neurosci. 2002;21(4):684–695. doi: 10.1006/mcne.2002.1205. [DOI] [PubMed] [Google Scholar]
- 37.Cho HJ, Kim JK, Zhou XF, et al. Increased brain-derived neurotrophic factor immunoreactivity in rat dorsal root ganglia and spinal cord following peripheral inflammation. Brain Res. 1997;764(1–2):269–272. doi: 10.1016/s0006-8993(97)00597-0. [DOI] [PubMed] [Google Scholar]
- 38.Dunbar SA, Yaksh TL. Spinal infusion of N-methyl-D-aspartate antagonist MK801 induces hypersensitivity to the spinal alpha-2 agonist ST91 in the rat. J Pharmacol Exp Ther. 1997;281(3):1219–1225. [PubMed] [Google Scholar]
- 39.Shu XQ, Llinas A, Mendell LM. Effects of trkB and trkC neurotrophin receptor agonists on thermal nociception: a behavioral and electrophysiological study. Pain. 1999;80(3):463–470. doi: 10.1016/S0304-3959(99)00042-1. [DOI] [PubMed] [Google Scholar]